

Abstract

We present a method to evaluate the free energies of ligand binding utilizing a Monte Carlo estimation of the configuration integrals concomitant with uncertainty quantification. Ensembles for integration are built through systematically perturbing an initial ligand conformation in a rigid binding pocket, which is optimized separately prior to incorporation of the ligand. We call the procedure producing the ensembles “blurring”, and it is carried out using an in-house developed code. The Boltzmann factor contribution of each pose to the configuration integral is computed and from there the free energy is obtained. Potential function uncertainties are estimated using a fragment-based error propagation method. This method has been applied to a set of small aromatic ligands complexed with T4 Lysozyme L99A mutant. Microstate energies have been determined with the force fields ff99SB and ff94, and the semiempirical method PM6DH2 in conjunction with continuum solvation models including Generalized Born (GB), the Conductor-like Screening Model (COSMO), and SMD. Of the methods studied, PM6DH2-based scoring gave binding free energy estimates, which yielded a good correlation to the experimental binding affinities (R2 = 0.7). All methods overestimated the calculated binding affinities. We trace this to insufficient sampling, the single static protein structure, and inaccuracies in the solvent models we have used in this study.
Introduction
Determination of binding affinities for protein–ligand complexes presents one of the most complicated and attractive problems of computational chemistry.1,2 The simplest methods attempting to suggest a solution to this problem (e.g., docking) mostly rely on end-point methods that estimate energies of single static complex structures.3 They usually neglect factors such as receptor flexibility, ligand strain upon binding, as well as various entropy effects.4−7 Some sampling is added on top of this strategy in methods such as Molecular Mechanics-Poisson–Boltzmann/Surface Area (MM-PBSA) and Molecular Mechanics-Generalized Born/Surface Area (MM-GBSA), which evaluate the absolute free energies of the protein and the ligand before and after binding.8−11 Both docking and the latter methods decompose the free energy into enthalpy and entropy contributions. In MM-PBSA and MM-GBSA, the enthalpies are obtained from the average energies from the molecular mechanics trajectories, which when combined with an entropy and a solvation free energy estimate using a continuum solvent model results in the final free energy estimate.8,12 In order to predict the free energy of binding accurately with this protocol, the first requirement is to correctly predict the absolute free energies of the bound and unbound species. However, these are typically large quantities, and we are interested in the small differences between them, so even a small error (percentage-wise) in their prediction might have a significant impact on the free energy difference ΔG. Hence, force field accuracy is very important in this exercise.
Another tool to calculate relative free energies of binding is alchemical free energy calculations, which modify a molecular entity into another via nonphysical (“alchemical”) pathways. Via thermodynamic cycles, incorporating these alchemical transformations, free energy changes of physical processes can be evaluated.13−17 It avoids decomposition of free energy change into individual thermodynamic terms by utilizing the ratio of the partition functions of the involved species, which eliminates the need to evaluate enthalpy and entropy contributions explicitly. One issue associated with alchemical calculations is obtaining enough sampling to generate a sufficient number of uncorrelated configurations and this is done using force fields coupled with molecular dynamics (MD) or Monte Carlo (MC) sampling.16 Thus, the sampling would always be based on the biases inherent in the force field employed. Moreover, to collect enough uncorrelated configurations to feed into the partition functions would be costly, especially for larger biological systems.16 Ending up with biased results is also possible with insufficient sampling when computed free energies depend on the choice of the initial receptor or ligand structure.18−20 The Mining Minima algorithm is worth noting here because it ties many aspects of the aforementioned free energy methods together. The attempt to systematically search for multiple local potential energy wells makes it unique among other end-point methods. Moreover, it computes the free energies of binding directly from the configuration integral contributions of the sampled local minima. However, just as the MM-PBSA and MM-GBSA protocols do, it estimates the absolute free energies of the protein, ligand, and their complex, which runs the risk of introducing errors when the difference between large numbers is taken.21,22
Recently, various types of restrained or unrestrained MD23−28 and potential of mean force simulations29−31 have appeared where observed ligand binding or ligand removal events23,25 provide another avenue to study protein–ligand binding. These simulations are very expensive and are subject to model uncertainties but do provide unique insight into binding events.
Regardless of their sophistication and practicability levels, all of these methods come with their intrinsic errors, which can be classified as systematic and random. Systematic errors in any measured or calculated quantity are rather easy to handle: They shift the result to a certain direction and, thus, are correctable. The random errors, on the other hand, may impact the determined energies in both fashions, up or down, and there is no way to correct for them in a post hoc manner.32 We have shown that one way to reduce the amount of this type of error is to include multiple microstates in the calculations. In other words, local sampling of the potential energy surface of interest decreases the random errors statistically.33
In this work, we propose a way to calculate the binding free energy of protein–ligand complexes directly from microstate energies utilizing a ratio of configuration integrals, and we then assess the uncertainty of our estimates with previously derived error propagation formulas.32,33 We predict the binding affinities directly from statistical mechanical definitions without splitting the free energy into enthalpic and entropic terms. This presents a huge advantage in terms of accuracy and uncertainty evaluation because introducing more terms into these calculations tends to propagate the errors originating from each component and understanding how these component errors do indeed propagate becomes a much more complicated task. Additionally, methods to directly calculate entropy are very challenging for a number of reasons.34−36 Moreover, commonly used entropy calculation methods such as normal-mode analysis utilized in methods such MM-PBSA and MM-GBSA rely on minimized snapshots from trajectories, which also introduce further uncertainties.35 Estimating the free energy of binding directly from microstate energies provides a straightforward way to estimate uncertainties in free energies directly from the computed energies. We understand how to propagate the errors contained in microstate energies, which allows us to directly determine the systematic and random errors associated with the calculated free energy of binding.32,33,36 With these aspects of uncertainty determination in mind, we aimed to create an unbiased ensemble of structures involved in modeling protein–ligand binding events and explored the effects of introducing such an ensemble on binding affinity prediction.
Similar to many other studies aiming to calculate ligand binding free energies to protein receptors,20,37−41 we apply our protocol to predict the binding affinities and the associated uncertainties to the experimentally well-characterized, engineered T4 Lysozyme L99A system.42,43 We are not the first ones to use this series of T4Lysozyme L99A inhibitors to explore the power of binding free energy determination methods. It was subject to several earlier docking and free energy perturbation (FEP) molecular dynamics (MD) studies.13,20,37,39,44,45 Moreover, an exhaustive docking study by Purisima et al. was recently conducted on this system using a very similar systematic ligand perturbation method to construct the protein–ligand complex and free ligand ensembles.41 The authors employed continuum solvent models that they developed, and this sets a limitation on the applicability and reproducibility of their protocol by others. Our work is novel in that it is the first study to offer error bars for binding free energy predictions, which are also corrected for their systematic errors. It uses the conventional force fields ff99SB46 and ff9447 in the AMBER1248 package, and the semiempirical PM6DH249−51 method in MOPAC201252 in conjunction with the readily available continuum solvent models Generalized Born (GB),53 Conductor-like Screening Model (COSMO),54 and SMD55 for scoring purposes, which enhances the extensibility of the present approach.
