

 Characterising the differences between oligomers formed from the amyloidogenic protein β2-microglobulin and its mutant H51A using ESI-IMS-MS.
Characterising the differences between oligomers formed from the amyloidogenic protein β2-microglobulin and its mutant H51A using ESI-IMS-MS.
Abstract
In vivo beta-2 microglobulin (β2m) forms amyloid fibrils that are associated with the disease dialysis-related amyloidosis. Here, electrospray ionisation-ion mobility spectrometry-mass spectrometry has been used to compare the oligomers formed from wild-type β2m with those formed from a variant of the protein containing a single point mutation in the D strand, H51A, during in vitro fibril assembly. Using the amyloid-binding fluorescent dye, Thioflavin T, to monitor fibrillation kinetics, H51A was shown to exhibit a two-fold increase in the lag-time of fibril formation. Despite this, comparison of the oligomeric species observed during the lag-time of self-aggregation indicated that H51A had a higher population of oligomers, and formed oligomers of higher order, than wild-type β2m. The cross-sectional areas of the oligomers arising from H51A and wild-type protein were indistinguishable, although the H51A oligomers were shown to have a significantly higher kinetic stability on account of their reluctance to undergo sub-unit exchange when mixed with 15N-labelled protein. Together the data reveal a significant effect of His51, and thus that of the D-strand sequence, on amyloid formation. The results also highlight the power of mass spectrometry in probing complex biochemical mechanisms in real-time.
Introduction
The correct folding of proteins into their native, functional states is highly dependent on protein sequence, the environment, and the presence of additional factors including chaperones.1,2 If the balance of these conditions is altered, protein misfolding and/or aggregation can occur resulting in some cases in long, straight, unbranched polymeric structures with a characteristic cross-β structure, termed amyloid.3,4 Here, we focus on the amyloid-forming protein beta-2 microglobulin (β2m) whose aggregation in humans is the primary causative agent of the disease dialysis-related amyloidosis.5,6 β2m is the non-covalently bound light chain of the major histocompatibility complex class 1 (MHC 1) and in healthy subjects is excreted through the kidneys after dissociation from the MHC 1. In patients undergoing dialysis, this excretion pathway is unavailable and β2m protein levels in the body rise, resulting eventually in their self-aggregation into amyloid fibrils.
β2m is a 99-residue, ∼12 kDa protein. In its native state, β2m consists of seven β-strands (A–G) arranged as two β-sheets involving the β-strands ABED and CFG in a β-sandwich fold connected by a disulphide bond between residues Cys25 and Cys80 (Fig. 1A).7 In vitro β2m remains soluble at pH 7.5 with no evidence of amyloid formation even after 100 days of incubation at 37 °C.8–10 However, insoluble amyloid fibrils do form in hours to days in vitro, dependent on the precise solution conditions and agitation rate, in low ionic strength (<100 mM) buffer at pH 2.5.11–13 In this environment, the highly dynamic acid-unfolded β2m protein self-aggregates into long, straight, unbranched, twisted fibrils displaying all the hallmarks of amyloid.13–18 Thus, the timescale of these experiments is ideally suited for model investigations into the self-aggregation process.
Fig. 1. Ribbon structures of wild-type human β2m highlighting the different conformations of the D strand (yellow) between (A) monomeric, apo-β2m (PDB file: ; 1LDS)7 and (B) the MHC 1-bound holo form (PDB file: ; 2X70). The hydrogen bond contacts (black dashed lines) between the D- (yellow) and E- (green) strands of β2m are shown in the case of both monomeric, apo-β2m (C) and the MHC 1-bound holo form (D). The location of His51 (red) is highlighted on each structure.

