

Abstract
While cigarette smoking (CS) and dysregulated complement are thought to play a central role in age-related macular degeneration (AMD), their exact roles are unknown. The aim of this study is to determine if CS activates complement and if the antioxidant transcription factor Nrf2 modulates this response. In AMD specimens, Nrf2 immunolabeling was strong in the cytoplasm with scattered nuclear labeling of macular retinal pigmented epithelial (RPE) cells that appeared normal, but was decreased and without nuclear labeling in dysmorphic cells overlying drusen, a hallmark AMD lesion. Cigarette smoke extract (CSE) induced Nrf2 nuclear translocation in RPE cells with increased antioxidant and complement gene expression. While CFH protein was not altered by CSE, cell membrane regulator proteins CD46, CD55, and CD59 were decreased, while C3a and C3b, but not iC3b, were increased compared to controls. C5b-9 was increased by CSE, but at sublytic levels only after addition of normal human serum. Nrf2-knockdown enhanced the increase of C3a and C3b from CSE, but not iC3b, C5a, or C5b-9. CSE also increased IL-1b expression and secretion after C3a generation, and was reduced by a C3aR antagonist. In contrast, the Nrf2 activator CDDO-Im restored complement gene expression in RPE cells exposed to CSE. We provide evidence of altered Nrf2 in human AMD, and that CSE induces a pro-inflammatory environment specifically by generating C3a and C3b, and Nrf2 deficiency magnifies this specific complement response.
Keywords: Aging-related disease, complement, innate immunity, Nrf2, oxidative stress
Introduction
Age-related macular degeneration (AMD), a prototypical aging-related disease, is a multifactorial, often blinding condition caused by progressive degeneration of the retinal pigmented epithelium (RPE) and neural retina. AMD affects 30–50 million individuals worldwide, and is a leading cause of legal blindness in developed countries[1]. Although the precise etiology of AMD remains elusive, a large body of evidence implicates the early involvement of oxidative stress[2]. The susceptibility of RPE cells to oxidative stress progressively increases with age, and cumulative oxidative damage contributes to RPE dysfunction and apoptosis, either directly or through inflammatory processes[2]. Complement has emerged as an important element in AMD pathophysiology since the identification of complement in drusen, a hallmark lesion[3]. Importantly, multiple laboratories have identified genetic susceptibility loci for AMD in the complement pathway, including factor H (CFH)[4–6], component 3 (C3)[7, 8], factor B (CFB)[9], component 2 (C2) [9], and factor I (CFI)[10]. Despite the growing awareness, triggers for complement and the specific mechanisms by which dysfunctional complement activation causes this disease remain unknown.
Besides aging, cigarette smoking (CS) is the strongest environmental risk factor associated with AMD[11]. CS is a potent, complex oxidant that contains over 4700 compounds[12]. The susceptibility therefore, appears to be mediated by oxidative processes. We reported that mice exposed to chronic CS develop oxidative damage, RPE apoptosis, and structural features of early AMD[13]. The exact role it plays in disease onset remains unresolved.
Nuclear factor-erythroid 2-related factor 2 (Nrf2), a CNC-basic leucine zipper transcription factor, regulates the inducible expression of antioxidant and cytoprotective enzymes via the cis-acting antioxidant response element (ARE)[14]. Not only does Nrf2 play a critical role in controlling the cellular redox status, but it influences the innate immune response[15]. Importantly, Nrf2 abundance and activity can decline with age and compromise antioxidant protection[16]. Furthermore, systemic complement activity increases with age[17], including increased complement gene expression in the RPE/choroid of old mice[18]. While its potential role in AMD has been previously introduced as reviewed in [19], and recent work suggests that Nrf2 deficiency in mice can lead to elements of an AMD phenotype[20], evidence for altered Nrf2 signaling in human AMD and its specific contribution toward disease development is lacking.
We suggest an integrative etiological model of AMD. The three established AMD susceptibility elements of aging, smoking, and complement, are intimately related and incrementally contribute to AMD, in which oxidative stress is a pivotal factor linking these three elements, and the decline in Nrf2 signaling will magnify this process. Given its central role in AMD, it is of great interest to determine if CS triggers oxidative stress and complement activation in the RPE, and if Nrf2 modulates these molecular events. Herein, we investigated how CS affects complement activation in RPE cells, and determined the influence of Nrf2 signaling on this response.
Materials and Methods
Immunohistochemistry
Human autopsy eyes (n=20) were obtained from the Ocular Pathology Division at the Wilmer Eye Institute after obtaining Institutional Review Board approval. Donors were classified as “unaffected” (n=8) if they had no history of AMD or histopathologic evidence of drusen. Early AMD donors (n=12) had an AMD history, and macular drusen (n=9), but no late stage disease such as geographic atrophy or choroidal neovascularization. Eyes were fixed in 4% formaldehyde, paraffin embedded, and sectioned at 4μm thickness. The sections contained both the macula and periphery so that comparisons in relative staining could be assessed between macula and periphery of the same section. Sections were deparaffinized. Antigens were retrieved using the Target Retrieval System (Dako, Inc., Carpinteria, CA) according to the manufacturer’s instructions. After blocking with horse serum, sections were incubated with rabbit monoclonal anti-human Nrf2 (1:200; Epitomics, Inc. Burlingame, CA) or isotype control rabbit IgG (1:200; Epitomics, Inc.) overnight at 4°C, with biotinylated anti-rabbit IgG (Vectastain®ABC-AP Kit; Vector labs, Burlingame, CA), and then with the ABC-AP reagent. Competition experiments were performed by pre-incubating the anti-Nrf2 antibody for 3h with a 1:200 excess by volume of human Nrf2 protein at 25°C, following our previously published protocol[22]. Full-length human Nrf2 cDNA (NM_006164) was expressed in DH5alpha bacteria, and Nrf2 protein was purified by gel filtration and dialyzed. Slides were incubated with levamizole added to blue substrate working solution (Vector labs) for 10min. Sections were imaged with a light microscope equipped with the Cri-Nuance system (Perkin Elmer Corp, Inc. Hopkinton, MA) to modify melanin pigmentation.
To exclude unintentional, confounding influences in immunostaining across donors, Nrf2 labeling was assessed for tissue distribution and relative intensity within cells of each section, differences between macula and periphery from the same sections, but not across different sections from different donors.
Cell Culture
The established human RPE cell line, ARPE-19,[23] was maintained in Dulbecco’s modified essential medium (DMEM)/Ham’s F12 50/50 mix (Invitrogen Corp, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen) and 2mM L-glutamine solution (Invitrogen) at 37°C in 5% CO2–95% air.
Cigarette smoke extract (CSE) was obtained from Murty Pharmaceuticals, Inc., Lexington, KY, and is 40mg/ml condensate, and 6% nicotine. According to the manufacturer, CSE was prepared by smoking University of Kentucky’s 3R4F Standard Research Cigarettes on an FTC Smoke Machine. The smoke on the filter is calculated by the weight gain of the filter after smoking. The amount of DMSO is calculated that will dissolve a 4% (40mg/mL) solution.