Methods
We calculated the binding free energies and their associated uncertainties on a series of T4 Lysozyme L99A inhibitors.42,43 T4 Lysozyme L99A is a very convenient system for this type of study due to the hydrophobic, entirely closed binding pocket facilitated by the L99A mutation. This isolated cavity accommodates a number of small hydrophobic molecules with experimentally measured binding affinities. Known experimental binding free energies allow us to judge the accuracy of our estimation, which in turn makes it possible for us to improve our methodology and workflow. Additionally, dealing with a completely closed, hydrophobic pocket mitigates some computational artifacts, which could emerge from computing solvation effects. The protein–ligand complexes employed have the Protein Data Bank (PDB) IDs 181L, 182L, 183L, 184L, 185L, 186L, 187L, and 188L and these contain benzene, 2,3-benzofuran, indene, i-butyl benzene, indole, n-butyl benzene, p-xylene, and o-xylene as ligands, respectively.43
After downloading the protein–ligand complex structures from the PDB, we separated each complex into their ligand and protein parts. The protonation states for each protein chain were obtained with the web server H++ at neutral pH.56 To prepare the binding pocket for each of these complexes, the amino acid residues within 5 Å of the ligand in its native PDB complex pose were relaxed using the ff99SB force field in the AMBER12 package while weak positional restraints of 10 kcal/mol·Å2 were applied on the rest of the structure. The relaxation protocol consisted of 25 000 steps of steepest descent minimization57 in the gas-phase and was performed in the absence of the ligand to prevent any structural bias which would favor certain poses over others. These minimized protein structures were kept rigid in the remainder of the analysis. The ligand structures, on the other hand, were optimized at the density functional theory (DFT) level using the M06L functional58 in conjunction with the Dunning type aug-cc-pVDZ basis set.59 The optimized geometries were utilized to obtain AM1-BCC partial charges and were used in the creation of the Generalized Amber Force Field (GAFF) parameters,60−63 which were employed in conjugation with both the ff99SB and ff94 force fields during scoring.
The optimized ligand structures were docked into their corresponding minimized binding pocket and their positions in the pocket were optimized using the ff99SB force field with the generalized Born (GB) solvent model. The ligands were then systematically translated and rotated using an in-house code. This process of systematically moving the ligand consisted of rotations of the whole ligand about its center of mass, rotating rotatable bonds within the ligand by 15° increments, and translating the ligand’s center of mass on an imaginary grid placed in the binding pocket with a spacing of 0.5 Å. We termed this process “blurring”. If the perturbation yielded a new, chemically meaningful pose that does not place the ligand atoms on top of the receptor atoms or induce other significant clashes, it was then appended to the ensemble of protein–ligand geometries. The free ligand structures (i.e., the unbound ligands) were also “blurred” by having their functional groups rotated incrementally, just as in complex structures and this lead to an unbiased ensemble for the ligands as well. For strictly aromatic ligands with no rotatable bonds only one single ligand pose made up the free ligand ensemble. Also, bond to methyl groups were not treated as rotatable bonds. In this way, we ended up with the following: an ensemble of protein–ligand complexes, an unbiased “ensemble” of free ligands, and a rigid protein.
The energies of each protein complex pose, each free ligand pose, and each rigid protein geometry were calculated utilizing several protocols: (1) with the ff99SB force field coupled with the GB implicit solvent model, (2) with the ff94 force field and GB implicit solvent model, (3) with the ff99SB force field in the gas-phase, (4) with the PM6DH2 semiempirical method in conjunction with the COSMO solvent model, (5) with the PM6DH2 semiempirical method in the gas-phase. Molecular mechanics scoring was carried out with the AMBER12 suite of programs while the PM6DH2 calculations were done with the MOPAC2012 package. When calculating the free energy of binding via eq 17, the temperature was set to 300 K and the concentration was taken to be 1 M.
The uncertainty and accuracy assessment for a particular free energy of binding relied on determining the uncertainty and accuracy of individual interaction energies associated with each protein–ligand complex pose making up that particular protein–ligand complex ensemble. The polar and nonpolar interactions between the ligand and the binding pocket in every pose were counted and the systematic and random errors per interaction were collected from the Biomolecular Fragment Database (BFDb).64 We used the “hsg” data set and acquired the systematic and random errors with respect to the “gold standard” CCSD(T)/CBS level of theory.32,65,66 Then, these individual errors were propagated to yield the cumulative error in the binding free energy as described in ref (22).33
Theory
The free energy of binding can be obtained directly with statistical mechanics without decomposing it into separate enthalpic and entropic terms. This is an advantage because it eliminates the need to estimate the errors introduced by individual enthalpic and entropic contributions, some of which are challenging to determine. Hence, we used the ratio of the partition functions of the protein–ligand complex (PL), ligand (L), and protein (P) to define the free energy of the protein–ligand binding, ΔGbind:
| 1 |
For the configuration integral affiliated with the PL complex, we assumed that the entire set of degrees of freedom (DOF) could be classified into six groups representing various physical contributions: (a) rigid translations (RT) of the PL complex, the entire complex could translate while keeping constant internal and rotational DOFs, (b) rigid rotations (RR) of the PL complex, the entire complex could rotate while keeping a constant position of center of mass and internal DOFs, (c) rigid docking translations (RDT), the ligand could translate while the ligand’s rotational and internal DOFs were constant and protein’s DOFs were fixed, (d) rigid docking rotations (RDR), the ligand could rotate while its center of mass, internal DOFs, and protein DOFs were constant, (e) internal protein DOFs (IP), and (f) internal ligand DOFs (IL).
In this expansion of DOFs, RT and RR deal with DOFs specific to the entire complex. RDT and RDR handle DOFs regarding the positioning of the ligand relative to the protein. IP and IL span the remaining DOFs and are specific to the protein and ligand DOFs, respectively. Hence, the total configuration space of the PL complex should be spanned by these six classes of DOFs, which when combined form the following integral:
| 2 |
Similarly, the configurational integral associated with the ligand was assumed to span (a) RT of the free ligand, it could translate while keeping constant internal and rotational DOFs, (b) RR of the free ligand, it could rotate while keeping a constant position of center of mass and internal DOFs, and (c) IL, internal DOFs of the free ligand. This yields
| 3 |
Likewise, three classes were identified to collect the protein’s DOFs: (a) RT of the unbound protein, it could translate while keeping constant internal and rotational DOFs, (b) RR of the unbound protein, it could rotate while keeping a constant position of center of mass and internal DOFs, and (c) IP, internal DOFs of the unbound protein, which results in
| 4 |
In all of these integrals RT and RR do not affect the energy, so the energy is constant with respect to them. We can evaluate the integrals as the volume of phase space associated with these DOFs times the value of the remainder of the integral. The PL complex, protein, and ligand can translate in a cubic box with the length equal to the average distance to another entity of their kind in a homogeneous solution, which allows us to assume the inverse concentration as the value of the RT phase-space volume. These integrals are thus reduced to
| 5 |
for the PL complex,
| 6 |
for the free ligand, and
| 7 |
for the unbound protein, respectively. If we placed an arbitrary unit vector on the center of mass of each of these species, this unit vector could rotate to point anywhere on a unit sphere with a surface area of 4π. Each of these possible vectors could serve as a rotational axis about which the molecule could rotate 2π. So the volume of phase space for RR terms is 8π2, which transforms the above integrals into
| 8 |
for the PL complex,
| 9 |
for the free ligand, and
| 10 |
for the unbound protein, respectively.