Electrospray ionisation-mass spectrometry and its combination with ion mobility spectrometry (ESI-IMS-MS) have led to significant advances in the understanding of the mechanism of amyloid fibril formation, in particular concerning the specific oligomeric intermediates formed during self-aggregation.19,20 The study of these transient oligomers is particularly important as their presence correlates well with amyloid disease progression21–23 and, furthermore, it has been suggested that these oligomers may well be the toxic species rather than the fibril end-products.24–26 Previous ESI-IMS-MS analyses have enabled the identification and separation of co-populated, non-covalently bound β2m oligomers based on their charge, shape and mass from within the highly heterogeneous mixtures present during amyloid formation.27,28 ESI-IMS-MS rotationally averaged collision cross-sectional area (CCS) estimations of the β2m oligomers formed at pH 2.5 have suggested that these species adopt a stacked conformation of end-to-end β2m monomers rather than an alternative, more compact, globular arrangement.27
The current study utilises ESI-IMS-MS, in parallel with mutagenesis and biochemical techniques, to focus on understanding the effect of the amino acid sequence comprising the native D β-strand of β2m on fibrillogenesis, a region which is known to form important structural contacts with the α-chain of the MHC 1.29 Interestingly, on release from the MHC 1, this β-strand becomes highly dynamic30 and is known to be aggregation prone,31 suggesting a key role for the D-strand sequence in the self-aggregation of the protein monomer.9,29,32 The D-strand of β2m is involved in the non-covalent binding of β2m to the heavy chain of the MHC 1 (Fig. 1). Available crystal structures show a bulged D-strand in complex with the heavy chain of the MHC 1 (Fig. 1B and D) with residue Asp53 (the β-bulge residue at the centre of the D-strand sequence) hydrogen-bonded to Arg35 on the heavy chain of the MHC 1.35 On dissociation from the MHC 1 complex, the D-strand becomes solvent exposed and thus may be involved in initiating β2m aggregation. Indeed, X-ray crystallography and NMR studies have indicated that the D-strand is highly dynamic, adopting multiple conformations in solution7,29,36–38 with molecular dynamic simulations showing a straight D-strand to be populated on decreasing pH.39 Although the exact β2m oligomeric structure at low pH still remains to be elucidated, experiments involving covalent labelling combined with mass spectrometry have suggested a role of the D-strand sequence in the oligomeric interface of the β2m dimer and tetramer at neutral pH in the presence of Cu2+.40,41 Mutations in the D-strand of β2m have been shown previously to alter the lag-time of fibril formation at pH 2.5 suggesting that the D-strand sequence may play a role in fibrillogenesis.33,42 A single point mutation, H51A, in particular, has been shown to increase the lag-time of fibrillogenesis in vitro by up to two-fold, even though the eventual fibril morphology, as judged by negative stain electron microscopy (EM), remained comparable with that of the WT β2m protein.33 Interestingly, on transition of the D-strand from the bent to the straight conformation (Fig. 1), His51 rotates through 180° and is no longer involved in H-bonding to Tyr66 (Fig. 1C and D). Thus, substitution of residue 51 to alanine is thought to cause slight structural perturbations in the D-strand conformation which result in the increased lag-time of amyloid formation observed.
Here, oligomers originating from wild-type (WT) β2m under in vitro fibril-forming conditions have been compared with oligomers formed from H51A, the construct resulting from a single point mutation within the D-strand of the WT sequence. Despite both proteins self-aggregating into fibrils of very similar morphology, the lag-times of assembly are different. Furthermore, the shape (in terms of CCS), population and dynamics of the oligomers observed during fibril formation have been investigated to provide insights into the role of the sequence that comprises the D-strand of β2m in the amyloid assembly process.
Materials and methods
Reagents
Ammonium formate and thioflavin T (ThT) were purchased from Sigma-Aldrich (Gillingham, Dorset, UK). 14N WT β2m, 14N H51A and 15N wild-type (WT) β2m were prepared as described previously.33 15N H51A was expressed in 15N-enriched minimal media and purified as described.33 All proteins were determined to be monomeric by gel filtration, shown to be pure by SDS-PAGE analysis, and their molecular mass determined using ESI-MS. The molecular masses for WT and H51A were accurate to within 0.01% of those predicted based on amino acid composition (11 860.3 Da and 11 794.3 Da, respectively). Protein concentrations were calculated from the A280 using the extinction coefficient 20 065 M–1 cm–1.
Formation of amyloid fibrils from β2m WT and H51A
β2m and H51A (0.4 mg mL–1) were incubated separately in 100 mM ammonium formate (pH 2.5; 37 °C) shaking at 600 rpm using a Thriller Thermoshaker Incubator (PEQLAB Ltd, Southampton, UK). Fibril growth kinetics were monitored using ThT fluorescence. 50 μL aliquots were removed at regular intervals and diluted with 950 μL ThT (20 μM) in 100 mM ammonium formate pH 2.5. ThT fluorescence was measured in a PTI Quantamaster C-61 spectrofluorimeter using an excitation wavelength of 444 nm and emission at 480 nm with slit-widths of 4.5 nm. At the end-point of the reaction, samples were taken for negative stain transmission electron microscopy (EM). Simultaneous to the ThT reading, a 10 μL sample of the β2m variant in 100 mM ammonium formate pH 2.5 was removed separately for mass spectrometric analysis. To ensure accuracy in lag-time estimations of fibril formation between experiments, WT β2m was analysed alongside the β2m variant H51A on each day of analysis. The lag-time was obtained by fitting a tangent to the steepest part of the normalised ThT growth phase and the time at which the line intersected the baseline was taken as the lag-time.33,34 The average fold change in lag-time compared with WT β2m is reported along with the standard deviation between five replicates.