Measurement of Cell Viability
Cell viability was evaluated with the LIVE/DEAD® Assay (Invitrogen) according to the manufacturer’s protocol. Cells were grown to visual confluence in 96-well plates, serum-starved for 24h, and treated with 0–500μg/ml CSE in DMSO for 24h. The number of live and dead cells was counted using the Cellomics ArrayScan VTI HCS Reader (Thermo Fisher Scientific, Waltham, MA). Hoechst staining was used to identify the total number of the cells.
Measurement of Cellular Reactive Oxygen Species
Cells were grown to confluence in 96-well plates and treated with CSE for 24h. To measure O2−, cells were labeled with 10μM dihydroethidium (DHE; Invitrogen) for 30min in the dark. Ethidium-DNA fluorescence was measured using a microplate reader (excitation 485 nm, emission 590 nm.)
Cellular Glutathione Assay
Glutathione was measured using a glutathione assay kit (Cayman Chemicals, Ann Arbor, MI), according to the manufacturer’s instructions. Cellular proteins (20–30μg) were deproteinized by phosphoric acid, and the amount of 5-thio-2-nitrobenzoic acid produced was measured in the supernatant. The absorbance was measured with a Synergy HT microplate reader (Synergy HT Biotek Laboratories, Winooski, VT).
Mitochondrial NADPH-dependent Oxidoreductase Activity
Mitochondrial function was evaluated by the CellTiter 96 Aqueous Cell Proliferation Assay Kit (Promega, Madison, WI), following the manufacturer’s instructions. Cells were grown to confluence in 96-well plates, serum-starved for 24h, and treated with 0–500μg/ml CSE for 24h.
Quantitative Real-time RT-PCR
Cells were grown to confluence in 6-well plates, serum-starved for 24h, and treated with 50–250μg/ml CSE or DMSO for 8–24h. Total RNA was isolated using an RNeasy Mini Kit (Qiagen, Valencia, CA), with on-column DNA digestion by RNase-free DNase (Qiagen), following the manufacturer’s instructions. cDNA was synthesized using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA). Quantitative real-time PCR were performed using TaqMan Gene Expression Assays (Applied Biosystems) on a StepOnePlus Real-Time PCR System (Applied Biosystems). Primer/probe catalogue numbers (Applied Biosystems) appear in Table S1. Data were analyzed by the comparative threshold cycle method with PPIA (peptidylprolyl isomerase A) as an internal control.
Immunoblotting
Whole cell lysates were prepared using RIPA buffer (Sigma, Inc., St. Louis, MO). Proteins were resolved on 4–12% Bis-Tris gels (NuPAGE precast gels, Invitrogen), transferred to a nitrocellulose membrane, and then probed with sheep polyclonal anti-CFH mouse (1:500; Abcam, Cambridge, MA), monoclonal rat anti-mouse C3a (1:500; BD Bioscience Inc., San Jose, CA), monoclonal anti-C3b (1:500; Abcam), goat anti-human C3 for iC3b (Complement Technology, Inc. Tyler, TX), mouse monoclonal anti-CD46 (1:500; Abcam), mouse monoclonal anti-CD55 (1:1000; Abcam), and goat polyclonal anti-CD59 (1:500; R&D, Minneapolis, MN), and rabbit monoclonal anti-GAPDH (1:5000; Abcam). Membranes were then incubated with HRP conjugated secondary antibody. Signal was visualized with ECL Plus WB Reagents (GE Healthcare, Buckinghamshire, UK).
Nuclear extracts were prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce, Rockford, IL) according to the manufacturer’s instructions. Nuclear proteins were resolved by SDS-PAGE, transferred onto nitrocellulose membranes, and incubated with polyclonal rabbit anti-Nrf2 (1:300 dilution, Santa Cruz Biotechnology, Santa Cruz, CA) or polyclonal goat anti-lamin B (1:200 dilution, Santa Cruz Biotechnology) followed by incubation with horseradish peroxidase-conjugated secondary antibody. Immune complexes were visualized with SuperSignal West Pico chemiluminescent substrate (Pierce).
Enzyme Immunoassay (ELISA)
Human C3a, iC3b, C5a, IL-1b, and SC5b-9 level in the medium was determined with a commercial ELISA kit (Quidel, Inc., San Diego, CA), according to the manufacturer’s instructions. Protein carbonyls were measured in the cell supernatant using an OxiSelect Protein Carbonyl ELISA kit following the manufacturer’s protocol (Cell Biolabs, San Diego, CA).
Transfection of Small Interfering RNA (siRNA)
siRNA targeting human Nrf2 and non-targeting control siRNA used in this study was previously described[24]. Cells were transfected when cultures were 70% confluent with either 15nM Nrf2 siRNA or control siRNA using 2.5μl/ml Lipofectamine 2000 (Invitrogen, Carlsbad, CA) for 24 h. Cells were serum starved for 24h, and treated with 50–125μg/ml CSE or DMSO. After 24h, cells were processed for RNA isolation and real-time PCR analysis or protein isolation for immunoblotting or ELISA.
Statistical Analysis
The difference between groups was statistically compared by Student’s t-test or analysis of variance (one-way ANOVA) for groups of more than two with post hoc testing using Tukey-Kramer HSD test. All data are expressed as the mean ± SEM. All statistical analyses were performed using JMP version 6.0.3 software (SAS, Cary, NC). A P-value of <0.05 was considered statistically significant. Experiments were conducted in duplicate or triplicate and performed at least 3 times.
Results
Nrf2 Immunolabeling Corresponds with AMD Histopathology
To demonstrate relevance of Nrf2 signaling in AMD, we assessed the distribution of Nrf2 immunolabeling in the RPE of unaffected controls from a wide age range (n=8; mean, 56yrs; range, 8 mo-84yrs) and in early AMD (n=12; mean, 80yrs, range, 60–96yrs; Table 1). In the absence of oxidative stress, Nrf2 is tethered to Kelch-like ECH-associating protein 1 (Keap1) in the cytoplasm and is degraded by the proteasome. With oxidative stress, Nrf2 expression is induced and importantly, Nrf2 translocates to the nucleus where it binds to ARE sequences of antioxidant genes to initiate transcription. Cytoplasmic Nrf2 staining of macular RPE was absent (n=6) or mild (n=2), and absent in all peripheral RPE of unaffected control eyes, suggesting absence of an Nrf2 inducing signal (Fig. S1). In contrast, all 12 AMD specimens displayed a mosaic distribution of Nrf2 staining in the RPE that inversely correlated with AMD histopathology (Fig. 1; Table 1). To demonstrate the specificity of the anti-Nrf2 immunostaining, the anti-Nrf2 antibody was preincubated with Nrf2 protein, which competed out the staining (Fig. S2). Cytoplasmic Nrf2 staining was consistently seen in morphologically preserved RPE, but was less intense in adjacent, dysmorphic RPE overlying drusen in all AMD specimens. Nuclear Nrf2 staining in the RPE, a sign of Nrf2 translocation to the nucleus, was seen in 83% of macular AMD samples at an average frequency of 5.3/200 RPE nuclei. When present, nuclear Nrf2 staining was more common in RPE with preserved morphology than in degenerated RPE overlying drusen. Cytoplasmic or nuclear Nrf2 labeling was not seen in peripheral RPE of any AMD specimen. These observations suggest that Nrf2 signaling is minimal in an unstimulated environment, increased in morphologically unaffected RPE in early AMD because of inciting oxidative stress, but Nrf2 is decreased and insufficient to provide adequate cytoprotection against oxidative stress in degenerating RPE overlying drusen.