We also assumed the protein’s internal DOFs would be constant as dictated by the rigid receptor approximation. The IP-bound piece would give the volume of phase space VP enclosing the internal DOFs affiliated with the protein within the PL complex times the remainder of the integral:
| 11 |
Monte Carlo (MC) integration was applied to the remaining DOFs for RDT, RDR and IL in the bound form. The volume of phase space comprising the RDT DOFs was the binding pocket volume in which the ligand’s center of mass could translate: Vpocket. The RDR DOFs span a volume of 8π2 with the same reasoning described above for the RR term. Finally, the volume of phase space containing the internal DOFs of the ligand in bound form was VL. Here, we emphasize that VL is not an actual volumetric entity but an abstract quantity, which expresses the phase space occupied by the internal ligand DOFs. Thus,
| 12 |
The pocket volume Vpocket was obtained by evaluating the smallest volume, which would fit the superimposition of the centers of mass of all the ligand conformations generated by the blur code within the binding pocket.
Applying MC integration to eq 9 leads to the following final expression for the configuration integral of the free ligand:
| 13 |
The same operation on eq 10 results in
| 14 |
for the unbound protein. Since we employed a single, static protein structure in our calculations, there was no need for an average:
| 15 |
By inserting eqs 13, 14, and 15 into eq 1, we obtain
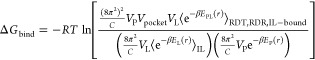 |
16 |
which reduces to our final expression for the free energy of binding in eq 17. Note that we have omitted symmetry corrections for the ligand because the symmetry number σ would be present in the ligand’s rotational degrees of freedom in both the free and bound states, and cancel in the derivation of eq 17.67 Furthermore, we assume the protein receptor is asymmetric.
| 17 |
In this expression, we sample over RDT, RDR, and IL DOF’s of the PL complex and the IL DOF’s of the free ligand. Then, using the conventional standard deviation formula,68 the sampling error associated with this free energy of binding expression would be
| 18 |
The single point energies calculated for each pose making up the PL complex and free ligand ensembles contained both systematic and random errors as proposed earlier by Faver et al.32,69 These individual errors would accumulate in the final free energy of binding estimation and reduce the reliability of our results. Faver et al. attacked this problem by first classifying and quantifying the molecular interactions present between the protein binding pocket and the ligand, then assessing the individual fragment-based errors with a probability density function built with a reference database of molecular fragment interactions, and finally propagating these errors as
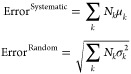 |
19 |
where k stands for different interaction types in the reference database (e.g., polar and nonpolar), Nk is the associated interaction count, μk and σk2 represent the mean error per interaction and variance about the mean error for interaction type k in the database.
A generic function f(xi) has a systematic error which could be, in its simplest model, evaluated as
| 20 |
and a random error which could be estimated as
| 21 |
where δxi expresses the uncertainty in the input variables. Faver et al. applied these operations in eqs 20 and 21 to the configuration integral Z and determined the total error of the free energies to be given as
| 22 |
where Z is the configuration integral, Ei stands for the microstate energies (here: energies of distinct PL poses), EiSys represents the systematic error within each microstate energy, and EiRand designates the random error contained in each microstate energy. This can also be written as
| 23 |
where Pi is the normalized weight of the microstate i. That is, the first part of this expression provides the systematic error in free energy of binding as the Boltzmann-weighted average of the systematic error of each microstate, while the second part gives the accompanying random error in form of a Pythagorean sum of the weighted random errors of each microstate.
Since systematic errors shift the results only in one, known direction, they can be corrected in a post hoc manner once their magnitudes are determined. Random errors, on the other hand, cause alterations in both directions, which are not predictable. This eliminates the possibility of a post hoc correction. The formula 23, however, suggests that if multiple microstates were considered in the binding free energy estimate, the second part related to the random errors would decay significantly because of the probability Pi within the Pythagorean sum. The impact of inclusion of numerous microstates on the cumulative systematic error is not as big as on the cumulative random error since the Pi values would add up to one eventually in the summation of the first part whereas the random error part would always get a coefficient, which is less than 1, due to the Pythagorean sum. This was demonstrated with several thought experiments:33 The higher the number of states included, the less the random error encountered.33
Our “blur” code accomplishes an exhaustive local sampling of the ligand within the protein binding pocket, in other words it creates the largest plausible PL complex ensemble given the input criteria, so that the uncertainty in the estimated free energy of binding is minimized as much as possible. As stated earlier, systematic errors can be accounted for completely assuming our reference “gold standards” are perfectly accurate. Hence, each pose making up the PL complex ensemble was analyzed for their protein–ligand interactions. Due to the strictly nonpolar nature of the binding pocket of interest, all the interactions detected were of van der Waals type. Then, utilizing the Biomolecular Fragment Database, the systematic and random errors present in the energy of each PL complex pose were estimated. Systematic errors were removed from these microstate energies and then the corrected PL complex energies were used to estimate the free energy of binding in the absence of systematic errors. The uncertainty of a particular binding free energy was computed as follows: once an error bar was obtained for each pose making up the PL complex ensemble, a random value of error was selected from the associated error distribution and added to the pose’s energy, which was already corrected for the systematic errors. These microstate energies were inserted into the final binding free energy formula shown in eq 17 to yield an estimate of the binding free energy. A distribution of binding free energies was acquired by calculating the free energy in this manner 10 000 times. Its standard deviation was used to measure the imprecision in the computed free energy due to microstate energy uncertainties.
Results and Discussion
Binding Free Energies
The protocol of exhaustively sampling the ligand configurations within and without the PL complex, that is, “blurring”, calculating the free energy of binding for the system of interest directly from eq 17, and estimating the uncertainty contained in the calculated binding free energy was tested on a congeneric series of eight T4Lysozyme L99A inhibitors: benzene, 2,3-benzofuran, indene, i-butyl benzene, indole, n-butyl benzene, p-xylene, and o-xylene with the PDB ID’s of 181L, 182L, 183L, 184L, 185L, 186L, 187L, and 188L, respectively. A sample blurring process involving n-butyl benzene (186L) is represented in Figure 1 using the Visual Molecular Dynamics (VMD) program.70 The “blurred” ensembles were checked for duplicate poses in order to prevent double counting, which alters the final binding affinity estimates. Assuming unique poses would have distinct energies, recurring energy scores were filtered out from the PL ensemble scores along with their error estimations. Thus, only structurally unique poses were retained in the ensemble.
Figure 1.
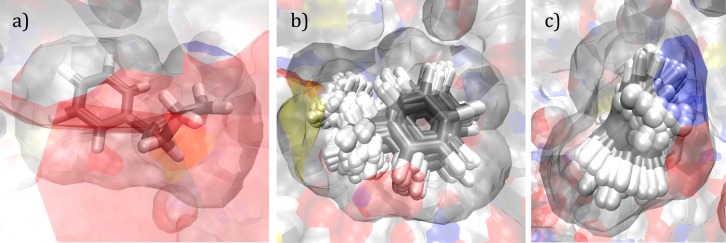
Process of “blurring”, that is, systematically perturbing the ligand within the protein binding pocket, shown for n-butyl benzene (186L). (a) Before blurring, single conformation in the binding pocket. (b) After blurring, numerous conformations superimposed in the binding pocket: front view, (c) side view. Carbon atoms are displayed in gray and hydrogen atoms in white while red symbolizes oxygen and blue represents nitrogen.