Fibril yield
β2m and H51A (0.4 mg mL–1) were incubated under the fibril forming conditions described above. After 46 h, samples were taken and the fibrils pelleted by centrifugation (13 000g, 30 min). The supernatant in each case was analysed by SDS-PAGE along with an identical sample taken prior to fibril formation.
Electron microscopy
10 μl of fibrils in 100 mM ammonium formate pH 2.5 (i.e., reaction end products) were placed on freshly ionised formvar- and carbon-coated EM grids for 30 s. The grids were then blotted with filter paper to remove excess solvent and the samples stained with 2% (w/v) uranyl acetate for 30 s. The grids were blotted again and air-dried before analysis. All images were taken using a CM10 electron microscope (Phillips Research, Eindoven, Netherlands) operating at 80 keV.
ESI-IMS-MS
ESI-IMS-MS experiments were performed on a Synapt HDMS hybrid quadrupole-IMS-oaTOF (Waters Ltd, Manchester, UK) mass spectrometer interfaced with a NanoMate Triversa nanoESI sample inlet and source (Advion Biosystems Inc. Ithaca, NY, USA). Positive nanoESI was performed using a capillary voltage of 1.75 kV and a nitrogen nebulising gas pressure of 0.7 p.s.i. A cone voltage of 170 V and a backing pressure of 3.6 mbar were used to observe the β2m oligomers. The ion accelerating voltages into the trap and transfer T-wave were set at 6 V and 20 V, respectively. The wave height was ramped from 4–12.5 V with a speed of 300 m s–1. For all mass spectrometry experiments, CsI clusters were used to calibrate the mass spectrometer and the raw data processed using MassLynx v.4.1 software (Waters Ltd, Manchester, UK) in combination with Driftscope v.3.0.
14N/15N-β2m oligomer exchange
For subunit exchange measurements carried out at 50% of the lag-time of fibril growth, the lag-time was estimated (using previously acquired data) and at the correct time-point during fibril growth, 100 μL of the 14N β2m WT or 14N H51A was mixed in a 1 : 1 v/v ratio with 100 μL of predominantly monomeric 15N WT β2m or 15N H51A (incubated on ice throughout the time course), respectively. Mass spectra were acquired after 1 min and 50 min of mixing. All samples were incubated in the 96-well plate within the NanoMate Triversa device at 20 °C throughout the subunit exchange experiments. The extent of subunit exchange was determined by comparing the area under the peaks corresponding to the 14N WT β2m/14N H51A and the 15N WT β2m/15N H51A oligomers analysed at t = 1 min with the mixed 14N WT β2m:15N WT β2m and 14N H51A:15N H51A subunit exchanged oligomers, respectively.
Sedimentation velocity analytical ultracentrifugation
Sedimentation velocity experiments were carried out at 25 °C using a Beckman Optima XL-I analytical ultracentrifuge (Beckman, Palo Alto, CA) using an An-60 Ti rotor with conventional aluminium double-sector centrepieces with a rotor speed of 40 000 rpm. Samples (0.4 mg mL–1) were prepared in the buffer used for MS analyses by overnight dialysis 4 °C. Radial absorbance scans at 280 nm were collected at 300 s intervals and the data were analysed using SEDFIT.43
Results and discussion
H51A increases the lag-time of in vitro fibril formation compared with wild-type β2m
Here, ESI-MS was used to investigate the nature of the oligomers formed during H51A fibril formation in order to probe how this single point mutation retards the rate of amyloid formation at low pH in vitro. To achieve this, fibril formation was performed independently for both wild-type (WT) β2m and H51A by incubating each protein at 37 °C with agitation (600 rpm) in 100 mM ammonium formate at pH 2.5.27,33 Under these conditions, WT β2m and H51A both form amyloid-like fibrils with lag-dependent kinetics, as shown by ThT fluorescence (Fig. 2A). In each case, negative stain EM analysis indicated that the fibrils produced exhibit long, straight morphologies (Fig. 2B), similar to the architecture of ex vivo fibrils. Consistent with previous results carried out using non-volatile (i.e., MS-incompatible) buffers,33 the lag-time of fibril formation is increased ∼two-fold for H51A compared with WT β2m (Fig. 2A and C), thus supporting a potential role of His51 in β2m amyloid fibril formation.