Table 1.
Nrf2 Immunolabeling in the RPE of Unaffected and Early AMD donors.
| Donor | Age (Yrs) | Gender | D-E (Hrs) | Nrf2 labeling
|
|||
|---|---|---|---|---|---|---|---|
| Macula | Periphery | ||||||
|
| |||||||
| Nucleus | Cytoplasm | Nucleus | Cytoplasm | ||||
|
| |||||||
|
AMD
| |||||||
| 1 | 60 | M | 28 | Yes | Yes | No | No |
| 2 | 61 | M | 20 | Yes | Yes | No | No |
| 3 | 68 | F | 20 | Yes | Yes | No | No |
| 4 | 69 | F | 19 | Yes | Yes | No | No |
| 5 | 83 | F | 12 | Yes | Yes | No | No |
| 6 | 83 | F | 14 | No | Yes | No | No |
| 7 | 84 | M | 44 | Yes | Yes | No | No |
| 8 | 87 | F | 70 | Yes | Yes | No | No |
| 9 | 87 | M | 24 | Yes | Yes | No | No |
| 10 | 90 | M | 20 | Yes | Yes | No | No |
| 11 | 94 | F | 8 | Yes | Yes | No | No |
| 12 | 96 | M | 5 | No | Yes | No | No |
|
| |||||||
|
Unaffected
| |||||||
| 1 | 8 months | M | 63 | No | No | No | No |
| 2 | 37 | M | 23 | No | No | No | No |
| 3 | 50 | F | 3 | No | No | No | No |
| 4 | 64 | M | 65 | No | No | No | No |
| 5 | 67 | F | 67 | No | No | No | No |
| 6 | 69 | M | 46 | No | Yes | No | No |
| 7 | 82 | F | 45 | No | No | No | No |
| 8 | 84 | F | 75 | Yes | Yes | No | No |
M: male; F: female; D-E: death to enucleation time.
Figure 1.
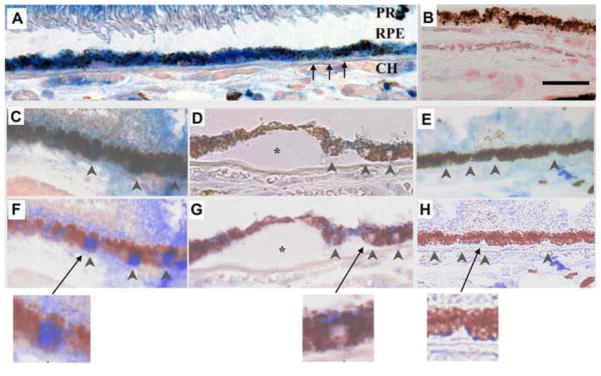
Nrf2 immunolabeling in a 60-year-old Caucasian female with early AMD. A) Macular RPE with normal, cuboidal morphology have prominent cytoplasmic Nrf2 labeling (blue). B) IgG control. C) Normal macular RPE with both cytoplasmic and nuclear Nrf2 labeling (arrowheads). D) Dysmorphic macular RPE overlying drusen have lighter cytoplasmic labeling than cuboidal RPE from the same section. Nuclei (arrowheads) do not stain for Nrf2. E) Peripheral RPE with minimal Nrf2 labeling in the cytoplasm and no nuclear labeling (arrowheads). F–H) Same images as C–E, respectively, after Nuance software has converted melanin to maroon to improve visualization of Nrf2 labeling in the RPE. The arrow points to the inset of an enlarged image showing nuclear Nrf2 staining in a normal macular RPE cell (F), lack of nuclear Nrf2 staining in a dysmorphic macular RPE cell overlying a druse (*) (G), and lack of Nrf2 staining in a peripheral RPE cell (H). CH, choroid, RPE, retinal pigment epithelium, Black arrows point to Bruch’s membrane. Bar=25μm.
CSE Induces Oxidative Stress, Nrf2 Signaling, and Oxidative Damage and Nrf2 Knockdown Magnifies Oxidative Damage
To examine how CS and Nrf2 deficiency influence complement, we first tested the response of Nrf2 competent RPE cells exposed to viable doses of cigarette smoke extract (CSE). Treatment with 0–250μg/ml CSE did not influence ARPE-19 cell viability (Fig. S3), but generated reactive oxygen species (ROS), as indicated by superoxide anion formation, in a dose dependent manner compared to controls (p<0.05; Fig. 2A). This increased oxidative stress by CSE induced Nrf2 signaling, as suggested by nuclear Nrf2 protein accumulation (Fig. 2B), and the expression of GCLM, HO1, and NQO1, three known Nrf2-responsive antioxidant genes, compared to vehicle treated cells (Fig. 2C). This Nrf2 signaling response provided incomplete protection because CSE reduced cellular glutathione (Fig. 2D) and increased protein carbonylation, a marker of protein oxidation (Fig. 2E), in a dose dependent fashion, compared to vehicle controls.
Figure 2.

CSE induces ROS and an Nrf2 signaling response. A) DHE assay showing dose dependent superoxide anion production by ARPE-19 cells after 0–250μg/ml CSE for 24h. B) Representative immunoblot of Nrf2 protein in nuclear extracts from ARPE-19 cells. Lamin B1 served as a loading control. Below, graph quantifying nuclear Nrf2 protein abundance. Data are presented as fold change over control. C) Gene expression of Nrf2-dependent antioxidant genes, GCLM, HO-1, and NQO1, as determined by RT-qPCR. D) Graph showing decreased cellular total glutathione after CSE compared to controls. E) Graph showing increased protein carbonylation after CSE compared to controls. *p<0.05, ** p< 0.01, ***, p<0.001 compared to control. For all assays, n=3 ind exp.