One disadvantage of this set is that the measured experimental binding affinities span a very narrow range of 2.1 kcal/mol and this is beyond the accuracy levels of most computational methods.42 Table 1 reports free energy of binding estimates obtained from force field scoring in implicit GB solvent and the semiempirical PM6DH2 method with the COSMO solvation model. Interestingly, we ended up with much more negative free energies of binding estimates regardless of the method employed. As seen in Figure 2, the binding free energy estimates acquired from PM6DH2 calculations correlated better with the experimental values indicated by the higher R2 values of 0.56 and 0.68 in conjunction with SMD and COSMO, respectively, compared to the calculated free energies of binding from the ff99SB microstate energies, which gave R2 values of 0.45 and 0.53 utilizing the GB and SMD solvent models, respectively. Considering how well PM6DH2 is parametrized for interaction energies, this is not surprising.32 The force fields ff94 and ff99SB showed very similar performances where the results changed by at most 0.8% from one to the other. That is why only one plot is given for the to force field approaches. Only the results from the more recent ff99SB force field are given.
Table 1. Experimental and Calculated Binding Free Energies Using Microstate Energies Obtained with ff94 Scoring in Conjunction with the Implicit GB Solvent Model, ff99SB Scoring with the GB Solvent Model, and Semiempirical PM6DH2 Scoring with COSMO Solvent Modela.
| ff94/GB |
ff99SB/GB |
PM6DH2/COSMO |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PDB | ligand | exptl. ΔGbind | ΔGbind | cor. | unc. | ΔGbind | cor. | unc. | ΔGbind | cor. | unc. |
| 181L | benzene | –5.2 | –8.63 | 0.43 | 3.61 | –8.64 | 0.43 | 3.46 | –10.45 | –0.39 | 0.77 |
| 182L | 2,3-benzofuran | –5.5 | –10.43 | 1.12 | 4.64 | –10.48 | 1.12 | 4.75 | –15.51 | –0.96 | 0.89 |
| 183L | indene | –5.1 | –12.17 | 0.99 | 4.51 | –12.10 | 0.99 | 4.58 | –15.86 | –0.47 | 0.77 |
| 184L | i-butyl benzene | –6.5 | –19.18 | 1.10 | 5.06 | –19.24 | 1.10 | 5.28 | –19.90 | –0.95 | 0.95 |
| 185L | indole | –4.9 | –9.70 | 1.19 | 5.00 | –9.68 | 1.19 | 5.04 | –14.04 | –0.94 | 0.99 |
| 186L | n-butyl benzene | –6.7 | –20.60 | 1.19 | 5.22 | –20.55 | 1.19 | 5.09 | –19.84 | –0.97 | 1.03 |
| 187L | p-xylene | –4.7 | –15.78 | 0.84 | 4.03 | –15.66 | 0.84 | 4.07 | –13.93 | –0.68 | 0.71 |
| 188L | o-xylene | –4.6 | –14.09 | 1.37 | 5.51 | –14.06 | 1.37 | 5.57 | –13.68 | –0.81 | 0.87 |
The systematic and random errors for each level of theory are also shown. “cor.” stands for the overall systematic correction to binding free energies, and “unc.” represents the uncertainty in those. All the numbers are in kcal/mol.
Figure 2.

Experimental free energies of binding plotted against calculated free energies of binding employing: (black) PM6DH2 with SMD solvation model, (red) PM6DH2 with COSMO solvation model, (green) ff99SB force field with GB implicit solvent model, (blue) ff99SB force field with SMD solvation model.
A correlation of 0.68 with the PM6DH2/COSMO method is quite reasonable in the face of the approximations made. We employed a static binding pocket conformation, in other words, we did not account for induced-fit interactions between the protein and its ligand. Mobley et al. found that considering a single, static protein conformation yields poorer binding free energy estimates rather than when several protein conformations are included.40 They remedied this problem to some extent by performing independent local geometry optimizations of the complex for each ligand and then simulating a rigid protein in its optimized geometry. For them, this was a more severe problem because they were sampling with MD methods and ligands were prone to be energetically trapped in certain conformations if conformational changes in the protein were required to overcome the local trapping. In our procedure, ligands are less likely to get trapped in particular orientations because the phase space is searched systematically. Another source of inaccuracy might have been the physical variables of “blurring”. If we made use of an infinitely small grid size for the translations within the binding pocket, we would end up with a larger ensemble covering a larger phase space. Likewise, if the rotation increments were decreased to infinitesimally small values, a higher number of conformations would be produced ensuring a better sampling. Both of these fine-tuning steps may yield improvements.
The origin of the consistent negative shift in the results from ff99SB/GB, ff94/GB, and PM6DH2/COSMO microstate energies might also have stemmed from the inaccuracies of the continuum solvation models we used (GB and COSMO).36 According to the thermodynamic cycle shown for protein–ligand binding in the gas-phase and solvent (Figure 3), a way to analyze this problem can be developed as follows:
| 24 |
Assuming the free energies of solvation for the protein P and the protein–ligand complex PL would have nearly identical solvation free energies due to the closed binding pocket, we can write
| 25 |
We tested this hypothesis of almost identical solvation free energies of the protein P and the PL complex on the protein structure used for the benzene system and a sample benzene–protein complex structure out of the benzene PL ensemble. Because we are concerned with single poses for both, free energy and energy would be interchangeable in this case. We calculated the solvation energies by extracting the polar contribution to their ff99SB/GB energies and summing that up with the nonpolar contribution obtained from the solvent accessible surface area using the LCPO algorithm.71 The solvation energy for the benzene–protein complex was −3075 kcal/mol, whereas the solvation energy for the unbound protein was −3073 kcal/mol. Hence, their difference is, indeed, negligible, which supports our hypothesis.
Figure 3.
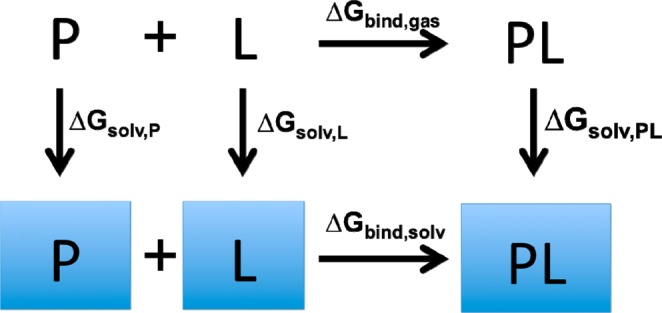
Thermodynamic cycle for protein–ligand binding in the gas and aqueous phases.
Hence, the microstate energies could be computed in the gas-phase and then the free energy of solvation for the ligand could be subtracted from the free energy of binding in the gas-phase. We utilized the experimental solvation free energies for the ligands, if they were available. The results collected using this protocol employed the same equations as before and are shown in Table 2.
Table 2. Comparison of the Solvation Energies with GBSA, COSMO, and SMD solvation Models against the Experimental Solvation Energies (exptl.) for the Congeneric Set of Ligands.
| ligand | GBSA solvation energy | COSMO Solvation energy | SMD | exptl. |
|---|---|---|---|---|
| benzene | –2.18 | –2.01 | –0.4 | –0.9 |
| 2,3-benzofuran | –5.69 | –3.41 | –0.8 | |
| indene | –3.48 | –3.06 | –1.3 | |
| i-butyl benzene | –0.84 | –2.35 | 0.7 | –0.4 |
| indole | –7.54 | –6.49 | –4.4 | |
| n-butyl benzene | –0.74 | –2.47 | 0.4 | 0.2 |
| p-xylene | –1.44 | –2.89 | 0.2 | –0.8 |
| o-xylene | –1.59 | –2.92 | –0.1 | –0.9 |
Using these approximations, we obtained estimates for the binding free energies from in vacuo microstate energies, and these calculated binding affinities were more negative than those we gathered from microstate energies incorporating a solvation model. Experimental solvation free energies were available only for benzene and benzene derivatives out of the set of eight ligands, which decreased our test set size. The solvation free energies were obtained from the Minnesota Solvation Database, version 2012.72 We are unable to report free energy of binding estimates for benzofuran, indene, and indole because of the missing ligand solvation free energies. The experimental solvation free energy values were smaller than the magnitude of the GB and COSMO solvation values (Table 2) yielding more negative free energies of binding (Table 3).