Fig. 2. (A) Graph showing ThT kinetics of fibril formation for WT β2m (black) and H51A (grey); one trace from five replicates is shown for each for simplicity. (B) Electron microscopy images of fibrils formed in 100 mM ammonium formate pH 2.5, 37 °C, 600 rpm, from WT β2m and the β2m variant H51A; the scale bar in each case represents 200 nm. (C) Fold change in lag-time of H51A compared with WT β2m; the average of five replicates is shown together with the standard deviation between replicates. (D) Final H51A ThT fluorescence signal expressed as a percentage of the WT β2m final ThT fluorescence signal; the average and standard deviation between five replicates is shown. (E) SDS-PAGE analysis of WT β2m (lane 1) and H51A (lane 3) prior to fibril formation compared with the remaining soluble fractions post-fibril formation (lanes 2 and 4 for WT β2m and H51A, respectively).

H51A populates higher order oligomers during the lag-time of in vitro fibril assembly compared with wild-type β2m
ESI-MS and ESI-IMS-MS were used to investigate the populations of, and any structural differences between, the oligomeric intermediates formed by H51A and WT β2m during fibril assembly, which may explain the difference in the kinetics observed and reveal insights into the amyloid formation mechanism. On initial dilution of WT β2m into the fibril-forming buffer (100 mM ammonium formate, pH 2.5 to give a final protein concentration of 0.4 mg mL–1), an aliquot was removed and analysed. At the earliest time-point measurable (1 min) the ESI mass spectrum was dominated by monomer ions (Fig. S1, ESI†) together with low intensity dimer, trimer and tetramer ions (the latter highlighted in Fig. 3A). In contrast, in addition to monomer, higher order oligomers from dimer to pentamer, inclusively, were observed at higher relative intensities in the ESI mass spectrum upon dilution of H51A into the fibril-forming buffer (Fig. 3B, Fig. S1, ESI†) suggesting that the H51A variant is significantly more prone to form oligomers compared with WT β2m at low pH, despite having a longer lag-time of fibril assembly.
Fig. 3. ESI-MS mass spectra highlighting the oligomers present (m/z 2500–5000) for (A) WT β2m and (B) H51A, immediately after dilution into 100 mM ammonium formate, pH 2.5. The charge states corresponding to the oligomers are highlighted: dimer (D), trimer (T), tetramer (Q) and pentamer (P). The majority of the ions originating from the WT β2m and H51A monomers appear ≤m/z 2500 (Fig. S1, ESI†). No noticeable differences in the charge state distributions for H51A and WT β2m monomers were observed. ESI-IMS-MS driftscope plots of (C) WT β2m and (D) H51A immediately after dilution of the proteins into the fibril-forming buffer (100 mM ammonium formate, pH 2.5) i.e., at 0% through their lag-phases of amyloid fibril formation; and (E) WT β2m and (F) H51A at 50% through their respective lag-phases of amyloid fibril formation (monomer = red, dimer = orange, trimer = yellow, tetramer = green, pentamer = blue, hexamer = purple).

The ESI-IMS-MS driftscope plots of WT β2m and the variant H51A acquired on initial dilution into fibril-forming buffer illustrate the added dimension of IMS in its ability to separate oligomers of different sizes including those ions of the same m/z but of both different mass and different charge (Fig. 3C and D, respectively). For both proteins, oligomers were separated within these highly heterogeneous populations, allowing each species to be assigned uniquely based on its m/z values and charge states. With the extra separation dimension, at the initial time-point at the start of the lag-phase oligomers up to and including the pentamer could be detected for WT β2m (as described previously27), and oligomers up to and including the hexamer were observed for H51A (Fig. 3C and D, respectively). Furthermore, the ESI-IMS-MS data show clearly that the higher order oligomers are significantly more populated in the case of H51A compared with WT β2m.