We next rendered RPE cells Nrf2 deficient using an siRNA strategy and tested their response to viable doses of CSE. Nrf2-knockdown (Nrf2-KD) decreased Nrf2 mRNA by 70% and downregulated NQO1 in the absence or presence of a viable dose of (125μg/ml; Fig. 3A). Nrf2-KD further increased CSE induced cellular superoxide anion (Fig. 3B), magnified the decrease in cellular glutathione (Fig. 3C) and the increase in protein carbonylation (Fig. 3D) beyond CSE alone. Because superoxide anion is produced predominantly in the mitochondria, we also tested mitochondrial function and found a magnified reduction in NADPH-dependent oxidoreductase activity, an indicator of mitochondrial injury with Nrf2-KD and 125μg/ml CSE (Fig. 3E), over that caused by 125μg/ml CSE alone with or without siRNA control. These changes induced by Nrf2-KD and 125μg/ml CSE however, were not sufficient to decrease cell viability (Fig S4).
Figure 3.

Nrf2-knockdown impairs the antioxidant response following CSE, with increased cellular superoxide anion. Cells transfected with 15nM non-targeting siRNA control (CTLR) or Nrf2 siRNA were treated with DMSO vehicle (control) or 125μg/ml CSE for 24h. A) Gene expression of Nrf2 and NQO-1. Changes are presented as fold change over vehicle control. B) Superoxide anion, measured by DHE assay, in ARPE-19 cells after 125μg/ml CSE. C) Graph showing decreased cellular total glutathione by 125μg/ml CSE is magnified by Nrf2-KD. D) Graph showing increased protein carbonylation by125μg/ml CSE is magnified by Nrf2-KD. E) Graph showing decreased NAPDH oxidoreductase activity by 125μg/ml CSE is magnified by Nrf2-KD compared to siRNA control with CSE. Note: B–E, since values for vehicle control and siRNA CTRL were similar, the vehicle control bars were omitted. For all assays, n=3 ind exp. *p<0.05, **p<0.01, ***p<0.001.
CSE Triggers Complement Activation, and Nrf2 knockdown magnified C3a and C3b generation in ARPE-19 Cells
While complement components in general, are produced in the liver, CS can influence complement expression in local tissues[25]. We therefore, surveyed the transcription of selected genes encoding complement components (C3 and C5) and an alternative pathway activator (CFB) in ARPE-19 cells, and found that all were upregulated in cells treated with 50–125μg/ml CSE for 24h (Fig. 4). While C5 was upregulated by CSE, its expression level was substantially lower than C3. These results prompted us to evaluate the extent of complement activation by measuring the generation of anaphylatoxins (C3a, C5a), opsonins (C3b, iC3b), and C5b-9 complexes. With preserved Nrf2 signaling, C3a is generated starting at 50μg/ml CSE in a dose dependent fashion, compared to vehicle controls (Fig. 5) while C5a was not detected using ELISA under our experimental conditions (data not shown). CSE (125μg/ml) increased the opsonin C3b 1.5 fold (p<0.05; n=3 ind exp) compared to vehicle controls by Western blot, but not iC3b in the supernatant by ELISA or in whole cell lysates by Western blot (data not shown). Using ELISA, C5b-9 was formed at very low levels after 125μg/ml CSE unless 20% normal human serum (NHS) was added to CSE treated cells, at which a 2.1 fold increase in C5b-9 was observed relative to vehicle controls that were supplemented with NHS (p<0.05; n=3 ind exp).
Figure 4.

CSE triggers the expression of complement-related genes in ARPE-19 cells. Cells were treated with DMSO vehicle (CTLR) or 50–125μg/ml CSE for 24h. Transcript levels were determined by RT-qPCR. Data are presented as fold change over vehicle control. n=3 ind exp. *p<0.05.
Figure 5.
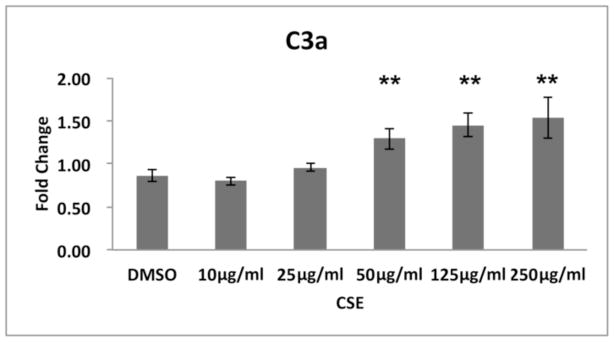
C3a is increased in a dose dependent manner by CSE. ARPE-19 cells were treated with DMSO vehicle or 10–500μg/ml CSE for 24h. C3a protein in the supernatant was measured by ELISA. Data are presented as fold change over vehicle control. n=3 ind exp. **p<0.01.
The generation of C3a and C3b after CSE was magnified by Nrf2-KD. Nrf2-KD magnified the CSE-induced expression of C3, C5, and CFB mRNAs, compared with control siRNA-transfected cells (Fig. 6). Since Nrf2-KD enhances CSE-induced complement activation, we examined whether pharmacological activation of Nrf2 attenuates the effects of CSE using the triterpenoid CDDO-Im. ARPE-19 cells pretreated with 100nM CDDO-Im for 24h exhibited higher expression of GCLM, HO-1, and NQO1 compared with vehicle-pretreated cells either with or without 125μg/ml CSE (Fig. 7A). CDDO-Im attenuated the CSE-induced expression of C3, C5, and CFB mRNA when compared with CSE alone (Fig. 7B). These data underscore a crucial role of Nrf2 in modulating the transcription of complement factors.
Figure 6.
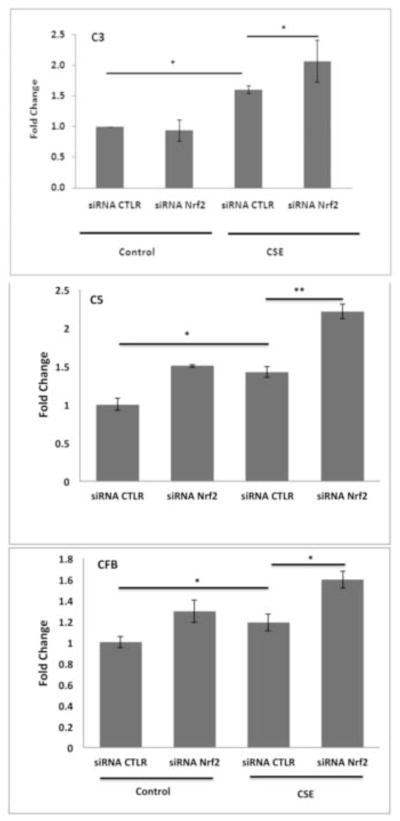
Nrf2-KD magnifies the increase of complement-related gene expression by CSE. Cells transfected with 15nM non-targeting control siRNA (CTRL) or siRNA Nrf2 were treated with DMSO vehicle control or 125μg/ml CSE for 24h. Transcript levels were determined by RT-qPCR. Data are presented as fold change over vehicle control n=3 ind exp. Since values for vehicle control and siRNA CTRL were similar, the vehicle control bars were omitted. *p<0.05; **p<0.01.