Table 3. Experimental and calculated Binding Free Energies Using Microstate Energies from Gas-Phase ff99SB and PM6DH2 Scoring with Experimental (exptl) Free Energies of Solvation for the Particular Ligand (If Any).
| ligand | exptl. ΔGbind | calcd. in vacuo ΔGbind ff99SB | calcd. in vacuo ΔGbind PM6DH2 | ΔGsolv/exp. for ligand | ΔGbind [ΔGsolv/exp. +ff99SB] | ΔGbind [ΔGsolv/exp. + PM6DH2] |
|---|---|---|---|---|---|---|
| benzene | –5.2 | –13.27 | –14.76 | –0.9 | –12.4 | –13.9 |
| 2,3-benzofuran | –5.5 | –19.20 | –21.84 | |||
| indene | –5.1 | –18.22 | –22.58 | |||
| i-butyl benzene | –6.5 | –23.23 | –26.35 | –0.4 | –22.8 | –26.0 |
| indole | –4.9 | –19.39 | –23.26 | |||
| n-butyl benzene | –6.7 | –25.46 | –27.25 | 0.2 | –25.7 | –27.5 |
| p-xylene | –4.7 | –20.29 | –20.82 | –0.8 | –19.5 | –20.0 |
| o-xylene | –4.6 | –18.48 | –20.88 | –0.9 | –17.6 | –20.0 |
In addition to these, we evaluated the free energy of solvation for all eight ligands with another continuum solvation model: the quantum mechanical SMD method by Truhlar et al.55 These values have been subtracted from the free energy of binding estimates in accordance to eq 25 to produce the final estimates for the free energy of binding for the whole set as shown in Table 4. Akin to the previous set of results using the experimental free energies of solvation for ligands, the free energies of binding turned out to again be too negative. The correlation to the experimental binding affinities improved considerably from 0.45 to 0.53 when the SMD values were incorporated along with the gas phase free energy of binding estimates from the ff99SB microstate energies compared to the results obtained with the GB solvent model (Table 1). The PM6DH2/COSMO combination, however, reproduced the trend in the measured binding affinities significantly better than the PM6DH2/SMD pair as the R2 values show: 0.68 vs 0.56. Figure 2 summarizes the information contained in Tables 1, 3, and 4 in the form of a plot.
Table 4. Experimental and Calculated Binding Free Energies Using Microstate Energies from Gas-Phase ff99SB and PM6DH2 Scoring with SMD Estimates for Free Energy of Solvation.
| ligand | exptl. ΔGbind | ΔGsolv/SMD for ligand | ΔGbind ff99SB/SMD | ΔGbind PM6DH2/SMD |
|---|---|---|---|---|
| benzene | –5.2 | –0.4 | –12.9 | –14.4 |
| 2,3-benzofuran | –5.5 | –0.8 | –18.4 | –21.0 |
| indene | –5.1 | –1.3 | –16.9 | –21.3 |
| i-butyl benzene | –6.5 | 0.7 | –23.9 | –27.1 |
| indole | –4.9 | –4.4 | –15.0 | –18.9 |
| n-butyl benzene | –6.7 | 0.4 | –25.9 | –27.7 |
| p-xylene | –4.7 | 0.2 | –20.5 | –21.0 |
| o-xylene | –4.6 | –0.1 | –18.4 | –20.8 |
For comparison purposes, we also performed a standard docking protocol on these systems and examined the correlation of the scores for the top hits to the experimental free energies. The Glide Standard Precision (SP) and Extra Precision (XP) algorithms73−75 were applied, which lead to R2 values of 0.28 and 0.37, respectively. The scores can be found in the Supporting Information, Table SI.1.
Binding Free Energy Error Analysis
Systematic error correction and uncertainty determination was conducted on the results obtained with ff99SB/GB, PM6DH2/COSMO, ff99SB/SMD, PM6DH2/SMD scoring methods. Using the Biomolecular Fragment Database and the iterative error bar assignment protocol, the systematic errors and uncertainties plotted in Figures 4 and 5 were collected. For the binding affinity estimates obtained from force field energy scoring, accounting for the systematic errors moved the binding free energy estimates in the wrong direction although these corrected errors were tiny. Depending on the Vpocket value, the completely entropic term – RT ln(VpocketC) contributes 0.5 to 1.5 kcal/mol to the free energy of binding at 300 K and a concentration of C = 1 M, which were used throughout in our calculations. Thus, its possible miscalculation cannot fully rationalize this systematic negative shift. There must be another source of inaccuracy arising from our binding free energy evaluation scheme which would shift the results to much more negative values. The calculated binding free energies with PM6DH2/COSMO suffer even from a bigger shift to more negative values (Figure 5) while they have considerably higher precision, which is not surprising given our earlier observations.32
Figure 4.
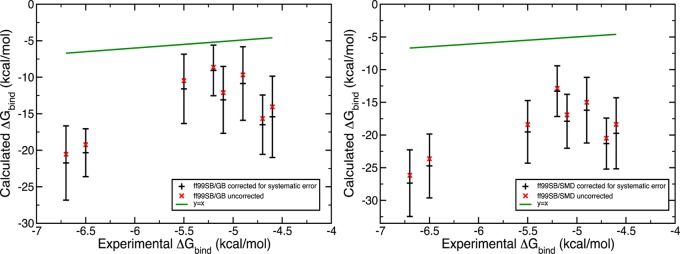
Systematic and random errors contained in the binding affinity estimates obtained from the ff99SB/GB (left) and ff99SB/SMD (right) microstate energies.
Figure 5.
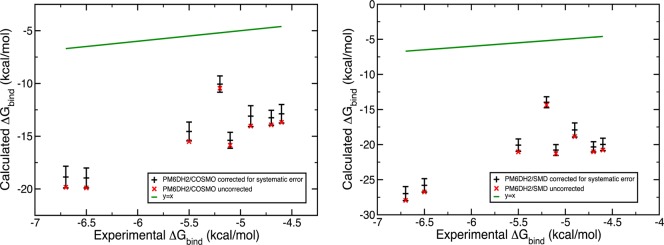
Systematic and random errors contained in the binding affinity estimates obtained from the PM6DH2/COSMO (left) and PM6DH2/SMD (right) microstate energies.
In addition to insufficient sampling, we suggest inaccuracies partially originate from the static protein binding pocket approximation and inaccuracies of the solvation free energy of the ligand since the inaccuracies in the solvation free energies of the PL complex and the free protein would mostly cancel out in this particular system. To test the effects of these two possible sources of error, we designed Gedanken experiments. First, we assumed omitting the local relaxation of the binding pocket upon its interaction with the ligand would result in an energetic penalty of 2.00 kcal/mol. This destabilization of 2 kcal/mol was reflected in the systematic errors of the PL microstate energies as −2 kcal/mol so that its application would yield a more stable microstate energy. We employed the same Monte Carlo error propagation protocol and observed a positive shift in all the energy values both with ff99SB/GB and PM6DH2/COSMO. We increased the hypothetical strain energy from 2 to 3 kcal/mol, which lead to a further improvement, that is, closer estimates to the experimental results. The estimates obtained are displayed in Figures 6 and 7.