A control experiment was performed to compare the extent of oligomerisation of WT β2m and H51A in solution and thus to verify the legitimate presence of the oligomers observed by use of gas-phase mass spectrometry analyses. Thus, solutions of both proteins in the buffers used for the mass spectrometry studies were subjected independently to sedimentation velocity analysis (Fig. S2, ESI†). In both cases, protein monomer together with a distribution of oligomers were observed. In agreement with the mass spectrometry data, oligomers were found in solution for both WT β2m and H51A, with the difference in peak widths of the distribution of species found suggestive of different rates of interconversion between different oligomeric forms for H51A compared with WT β2m. These results are consistent with our previous WT β2m studies in which protein aggregates observed in the gas-phase were shown to reflect the oligomers detected in solution using a similar analysis.44
To monitor the progression of self-assembly and fibril formation, WT β2m and H51A were incubated separately at 37 °C in 100 mM ammonium formate, pH 2.5, with agitation at 600 rpm. At various time-points, aliquots were removed for ESI-IMS-MS analysis, with simultaneous ThT fluorescence analysis to confirm the duration of the lag-phase of fibril formation. The ESI-IMS-MS data for WT β2m and H51A at 50% of their individual lag-phases of fibril formation are shown in Fig. 3E and F, respectively. These data indicate that the population of oligomeric species for the WT protein decreases during the lag-phase, with ions corresponding to the 12+ to 16+ charge states of the pentamer no longer being observed. This suggests that the higher order oligomers observed at the start of fibril formation have extended rapidly into pre-fibrillar species and/or into amyloid fibrils that are undetectable using ESI-IMS-MS analysis alone. By the end of the lag-phase, all of the oligomer peaks have disappeared and the sample becomes very challenging to electrospray into the gas-phase due to the high population of insoluble amyloid fibrils present (data not shown). The short lifetime of the oligomers observed suggest that they are either on-pathway species, or that they are transient off-pathway intermediates of fibrillation.
By contrast with the population of WT β2m oligomers, the H51A higher order oligomers are still highly populated at 50% of the lag-phase (Fig. 3F). On extraction of the lowest charge state ions corresponding to the WT β2m and H51A monomer (i.e., 3+ ions) and oligomers (i.e., dimer 5+, trimer 7+, tetramer 9+, pentamer 11+) from the IMS driftscope plots, the reproduced ESI mass spectra show clearly that a significantly lower abundance of oligomers are present for WT β2m (Fig. 4A) compared with the abundance of oligomers observed for the H51A variant (Fig. 4B) at this time. In addition, a comparison of the separated isobaric monomer 3+, dimer 6+ and trimer 9+ ions originating from H51A at m/z ∼4000 indicate that the dimer and trimer are more abundant than the monomer in contrast to the respective abundances of these three species in the case of WT β2m at 50% lag-phase (Fig. S3, ESI†). One possible explanation for this observation is that H51A oligomers are less efficient at converting into amyloid fibrils compared with the WT β2m oligomers, and hence the H51A oligomers accumulate during the lag-phase. Interestingly, the IMS arrival time distributions of the H51A monomer 3+, dimer 5+, trimer 7+, tetramer 9+ and pentamer 11+ ions (i.e., the lowest charge state ions in each case) are indistinguishable (<2% different) from those observed for the WT β2m monomer and oligomers indicating very similar CCSs (Fig. 4C and D). Overall, the extended lag-time of H51A fibril formation is accompanied by a higher population of oligomeric species that persist for longer periods than their WT β2m counterparts, but are indistinguishable based on their CCS values. This behaviour is consistent with the presence of on-pathway, or transient off-pathway, species that undergo relatively slow conversion to amyloid fibrils.
Fig. 4. ESI-IMS-MS mass spectra of the monomer (M) 3+ (red), dimer (D) 5+ (orange), trimer (T) 7+ (yellow), tetramer (Q) 9+ (green) and pentamer (P) 11+ (blue) ions extracted from the IMS driftscope plot are shown for (A) WT β2m and (B) H51A at 50% of their respective lag-phases together with their individual ESI-IMS-MS arrival time distributions for (C) WT β2m oligomers and (D) H51A oligomers.