Figure 7.
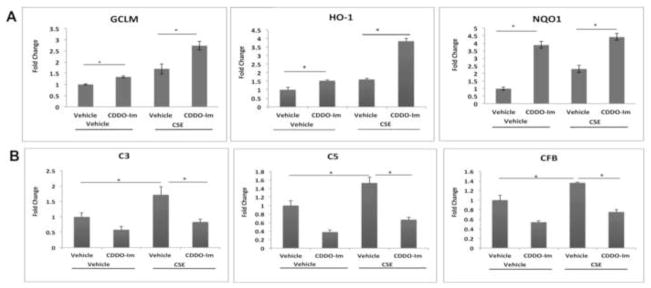
CDDO-Im potentiates the antioxidant response and reduces complement gene expression. ARPE-19 cells were pretreated with DMSO vehicle control or 100nM CDDO-Im for 24h, and then with DMSO vehicle control or 125μg/ml CSE for an additional 24h. Gene expression was assessed by RT-qPCR. Data are presented as fold change over vehicle control. n=3 ind exp. A) Nrf2-dependent antioxidant gene expression. B) Complement factor gene expression. *p<0.05.
While 25μg/ml CSE alone did not generate C3a, Nrf2-KD cells treated with 25μg/ml CSE generated C3a above that of siRNA controls treated with CSE (Fig. 8A). With 125μg/ml CSE, Nrf2 competent cells generated C3a, an effect that was magnified by Nrf2-KD compared to siRNA controls treated with CSE (Fig 8B). Likewise, the degree of Nrf2-KD influenced C3a generation. With more complete Nrf2-KD (93%), cells generated more C3a compared to cells transfected with non-targeting siRNA, in the absence of CSE treatment (Fig. S5). This is in contrast to cells with 70% Nrf2-KD that did not produce detectable C3a. Nrf2-KD also increased the opsonin C3b, another product of C3 convertase above that induced by 125μg/ml CSE with siRNA control (Fig. 8C,D). iC3b, a downstream product of C3b and an opsonin, was not detected in the supernatant by ELISA or in whole cell lysates by Western blot after CSE with or without Nrf2-KD (Fig 8E). C5a remained undetectable by ELISA in the supernatant of Nrf2-KD cells treated with 125μg/ml CSE (data not shown), and the degree of C5b-9 formation by CSE in the presence of NHS was not increased by Nrf2-KD (Fig. 9). This degree of C5b-9 formation was not sufficient to decrease cell viability (Fig S4).
Figure 8.

Nrf2-knockdown magnifies the increase in C3a and C3b activation products after CSE. Cells transfected with 15nM non-targeting siRNA control (CTRL) or siRNA Nrf2 were treated with DMSO vehicle control or CSE. A) Graph showing no C3a generation after 25μg/ml CSE alone, but an increase in C3a, as measured by ELISA, when combined with Nrf2-KD. B) Graph showing C3a generation after 125μg/ml CSE alone, which is magnified when combined with Nrf2-KD. C. Representative immunoblot of C3b after treatment with 125μg/ml CSE or vehicle control with siRNA CTRL or siRNA Nrf2. GAPDH was used as a loading control. D. Graph of increased C3b after 125μg/ml CSE and Nrf2-KD over CSE alone. E. Representative immunoblot of iC3b after treatment with either siRNA CTLR or Nrf2 siRNA, and then DMSO vehicle control or 125μg/ml CSE for 24h. Recombinant iC3b (2ng) served as a positive control. GAPDH was used as a loading control. *p<0.05. For all assays, n=3 ind exp.
Figure 9.

C5b-9 is induced by CSE in the presence of human serum supplementation, but it is not further increased by Nrf2-KD. ARPE-19 cells transfected with 15nM non-targeting siRNA control (CTRL) or Nrf2 siRNA were treated with DMSO vehicle control or 125μg/ml CSE with NHS for 24h. C5b-9 complexes are increased by CSE, as measured by ELISA, but the increase is not magnified by Nrf2-KD. n=3 ind exp. *p<0.05.
Complement activity is influenced by its regulators. Thurman et al showed that oxidative stress reduces complement regulator abundance[26]. We therefore quantified complement regulators that might influence complement activation, and evaluated the impact of Nrf2 KD. While CFH protein was unchanged by Western analysis (data not shown), CD46, CD55, and CD59 were decreased by 125μg/ml CSE, an effect that was not altered by Nrf2-KD (Fig. 10). These results indicate that CSE activates complement in part, by transcriptional activation and in part, by reducing complement regulator protein abundance. Under our experimental conditions, CSE primarily targets C3, with the generation of C3a and C3b, and Nrf2 KD magnifies this response without influencing C5, C5b-9, and complement regulators.
Figure 10.

CSE decreases RPE cell membrane complement regulators. A) Representative immunoblots of CD46, CD55, and CD59 after treatment with non-targeting siRNA control (CTLR) or Nrf2 siRNA, and then DMSO vehicle control or 125μg/ml CSE for 24 h. GAPDH was used as a loading control. Graphs of B) CD46, C) CD55, and D) CD59 after and siRNA Nrf2 and CSE treatment showing a decrease by CSE, but no further reduction by Nrf2-KD. n=3 ind exp. *p<0.05.
C3a induces IL-1b through the C3aR
C3a promotes a local proinflammatory environment by inducing potent cytokines such as IL-1b, through its receptor C3aR. CSE (125μg/ml) increased IL-1b mRNA (Fig. 11A) and protein (Fig. 11B), an effect that was magnified by Nrf2-KD. IL-1b expression was decreased after treatment with 1μM SB290157, an established C3aR blocker, with or without Nrf2-KD in CSE treated cells (Fig. 11C), which suggests that IL-1b production by CSE and Nrf2-KD is at least partially mediated through C3a-C3aR interaction.
Figure 11.

IL-1b is induced by CSE through C3a interacting with the receptor C3aR, and this induction is magnified by Nrf2-KD. A) Graph showing increased IL-1b mRNA, as measured by RT-qPCR, after 125μg/ml CSE, with magnified expression when combined with Nrf2-KD. B) Graph showing increased secreted IL-1b protein, as measured by ELISA, after 125μg/ml CSE, with magnified expression when combined with Nrf2-KD. C) Graph showing IL-1b mRNA induction is mediated by C3aR. ARPE-19 cells were transfected with siRNA control siRNA or siRNA Nrf2 and then DMSO vehicle control or 125μg/ml CSE for 24h. Some cells were treated with the C3aR antagonist SB290157 (1μM; C3aRA). n=3 ind exp for all assays. *p<0.05, **p<0.01, ***p<0.001.