Figure 6.
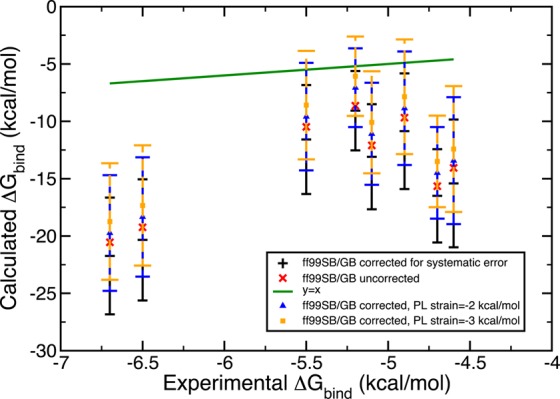
Free energy of binding estimates obtained with a Gedanken experiment, which assumes the static protein binding pocket approximation introduces a strain energy of 2 kcal/mol (blue) and 3 kcal/mol (orange) per PL complex pose. The original results with the static receptor are shown in black. Microstate scoring was done with ff99SB/GB.
Figure 7.
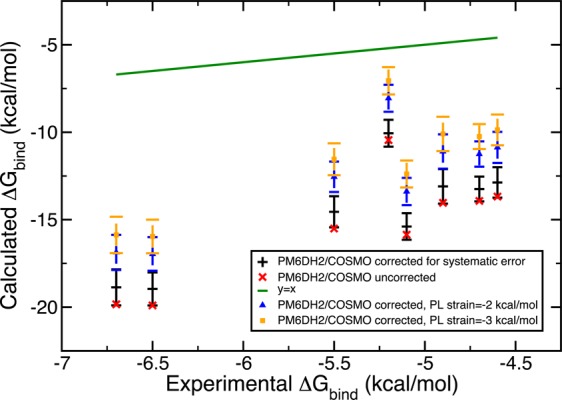
Free energy of binding estimates obtained with a Gedanken experiment, which assumes the static protein binding pocket approximation introduces a strain energy of 2 kcal/mol (blue) and 3 kcal/mol (orange) per PL complex pose. The original results with the static receptor are shown in black. Microstate scoring was done with PM6DH2/COSMO.
A second Gedanken experiment aimed to reduce the errors arising from the solvation models employed in this study. As we pointed out earlier, due to the completely closed nature of the receptor pocket, the solvation free energy of the PL complex and of the free protein P largely cancel out. Thus, the only source of solvation model errors could be the solvation energy of the free ligand L. To quantify its inaccuracy, we compared the ligand’s experimental free energy of solvation to the solvation contribution to the absolute ligand energy. The difference was assumed to be the systematic error and this was combined with a hypothetical error bar of 1.00 kcal/mol. If the experimental solvation free energies did not exist, we assumed the average of the systematic errors obtained for the ligands with experimental solvation free energies would give an acceptable estimate of the systematic error. Monte Carlo error estimation was extended such that it also iteratively assigns errors to the ligand piece in eq 17. The results were surprising in the amount of improvement they yielded. Figures 8 and 9 demonstrate the trends. This solvation correction to the ligand was as effective as accounting for strain in the binding pocket. Both experiments were coupled where a hypothetical receptor strain of 3 kcal/mol and solvation free energy corrections for the ligand L were considered. Consequently, the binding free energy estimates were significantly enhanced to the point where half of the ff99SB/GB estimates contained the experimental results in their final error bars. With these two small thought exercises we identified qualitatively two significant sources of error in our procedure.
Figure 8.
Gedanken experiments 1 and 2 combined. Blue represents the results with corrected solvation energy for the ligand and orange displays the results with both corrected solvation energy of the ligand and the receptor strain accounted for. Scoring was done with ff99SB/GB.
Figure 9.
Gedanken experiments 1 and 2 combined. Blue represents the results with corrected solvation energy for the ligand and orange displays the results with both corrected solvation energy of the ligand and the receptor strain accounted for. Scoring was done with PM6DH2/COSMO.
As stated earlier, the precision of calculated binding free energies benefits from sampling. Since uncertainties cannot be eliminated in a posthoc manner, sampling actually presents the only way to improve uncertainties of calculated quantities, in this case, protein–ligand binding affinities. This was already demonstrated by Faver et al. via a thought experiment where they showed even increasing the sample size from N = 1 to N = 2 enhances the precision considerably.33 We applied this to our ff99SB/GB test set where we varied the sample size N from 1 to 5, 10, 25, 50, 75, 100 and increased it by 25 from there on. The free energies of binding were calculated every time along with the associated uncertainty, which demonstrated the convergence of our binding affinity estimates and also displayed the rapid decay of the error bars with growing sample size.
For our convergence analysis, the energies for each pose of the “blurred” ensembles were ranked and the poses associated with lowest energies were assigned to the smallest samples. In other words, the sample of N = 1 consisted of the minimum-energy pose, the N = 5 sample contained the lowest 5 energies, the N = 10 comprised the lowest 10 energies, and so on. Therefore, the free energy of binding estimates rose as we included more and more microstates in our calculation. The maximum sample size was dictated by how compact the particular ligand was. For this system, N = 50 seems to be an acceptable sample size for a good binding affinity assessment. Unfortunately, the sample size of n-butyl benzene reaches only N = 42, which suggests that we might have under-sampled it or that it was packed very tightly into the binding pocket. With its large side chain, most of the produced poses lead to serious collisions with the binding pocket residues and hence had to be filtered out. Both the binding free energies and the error bars do not appear to change after N = 50 as seen in Figure 10. A better presentation of the effect of sample size on the error bars is given in Figure 11. The biggest impact on the random errors occurs when the ensemble size changes from N = 1 to N > 1 and the size of the error bars decrease drastically until N = 25. After N = 75, the uncertainties converge to their final values, which are at least 3 kcal/mol less than what they were using only a single microstate.
Figure 10.

Convergence of binding free energy calculations for the ff99SB/GB test set. Blurred poses were sorted as such the ones with the most negative energies were included in the smallest samples, hence the increase in the binding free energy estimate. Sample size was increased gradually and its maximum depends on the compactness of the particular ligand.
Figure 11.
Error bar propagation with growing sample size in ff99SB/GB calculations. Sample sizes of N = (1, 5, 10, 25, 50, 75, 100, 125, ...) were employed. N reached larger numbers for more compact ligands as the “blurred” ensembles for those were naturally bigger. After N = 75, the random errors did not change by much in size. Enlarging the sample size decreased the uncertainties in the final free energy estimation by at least 3 kcal/mol. The biggest improvement, namely the sharpest decrease, occurred when transiting from N = 1 to N > 1.
These two analyses prove the benefits of utilizing sampling rather than using single microstates. Over- or underestimating the final free energy values is almost inevitable and the obtained precision is the poorest with an ensemble of size N = 1. Even adding just a few microstates with local sampling would result in a more accurate and precise binding free energy. If one found the most stable conformation of a particular ligand bound to the receptor and locally sampled from that minimum-energy conformation such that only the bottom of the deepest potential energy well was sampled, even this practice would account for a large part of the significant contributions to the configuration integral of the PL complexes in eq 1, and we find that this would yield a much better free energy of binding estimate.