H51A forms more stable, less dynamic oligomers compared with WT β2m
As the WT β2m and H51A oligomers showed no significant differences in their ESI-IMS-MS CCS values indicating that they cannot be distinguished in terms of their rotationally averaged shapes, the stabilities and dynamics of these oligomers were investigated further to determine whether other features could explain the differences in fibril formation kinetics of the two closely-related proteins. The dynamics of β2m oligomers formed during fibrillogenesis in vitro can be determined using subunit exchange experiments whereby 14N- and 15N-β2m oligomers are mixed together and the rate of subunit exchange between the two species is monitored using ESI-IMS-MS.27,28 If the oligomers are highly dynamic entities and undergo rapid exchange with each other, then mixed oligomers containing both 14N- and 15N-protein will be observed using mass spectrometry. For example, three peaks in a 1 : 2 : 1 intensity ratio would be observed for a rapidly exchanging protein dimer corresponding to 14N:14N, 14N:15N and 15N:15N dimers. Similarly, four peaks in a 1 : 3 : 3 : 1 ratio would be observed for a rapidly exchanging trimer, and five peaks in a 1 : 4 : 6 : 4 : 1 ratio for a rapidly exchanging tetramer (Fig. S4, ESI†). Alternatively, if the oligomers are static entities, no exchange will occur and ions corresponding to all 14N- and all 15N-proteins will be observed at a 1 : 1 intensity ratio (Fig. S4, ESI†).
To determine whether the H51A oligomers are more kinetically stable (less dynamic) than their WT β2m counterparts, 14N/15N subunit exchange experiments were investigated for both WT β2m and H51A at 50% of their respective lag-phases. Hence, 14N-WT β2m and 14N-H51A were incubated separately in 100 mM ammonium formate, pH 2.5 with agitation at 37 °C. At times corresponding to 50% of their lag-phases, 15N-WT β2m or 15N-H51A, respectively (held on ice throughout and therefore not aggregating), were added to their 14N-labelled counterpart and any subunit exchange taking place was monitored using ESI-MS over a 50 minute time-course at room temperature. In all cases, the lag-phase was monitored in parallel using ThT fluorescence, and the presence of fibrils at the end of the lag-phase was confirmed by use of negative stain EM.
The extent of subunit exchange for 14N-WT β2m and 14N-H51A after 1 and 50 min of mixing with 15N-WT β2m or 15N-H51A at 50% lag-phase, respectively, is shown in Fig. 5. For WT β2m, the extent of 14N/15N-subunit exchange was complete as soon as the 14N- and 15N-WT proteins were mixed together, suggesting that all of the WT β2m oligomers are dynamic in nature, in rapid equilibrium with exchanging monomeric subunits (Fig. 5A and C). Interestingly, for the H51A oligomers formed during the lag-phase, very little subunit exchange occurred on initial mixing of the 14N- and 15N-H51A proteins (Fig. 5B), suggesting that these oligomers are more stable than the WT β2m oligomers, consistent with the results obtained using analytical ultracentrifugation. Indeed, no noticeable exchange was detected even after 50 minutes of incubation with 15N-H51A (Fig. 5D). Importantly, the lack of significant subunit exchange for the H51A oligomers supports our conclusion that the H51A oligomers observed in the gas-phase are reflective of oligomers present in solution and do not occur during the ESI process. Indeed, if oligomer formation in H51A were to arise from non-specific interactions occurring during the ESI process then complete subunit exchange would be expected to occur instantaneously upon mixing differently labelled protein subunits. The striking difference in subunit exchange kinetics for H51A oligomers compared with WT β2m is consistent with the increase observed in the lag-time of fibril formation (Fig. 2). Although the dynamics of the WT β2m and H51A oligomers differ considerably, their ESI-IMS-MS CCS values are very similar, suggesting that any structural differences in their ternary or quaternary structures that may affect their kinetic stability do not change the shape of the oligomers significantly.
Fig. 5. 14N- and 15N-labelled oligomeric β2m subunit exchange experiments illustrating the dimer (D) 7+, trimer (T) 10+ and 11+, and tetramer (Q) 13+ ions. Samples of 14N-WT β2m (A, C) or 14N-H51A (B, D) were mixed with 15N-WT β2m or 15N-H51A monomer samples, respectively in a 1 : 1 (v/v) ratio for 1 min (A, B) or 50 min (C, D) at 50% of the individual lag-phases for the two proteins. Peaks corresponding to all 14N- and all 15N-oligomers are shown in red, and peaks corresponding to mixed 14N/15N oligomers are shown in blue.