Discussion
Cigarette smoking is a powerful risk factor for AMD, and Nrf2 signaling has been hypothesized to play a protective role against this complex chemical oxidant. Evidence linking impaired Nrf2 signaling with human AMD has been lacking. Our study addresses this deficiency by observing Nrf2 immunolabeling in human maculas that correlated with AMD histopathology. Our work starts to unravel the complex protective effect of Nrf2 signaling in RPE cells against the potential toxicity of a complex chemical oxidant like cigarette smoke. We demonstrate that RPE cells exposed to CSE can activate complement by both transcriptionally upregulating complement components and reducing complement regulator protein abundance, which activates complement especially at C3, generating the anaphylatoxin C3a and the pro-inflammatory opsonin C3b without inducing the anti-inflammatory opsonin iC3b. We find that Nrf2 deficiency magnifies this C3 specific response without augmenting late activated complement components (C5a, C5b-9). Importantly, C3a generation was linked with induction of IL-1b, a powerful pro-inflammatory cytokine, through interaction with its receptor C3aR. Collectively, these results indicate that in RPE cells, CSE activates complement specifically at C3, and Nrf2 deficiency magnifies this effect to promote a pro-inflammatory microenvironment.
The Nrf2 immunolabeling pattern in the RPE of human eyes correlated with AMD histopathology. In early AMD maculas, Nrf2 labeling was stronger in the cytoplasm and had some nuclear Nrf2 labeling, which is suggestive of nuclear translocation during Nrf2 signaling, in morphologically normal RPE compared to dysmorphic RPE overlying drusen within the same tissue sections where cytoplasmic labeling was weak, and nuclear Nrf2 labeling was absent. These findings suggest that in early AMD, the RPE is stressed and elicits a protective response to preserve cellular function that includes Nrf2 induction and nuclear translocation. In pathologic areas such as overlying drusen, Nrf2 labeling was relatively decreased and nuclear staining was absent, which suggests an inadequate Nrf2 signaling response. Both aging and oxidative stress mediated diseases are associated with decreased Nrf2 signaling[16, 27]. AMD joins these chronic, oxidative stress diseases influenced by Nrf2 signaling.
In our experiments, sublethal oxidative stress activated complement. Several factors likely contribute to this activation. First, oxidative stress can trigger NF-κB, as reviewed in[28], which in turn, can activate innate immunity by degrading iκB-α through MyD88 dependent and independent signaling[15]. We found that CSE caused a dose dependent decrease in glutathione of up to 60%, exceeding the 40% depletion level that render cells vulnerable to oxidative injury[29], and was sufficient to induce transcriptional upregulation of complement factors. Since Thimmulappa et al showed that glutathione reduces NF-κB activation[15], we suggest that the decrease in glutathione by CSE contributed to the transcriptional activation of complement components by altering the cell’s redox status. Secondly, we found that CSE reduced the protein abundance of complement regulators CD46, CD55, and CD59, which would promote complement activation. This decrease may be a general response to oxidative stress since Thurman et al found that H2O2 depleted CD46, CD55, and CD59[26]. Likewise, we previously reported that CD46 and CD59 were decreased due to their release in exosomes and apoptotic blebs after RPE cells were exposed to oxidized lipids[30]. We suggest that the release of these regulators in exosomes and blebs could explain their decline after cells are exposed to oxidative stress. Lastly, cigarette smoke can directly activate the alternative pathway by cleaving an internal thiolester bond in C3,[31] which both diminishes C3’s ability to bind to Factor H[32] and increases Factor B cleavage[31].
We simulated a heightened oxidative stress environment by Nrf2 knockdown, which had a very targeted impact on complement by specifically magnifying the generation of C3a and C3b over CSE alone, without influencing other biologic consequences of complement such as iC3b, C5a, and C5b-9 formation. While the transcription of C3, C5, and CFB was magnified over CSE alone, Nrf2 deficiency did not augment the reduction of complement regulator proteins by CSE. Together, these results indicate that Nrf2 deficiency magnifies the complement response by augmenting the transcription of complement components, but that complement regulator abundance was sufficient to control C5b-9 generation at sublytic levels.
The impact at C3 induced by CSE and magnified by Nrf2 deficiency, has significant relevance to AMD because C3 activation products promote a pro-inflammatory microenvironment. While the anaphylatoxin C3a has been identified in drusen of AMD specimens[33], it has not been a focus of many studies. C3a promotes inflammation by transcriptionally inducing a number of cytokines including the powerful IL-1b[34]. Our results suggest that C3a could provide directional cues for innate immunity by inducing IL-1b through interaction with C3aR on either dendritic cells, or as in our experiments, through an autocrine pathway since the RPE expresses C3aR[33]. This finding is strengthened by Wu et al, who showed that IL-1b production is linked to interaction of C3a with C3aR in a rodent intestinal ischemia-reperfusion model[35]. IL-1b also increases Factor B in RPE cells[36], a key alternative cascade activator, as was found in our experiments, which could additionally promote a positive feedback loop, leading to unwanted chronic complement activation. Doyle et al demonstrated that drusen can activate NLRP3 inflammasomes, which through caspase-1, cleaved and secreted IL-18 as protection against AMD[37]. In their study, IL-1b, which is also activated by the inflammasome, was not transcriptionally induced. Since IL-18, which is constitutively expressed and activated by the inflammasome, restrains the effects of IL-1b, a shift in the abundance of either cytokine could initiate disease[38]. If the RPE is exposed to a trigger like CSE that transcriptionally activates IL-1b, the protective effect of IL-18 might be compromised due to an imbalance between IL-1b and IL-18.
The increase in the opsonin C3b, but not iC3b, likewise, promotes inflammation by inducing pro-inflammatory cytokines such as IL-8 and TNF-α during the removal of apoptotic debris[39]. In contrast, iC3b opsonins foster an anti-inflammatory environment by stimulating TGF-b and IL-10[39]. RPE apoptosis is a documented event in AMD[40]. In late-onset diseases such as AMD, local alternative pathway regulation may gradually be overwhelmed by cellular injury or the accumulation of debris[41]. Chronic or excessive inflammation triggered by C3b rather than iC3b opsonins, during the removal of apoptotic debris could produce unintended tissue injury.
Sublytic C5b-9 generated by CSE exposure can lead to two consequences that can promote AMD. First, Thurman et al reported that C5b-9 mediated upregulation of VEGF, the central pro-angiogenic cytokine involved in neovascular AMD[26]. Secondly, Laudisi et al recently reported that C5b-9 activates the NLRP3 inflammasome[42]. Thus, the inflammasome, which also has been causally linked with geographic atrophy[43], is now known to intimately interact with complement. These innate immunity components must be kept in delicate balance in order to maintain a protective response. We raise the possibility that a trigger such as CSE could induce IL-1b via C3a, which could imbalance this protective response and instead, mediate pathologic changes during AMD, an effect that can be magnified by Nrf2 deficiency.