Conclusions
We evaluated the binding free energies and their associated uncertainty for a congeneric series of T4Lysozyme L99A inhibitors. These were calculated directly from the ratio of the configuration integrals of the protein–ligand (PL) complex, the protein (P), and the ligand (L). Decomposition into entropic and enthalpic terms was not performed which presents an advantage: we did not have to work around the uncertainties arising from these separate terms. Especially computing entropy accurately would be a challenge. Importantly, we introduced for the first time a way to estimate the systematic and random errors in computed free energies of binding on the fly. This is a step forward for protein–ligand binding affinity calculation efforts because it allows for the understanding of the reliability of the computed quantity of interest. A workflow was laid out that involves creation of unbiased ensembles for protein–ligand complexes and unbound ligands via our in-house “blur” code, a direct binding free energy calculation formula from statistical mechanics, systematic error correction for microstate energies, and an error bar estimation protocol for the final binding free energy estimates. The microstate energies were obtained with the ff99SB force field and the PM6DH2 semiempirical method. Continuum solvent models were employed where the ff99SB force field was combined with the Generalized Born (GB) model and the PM6DH2 approach with the Conductor-like Screening Model (COSMO) model. In vacuo binding free energies were also calculated with the same methods, and they were combined with the experimental and SMD-solvation free energies of the ligands. The systematic and random errors for each microstate were collected from the Biomolecular Fragment Database (BFDb) of Faver et al., and they were propagated as described elsewhere.33 The results were promising: they yielded reasonable correlation values (R2 = 0.45 with ff99SB/GB and 0.68 with PM6DH2/COSMO) to the experimental binding affinities. However, they were all shifted to more negative values. We suggest that this artifact arises from the use of a single, static protein conformation, and the inaccuracies contained within the solvent models as demonstrated via Gedanken experiments which showed improvements once these two sources of error were approximately accounted for. Insufficient sampling due to the dependency on the initial pose is another probable source of error. Hence, there is definitely room to improve our protocol. “Blurring” the protein would likely improve our estimates to include receptor flexibility. Likewise, exploiting explicit solvent for the force field scoring would enhance the results. Moreover, a bigger ensemble could be developed by utilizing finer settings during “blurring”, that is, systematic perturbation of the ligand in or out of the binding pocket and potentially of the protein receptor itself.
This work demonstrates the significant advantages of local sampling in enhancing precision of binding affinity estimates. It establishes the grounds for more sophisticated protein–ligand binding free energy calculations, which correct for the systematic errors contained in microstate energies, hence leading to a more accurate free energy of binding estimation, and at the same time determining error bars for binding free energies by propagating the random errors contained in microstate energies. We believe presenting error bars with binding affinity calculations should become common practice in our community. Hopefully, this study would contribute to improving the reliability of the calculated binding free energies.
Acknowledgments
We thank Dr. Danial Sabri Dashti, Dr. Jason Swails, and Prof. Christopher Cramer for fruitful discussions and insight. We acknowledge National Institute of Health for funding (GM044974 and GM066859) and University of Florida’s High Performance Computing Center for technical support.
Supporting Information Available
Coordinates for the initial ligand poses and the parameter files. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Present Address
† J.C.F., Department of Chemistry, Yale University, New Haven, Connecticut 06520-8107, United States
Author Present Address
‡ K.M.M., Institute for Cyber Enabled Research, Department of Chemistry and Department of Biochemistry and Molecular Biology, Michigan State University, 578 South Shaw Lane, East Lansing, Michigan 48824-1322, United States
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Gilson M. K.; Zhou H. X. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 21. [DOI] [PubMed] [Google Scholar]
- Leach A. R.; Shoichet B. K.; Peishoff C. E. J. Med. Chem. 2006, 49, 5851. [DOI] [PubMed] [Google Scholar]
- Halperin I.; Ma B. Y.; Wolfson H.; Nussinov R. Proteins: Struct., Funct., Genet. 2002, 47, 409. [DOI] [PubMed] [Google Scholar]
- Kitchen D. B.; Decornez H.; Furr J. R.; Bajorath J. Nat. Rev. Drug Discovery 2004, 3, 935. [DOI] [PubMed] [Google Scholar]
- Sotriffer C. A. Curr. Top. Med. Chem. 2011, 11, 179. [DOI] [PubMed] [Google Scholar]
- Yuriev E.; Agostino M.; Ramsland P. A. J. Mol. Recognit. 2011, 24, 149. [DOI] [PubMed] [Google Scholar]
- Yuriev E.; Ramsland P. A. J. Mol. Recognit. 2013, 26, 215. [DOI] [PubMed] [Google Scholar]
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S. H.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; Donini O.; Cieplak P.; Srinivasan J.; Case D. A.; Cheatham T. E. Acc. Chem. Res. 2000, 33, 889. [DOI] [PubMed] [Google Scholar]
- Kuhn B.; Kollman P. A. J. Med. Chem. 2000, 43, 3786. [DOI] [PubMed] [Google Scholar]
- Li Y.; Liu Z. H.; Wang R. X. J. Chem. Inf. Model. 2010, 50, 1682. [DOI] [PubMed] [Google Scholar]
- Rastelli G.; Del Rio A.; Degliesposti G.; Sgobba M. J. Comput. Chem. 2010, 31, 797. [DOI] [PubMed] [Google Scholar]
- Homeyer N.; Gohlke H. Mol. Inf. 2012, 31, 114. [DOI] [PubMed] [Google Scholar]
- Chodera J. D.; Mobley D. L.; Shirts M. R.; Dixon R. W.; Branson K.; Pande V. S. Curr. Opin. Struct. Biol. 2011, 21, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ C. D.; Mark A. E.; van Gunsteren W. F. J. Comput. Chem. 2010, 31, 1569. [DOI] [PubMed] [Google Scholar]
- Rodinger T.; Pomes R. Curr. Opin. Struct. Biol. 2005, 15, 164. [DOI] [PubMed] [Google Scholar]
- Shirts M. R.; Mobley D. L.; Chodera J. D. Annu. Rep. Comput. Chem. 2007, 3, 41. [Google Scholar]
- Woo H. J.; Roux B. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley D. L.; Chodera J. D.; Dill K. A. J. Chem. Phys. 2006, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. Y.; Deng Y. Q.; Roux B. Biophys. J. 2006, 91, 2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y. Q.; Roux B. J. Chem. Theory Comput. 2006, 2, 1255. [DOI] [PubMed] [Google Scholar]