Joseph et al recently found that H2O2 activated the lectin pathway through natural IgM antibodies (IgM-C2) that recognize phospholipid cell surface modifications on oxidatively stressed RPE cells[44]. Together with our findings, we propose that oxidative stress, through cigarette smoking for example, will activate both early (C3a and C3b) and late (C5b-9) components that can contribute to cell damage, especially since complement regulator abundance is reduced, or activate the lectin pathway. The augmented oxidative stress generated by Nrf2 deficiency will magnify C3a and C3b generation, or amplify oxidized phospholipid modifications on the RPE that could ultimately heighten lectin pathway activation, and subsequent RPE injury. The mechanism by which oxidative stress activates complement appears to be complex, and due to our incomplete understanding, further study is warranted.
In conclusion, we provide the first evidence that Nrf2 is altered in human AMD specimens, and that Nrf2 deficiency promotes cellular oxidative damage and a pro-inflammatory environment by selectively and unambiguously targeting the generation of the anaphylatoxin C3a, which led to increased IL-1b secretion, and the pro-inflammatory opsonin C3b. While the RPE cell has significant cytoprotective capability due in part to Nrf2 signaling[44], aging and/or chronic exposure to a complex and powerful oxidant like CS could impair Nrf2 signaling and promote inflammation sufficient to permit AMD lesion formation. Given the delicate balance of the complement system, a logical treatment strategy would be to neutralize oxidative stress, as suggested by CDDO-Im in our studies, so that complement remains controlled and protective.
Supplementary Material
Highlights.
CSE specifically induces C3a and C3b, and reduces complement regulators in RPE cells.
C3a induces IL-1b through its C3a receptor.
Nrf2 deficiency magnifies this specific pro-inflammatory C3a and C3b generation.
Nrf2 labeling is decreased in diseased RPE overlying drusen in human AMD
Acknowledgments
Mike Sporn, MD, Dartmouth School of Medicine, for providing CDDO-Im; EY14005 (JTH), EY019904 (JTH), P50HL107169 (SB), R01CA140492 (SB), Thome Foundation Award in AMD (JTH), Research to Prevent Blindness Senior Scientist Award (JTH), Wilmer Core Grant, EY001765, Unrestricted grant from RPB, Gifts from the Merlau family and Aleda Wright. JTH is the Robert Bond Welch Professor.
Abbreviations
- AMD
age-related macular degeneration
- CDDO-Im
2-Cyano-3,12-dioxooleana-1,9-dien-28-imidazolide
- GCLM
glutamate-cysteine ligase, modifier subunit
- HO1
heme oxygenase-1
- NQO1
NAD(P)H dehydrogenase, quinone-1
- Nrf2
Nuclear factor-erythroid 2-related factor 2
- RPE
retinal pigmented epithelium
Footnotes
Conflict of interest: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Lei Wang, Email: lwang9@jhmi.edu.
Naoshi Kondo, Email: nskondo@gmail.com.
Marisol Cano, Email: mcano1@jhmi.edu.
Katayoon Ebrahimi, Email: kebrahi2@jhmi.edu.
Takeshi Yoshida, Email: TYoshida@doheny.org.
Bradley P. Barnett, Email: bradpbarnett@gmail.com.
Shyam Biswal, Email: sbiswal@jhsph.edu.
References
- 1.Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration--emerging pathogenetic and therapeutic concepts. Ann Med. 2006;38:450–471. doi: 10.1080/07853890600946724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–134. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 3.van der Schaft TL, Mooy CM, de Bruijn WC, de Jong PT. Early stages of age-related macular degeneration: an immunofluorescence and electron microscopy study. Br J Ophthalmol. 1993;77:657–661. doi: 10.1136/bjo.77.10.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edwards AO, Ritter R, III, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement Factor H Polymorphism and Age-Related Macular Degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 5.Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA. Complement Factor H Variant Increases the Risk of Age-Related Macular Degeneration. Science. 2005 doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 6.Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, Sangiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement Factor H Polymorphism in Age-Related Macular Degeneration. Science. 2005 doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, Clayton DG, Hayward C, Morgan J, Wright AF, Armbrecht AM, Dhillon B, Deary IJ, Redmond E, Bird AC, Moore AT. Complement C3 Variant and the Risk of Age-Related Macular Degeneration. N Engl J Med. 2007 doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- 8.Maller JB, Fagerness JA, Reynolds RC, Neale BM, Daly MJ, Seddon JM. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet. 2007;39:1200–1201. doi: 10.1038/ng2131. [DOI] [PubMed] [Google Scholar]
- 9.Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, Cramer K, Neel J, Bergeron J, Barile GR, Smith RT, Hageman GS, Dean M, Allikmets R. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–462. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kondo N, Bessho H, Honda S, Negi A. Additional evidence to support the role of a common variant near the complement factor I gene in susceptibility to age-related macular degeneration. Eur J Hum Genet. 2010;18:634–635. doi: 10.1038/ejhg.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomany SC, Wang JJ, Van Leeuwen R, Klein R, Mitchell P, Vingerling JR, Klein BE, Smith W, De Jong PT. Risk factors for incident age-related macular degeneration: pooled findings from 3 continents. Ophthalmology. 2004;111:1280–1287. doi: 10.1016/j.ophtha.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 12.Church DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect. 1985;64:111–126. doi: 10.1289/ehp.8564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujihara M, Nagai N, Sussan TE, Biswal S, Handa JT. Chronic cigarette smoke causes oxidative damage and apoptosis to retinal pigmented epithelial cells in mice. PLoS ONE. 2008;3:e3119. doi: 10.1371/journal.pone.0003119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sykiotis GP, Bohmann D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci Signal. 2010;3:re3. doi: 10.1126/scisignal.3112re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu RM, Hagen TM. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A. 2004;101:3381–3386. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagaki K, Hiramatsu S, Inai S, Sasaki A. The effect of aging on complement activity (CH50) and complement protein levels. J Clin Lab Immunol. 1980;3:45–50. [PubMed] [Google Scholar]