- Huang Y. M. M.; Chen W.; Potter M. J.; Chang C. E. A. Biophys. J. 2012, 103, 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.; Gilson M. K.; Webb S. P.; Potter M. J. J. Chem. Theory Comput. 2010, 6, 3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buch I.; Giorgino T.; De Fabritiis G. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgino T.; De Fabritiis G. J. Chem. Theory Comput. 2011, 7, 1943. [DOI] [PubMed] [Google Scholar]
- Shan Y. B.; Kim E. T.; Eastwood M. P.; Dror R. O.; Seeliger M. A.; Shaw D. E. J. Am. Chem. Soc. 2011, 133, 9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannigan G.; LeBard D. N.; Henin J.; Eckenhoff R. G.; Klein M. L. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buch I.; Harvey M. J.; Giorgino T.; Anderson D. P.; De Fabritiis G. J. Chem. Inf. Model. 2010, 50, 397. [DOI] [PubMed] [Google Scholar]
- Wu C.; Biancalana M.; Koide S.; Shea J. E. J. Mol. Biol. 2009, 394, 627. [DOI] [PubMed] [Google Scholar]
- Buch I.; Sadiq S. K.; De Fabritiis G. J. Chem. Theory Comput. 2011, 7, 1765. [DOI] [PubMed] [Google Scholar]
- Essex J. W.; Severance D. L.; TiradoRives J.; Jorgensen W. L. J. Phys. Chem. B 1997, 101, 9663. [Google Scholar]
- Gumbart J. C.; Roux B.; Chipot C. J. Chem. Theory Comput. 2013, 9, 794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faver J. C.; Benson M. L.; He X. A.; Roberts B. P.; Wang B.; Marshall M. S.; Kennedy M. R.; Sherrill C. D.; Merz K. M. J. Chem. Theory Comput. 2011, 7, 790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faver J. C.; Yang W.; Merz K. M. J. Chem. Theory Comput. 2012, 8, 3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genheden S.; Ryde U. Phys. Chem. Chem. Phys. 2012, 14, 8662. [DOI] [PubMed] [Google Scholar]
- Hou T. J.; Wang J. M.; Li Y. Y.; Wang W. J. Chem. Inf. Model. 2011, 51, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N.; Warshel A. Proteins: Struct., Funct., Bioinf. 2010, 78, 1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boresch S.; Tettinger F.; Leitgeb M.; Karplus M. J. Phys. Chem. B 2003, 107, 9535. [Google Scholar]
- Deng Y. Q.; Roux B. J. Phys. Chem. B 2009, 113, 2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans J.; Wang L. J. Am. Chem. Soc. 1997, 119, 2707. [Google Scholar]
- Mobley D. L.; Graves A. P.; Chodera J. D.; McReynolds A. C.; Shoichet B. K.; Dill K. A. J. Mol. Biol. 2007, 371, 1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purisima E. O.; Hogues H. J. Phys. Chem. B 2012, 116, 6872. [DOI] [PubMed] [Google Scholar]
- Morton A.; Baase W. A.; Matthews B. W. Biochemistry 1995, 34, 8564. [DOI] [PubMed] [Google Scholar]
- Morton A.; Matthews B. W. Biochemistry 1995, 34, 8576. [DOI] [PubMed] [Google Scholar]
- Wei B. Q. Q.; Baase W. A.; Weaver L. H.; Matthews B. W.; Shoichet B. K. J. Mol. Biol. 2002, 322, 339. [DOI] [PubMed] [Google Scholar]
- Graves A. P.; Brenk R.; Shoichet B. K. J. Med. Chem. 2005, 48, 3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. Proteins: Struct., Funct., Bioinf. 2006, 65, 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. J. Am. Chem. Soc. 1995, 117, 5179. [Google Scholar]
- Case D. A.; Darden T. A.; Cheatham T. E. I; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Walker R. C.; Zhang W.; Merz K. M.; Roberts B.; Hayik S.; Roitberg A.; Seabra G.; Swails J.; Goetz A. W.; Kolossvai I.; Wong K. F.; Paesani F.; Vanicek J.; Wolf R. M.; Liu J.; Wu X.; Brozell S. R.; Steinbrecher T.; Gohlke H.; Cai Q.; Ye X.; Wang J.; Hsieh M.-J.; Cui G.; Roe D. R.; Mathews D. H.; Seetin M. G.; Salomon-Ferrer R.; Sagui C.; Babin V.; Luchko T.; Gusarov S.; Kovalenko A.; Kollman P. A.. AMBER 12; University of California: San Francisco, 2012. [Google Scholar]
- Fanfrlik J.; Bronowska A. K.; Rezac J.; Prenosil O.; Konvalinka J.; Hobza P. J. Phys. Chem. B 2010, 114, 12666. [DOI] [PubMed] [Google Scholar]
- Korth M.; Pitonak M.; Rezac J.; Hobza P. J. Chem. Theory Comput. 2010, 6, 344. [DOI] [PubMed] [Google Scholar]
- Rezac J.; Fanfrlik J.; Salahub D.; Hobza P. J. Chem. Theory Comput. 2009, 5, 1749. [DOI] [PubMed] [Google Scholar]
- Stewart J. J. P.MOPAC2012; Stewart Computational Chemistry: Colorado Springs, CO, 2012. [Google Scholar]
- Tsui V.; Case D. A. Biopolymers 2001, 56, 275. [DOI] [PubMed] [Google Scholar]
- Klamt A.; Schuurmann G. J. Chem. Soc., Perkin Trans. 2 1993, 799. [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. J. Phys. Chem. B 2009, 113, 6378. [DOI] [PubMed] [Google Scholar]
- Gordon J. C.; Myers J. B.; Folta T.; Shoja V.; Heath L. S.; Onufriev A. Nucleic Acids Res. 2005, 33, W368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach A. R.Molecular Modelling: Principles and Applications, 2nd ed.; Pearson Education Limited: Essex, 2001. [Google Scholar]
- Zhao Y.; Truhlar D. G. J. Chem. Phys. 2006, 125, 194101. [DOI] [PubMed] [Google Scholar]
- Woon D. E.; Dunning T. H. J. Chem. Phys. 1993, 98, 1358. [Google Scholar]
- Wang J. M.; Wang W.; Kollman P. A.; Case D. A. J. Mol. Graphics Modell. 2006, 25, 247. [DOI] [PubMed] [Google Scholar]
- Wang J. M.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. J. Comput. Chem. 2004, 25, 1157. [DOI] [PubMed] [Google Scholar]
- Jakalian A.; Bush B. L.; Jack D. B.; Bayly C. I. J. Comput. Chem. 2000, 21, 132. [Google Scholar]
- Jakalian A.; Jack D. B.; Bayly C. I. J. Comput. Chem. 2002, 23, 1623. [DOI] [PubMed] [Google Scholar]
- Faver J. C. Biomolecular Fragment Database (BFDb). http://www.merzgroup.org (accessed Apr. 1, 2013).
- Bartlett R. J.; Musial M. Rev. Mod. Phys. 2007, 79, 291. [Google Scholar]
- Halkier A.; Helgaker T.; Jorgensen P.; Klopper W.; Koch H.; Olsen J.; Wilson A. K. Chem. Phys. Lett. 1998, 286, 243. [Google Scholar]
- Gilson M. K.; Irikura K. K. J. Phys. Chem. B 2010, 114, 16304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonnacott R. J.; Wonnacott T. H.. Introductory Statistics; John Wiley & Sons, Inc.: New York, 1985. [Google Scholar]
- Faver J. C.; Benson M. L.; He X.; Roberts B. P.; Wang B.; Marshall M. S.; Sherrill C. D.; Merz K. M. PLoS One 2011, 6, e18868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. J. Mol. Graphics 1996, 14, 33. [DOI] [PubMed] [Google Scholar]
- Weiser J.; Shenkin P. S.; Still W. C. J. Comput. Chem. 1999, 20, 217. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Kelly C. P.; Thompson J. D.; Hawkins G. D.; Chambers C. C.; Giesen D. J.; Winget P.; Cramer C. J.; Truhlar D. G.. Minnesota Solvation Database, version 2012; University of Minnesota Minneapolis: Minneapolis, 2012. [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. J. Med. Chem. 2004, 47, 1739. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. J. Med. Chem. 2006, 49, 6177. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. J. Med. Chem. 2004, 47, 1750. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.