- 18.Chen H, Liu B, Lukas TJ, Neufeld AH. The aged retinal pigment epithelium/choroid: a potential substratum for the pathogenesis of age-related macular degeneration. PLoS One. 2008;3:e2339. doi: 10.1371/journal.pone.0002339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cano M, Thimmalappula R, Fujihara M, Nagai N, Sporn M, Wang AL, Neufeld AH, Biswal S, Handa JT. Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and Age-related Macular Degeneration. Vision Res. 2010;50:652–664. doi: 10.1016/j.visres.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Z, Chen Y, Wang J, Sternberg P, Freeman ML, Grossniklaus HE, Cai J. Age-related retinopathy in NRF2-deficient mice. PLoS One. 2011;6:e19456. doi: 10.1371/journal.pone.0019456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farboud B, Aotaki-Keen A, Miyata T, Hjelmeland LM, Handa JT. Development of a polyclonal antibody with broad epitope specificity for advanced glycation endproducts and localization of these epitopes in Bruch’s membrane of the aging eye. Mol Vis. 1999;5:11. [PubMed] [Google Scholar]
- 22.Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res. 1996;62:155–169. doi: 10.1006/exer.1996.0020. [DOI] [PubMed] [Google Scholar]
- 23.Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, Herman JG, Baylin SB, Sidransky D, Gabrielson E, Brock MV, Biswal S. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vandermeer J, Sha Q, Lane AP, Schleimer RP. Innate immunity of the sinonasal cavity: expression of messenger RNA for complement cascade components and toll-like receptors. Arch Otolaryngol Head Neck Surg. 2004;130:1374–1380. doi: 10.1001/archotol.130.12.1374. [DOI] [PubMed] [Google Scholar]
- 25.Thurman JM, Renner B, Kunchithapautham K, Ferreira VP, Pangburn MK, Ablonczy Z, Tomlinson S, Holers VM, Rohrer B. Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J Biol Chem. 2009;284:16939–16947. doi: 10.1074/jbc.M808166200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki M, Betsuyaku T, Ito Y, Nagai K, Nasuhara Y, Kaga K, Kondo S, Nishimura M. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am J Respir Cell Molec Biol. 2008;39:673–682. doi: 10.1165/rcmb.2007-0424OC. [DOI] [PubMed] [Google Scholar]
- 27.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Ann Review Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 28.Han D, Canali R, Rettori D, Kaplowitz N. Effect of glutathione depletion on sites and topology of superoxide and hydrogen peroxide production in mitochondria. Molec Pharmacol. 2003;64:1136–1144. doi: 10.1124/mol.64.5.1136. [DOI] [PubMed] [Google Scholar]
- 29.Ebrahimi KB, Fijalkowski N, Cano M, Handa JT. Decreased Membrane Complement Regulators in the Retinal Pigmented Epithelium Contributes to Age-Related Macular Degeneration. J Pathol. 2012 doi: 10.1002/path.4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kew RR, Ghebrehiwet B, Janoff A. Characterization of the third component of complement (C3) after activation by cigarette smoke. Clin Immunol Immunopathol. 1987;44:248–258. doi: 10.1016/0090-1229(87)90069-9. [DOI] [PubMed] [Google Scholar]
- 31.Kew RR, Ghebrehiwet B, Janoff A. Cigarette smoke can activate the alternative pathway of complement in vitro by modifying the third component of complement. J Clin Invest. 1985;75:1000–1007. doi: 10.1172/JCI111760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD, Chen Y, Zhang K, Ambati BK, Baffi JZ, Ambati J. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006;103:2328–2333. doi: 10.1073/pnas.0408835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monsinjon T, Gasque P, Chan P, Ischenko A, Brady JJ, Fontaine MC. Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J. 2003;17:1003–1014. doi: 10.1096/fj.02-0737com. [DOI] [PubMed] [Google Scholar]
- 34.Wu MC, Brennan FH, Lynch JP, Mantovani S, Phipps S, Wetsel RA, Ruitenberg MJ, Taylor SM, Woodruff TM. The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc Natl Acad Sci U S A. 2013;110:9439–9444. doi: 10.1073/pnas.1218815110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Ohno-Matsui K, Yoshida T, Shimada N, Ichinose S, Sato T, Mochizuki M, Morita I. Amyloid-beta up-regulates complement factor B in retinal pigment epithelial cells through cytokines released from recruited macrophages/microglia: Another mechanism of complement activation in age-related macular degeneration. J Cell Physiol. 2009;220:119–128. doi: 10.1002/jcp.21742. [DOI] [PubMed] [Google Scholar]
- 36.Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, Farrar GJ, Kiang AS, Humphries MM, Lavelle EC, O’Neill LA, Hollyfield JG, Humphries P. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med. 2012 doi: 10.1038/nm.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hitzler I, Sayi A, Kohler E, Engler DB, Koch KN, Hardt WD, Muller A. Caspase-1 has both proinflammatory and regulatory properties in Helicobacter infections, which are differentially mediated by its substrates IL-1beta and IL-18. J Immunol. 2012;188:3594–3602. doi: 10.4049/jimmunol.1103212. [DOI] [PubMed] [Google Scholar]
- 38.Amarilyo G, Verbovetski I, Atallah M, Grau A, Wiser G, Gil O, Ben-Neriah Y, Mevorach D. iC3b-opsonized apoptotic cells mediate a distinct anti-inflammatory response and transcriptional NF-kappaB-dependent blockade. Eur J Immunol. 2010;40:699–709. doi: 10.1002/eji.200838951. [DOI] [PubMed] [Google Scholar]
- 39.Dunaief JL, Dentchev T, Ying GS, Milam AH. The role of apoptosis in age-related macular degeneration. Arch Ophthalmol. 2002;120:1435–1442. doi: 10.1001/archopht.120.11.1435. [DOI] [PubMed] [Google Scholar]
- 40.Elward K, Griffiths M, Mizuno M, Harris CL, Neal JW, Morgan BP, Gasque P. CD46 plays a key role in tailoring innate immune recognition of apoptotic and necrotic cells. J Biol Chem. 2005;280:36342–36354. doi: 10.1074/jbc.M506579200. [DOI] [PubMed] [Google Scholar]
- 41.Laudisi F, Spreafico R, Evrard M, Hughes TR, Mandriani B, Kandasamy M, Morgan BP, Sivasankar B, Mortellaro A. Cutting edge: The NLRP3 inflammasome links complement-mediated inflammation and IL-1beta release. J Immunol. 2013;191:1006–1010. doi: 10.4049/jimmunol.1300489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, Cho WG, Kaneko H, Fowler BJ, Bogdanovich S, Albuquerque RJ, Hauswirth WW, Chiodo VA, Kugel JF, Goodrich JA, Ponicsan SL, Chaudhuri G, Murphy MP, Dunaief JL, Ambati BK, Ogura Y, Yoo JW, Lee DK, Provost P, Hinton DR, Nunez G, Baffi JZ, Kleinman ME, Ambati J. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell. 2012;149:847–859. doi: 10.1016/j.cell.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Joseph K, Kulik L, Coughlin B, Kunchithapautham K, Bandyopadhyay M, Thiel S, Thielens NM, Holers VM, Rohrer B. Oxidative stress sensitizes retinal pigmented epithelial (RPE) cells to complement-mediated injury in a natural antibody-, lectin pathway-, and phospholipid epitope-dependent manner. J Biol Chem. 2013;288:12753–12765. doi: 10.1074/jbc.M112.421891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ha KN, Chen Y, Cai J, Sternberg P., Jr Increased glutathione synthesis through an ARE-Nrf2-dependent pathway by zinc in the RPE: implication for protection against oxidative stress. Invest Ophthalmol Vis Sci. 2006;47:2709–2715. doi: 10.1167/iovs.05-1322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.