The amino acid sequence corresponding to the D-strand of native β2m has been implicated in fibril formation based on structural,7,36,45 kinetic33 and peptide studies.31 Here we have shown that although mutation of His51 to alanine decreases the kinetics of fibril formation by increasing the lag-time, the resulting fibrils have a morphology that is similar to that of fibrils formed from WT β2m in vitro under the same solution conditions. The increase in the lag-time of fibril formation brought about by H51A is consistent with previous results on the involvement of this residue in amyloid fibril formation.33,42 Detailed ESI-IMS-MS analyses of the H51A oligomeric intermediates observed during the lag-time of fibril formation have revealed for the first time significant differences in the populations and subunit exchange dynamics of these oligomers compared with the corresponding oligomers arising from the WT β2m protein. Not only are higher order oligomers observed for H51A, but all of the oligomers detected are significantly more populated and have increased stability compared with their WT counterparts.
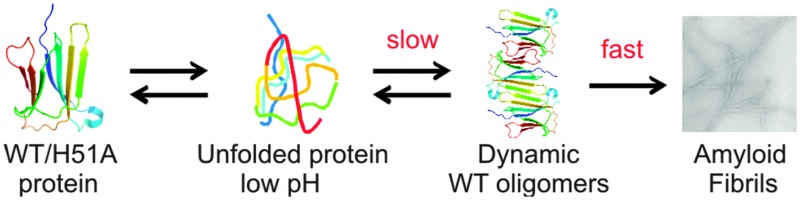
One possible explanation for these differences is that the H51A oligomers observed here may be off-pathway species, thus slowing the rate of fibril formation by diverting H51A monomers to dead-end products. Alternatively the more stable, less dynamic H51A oligomers could be slower at converting to the amyloidogenic state compared with the oligomers formed from WT β2m (Fig. 6). Interestingly, although ESI-IMS-MS was required to separate, identify and estimate the populations of the individual oligomers from within heterogeneous mixtures, any differences in the conformational properties of the oligomers arising from the two different proteins were not evident from the ESI-IMS-MS data, with the β2m WT and H51A oligomeric species having similar CCS values indicative of shapes with similar overall dimensions.
Fig. 6. Model of β2m amyloid fibril formation whereby WT β2m unfolds at low pH and forms dynamic oligomers that reach a critical nucleus and then convert rapidly into mature amyloid fibrils. In the case of H51A, the point mutation causes oligomers to form rapidly, although these more abundant, highly populated and kinetically stable oligomers are significantly more stable and less dynamic than the WT β2m oligomers and thus slower at converting into the critical nucleus that results in an increased lag-time of amyloid fibril formation.

Despite differences in the kinetics of amyloid fibril formation, the fibril yield of H51A and WT β2m remain comparable as analysed by SDS-PAGE analysis (Fig. 2E). This suggests that although more stable oligomers form extremely rapidly in the case of H51A compared with WT β2m, at the end of the lag-time and exponential growth phase these oligomers are all converted to fibrils with the same morphology as WT β2m (Fig. 2B).
Conclusions
Numerous studies have investigated the effects of small molecule binding to amyloid-forming protein monomers,45–48 in addition to probing the effect of point mutations in the starting monomer sequence on amyloid fibril formation.9,49 However, to our knowledge, this is the first detailed study reporting on the correlation between the lag-time and the population and kinetic stability of oligomeric species. Thus, the H51A oligomers become kinetically trapped, increasing in population and converting slowly into amyloid fibrils. These results illustrate the importance of H51 in β2m fibril formation at low pH. They also highlight the power of ESI-IMS-MS combined with subunit exchange dynamics in characterising the behaviour of individual oligomeric intermediates from a heterogeneous ensemble in real-time, revealing detailed insights into the nature and potential role of oligomeric intermediates in amyloid fibril formation.
Acknowledgments
ACL was funded by a BBSRC CASE PhD studentship in collaboration with Dr John Hoyes and Dr Mike Morris, Micromass UK Ltd/Waters Corpn, Manchester, UK (BB/526502/1). CLP is funded by the Wellcome Trust (WT092896MA) and CAS by the BBSRC (BB/H024875/1). We acknowledge the BBSRC for funding the purchase of the Synapt HDMS mass spectrometer (BB/E012558/1). We also thank Dr Katy Routledge for the expression and purification of the 14N-H51A protein used throughout these experiments and all members of the Ashcroft and Radford groups for useful discussions.
Footnotes
References
- Dobson C. M. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- Hartl F. U. Nature. 1996;381:571–580. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- Eichner T., Radford S. E. Mol. Cell. 2011;43:8–18. doi: 10.1016/j.molcel.2011.05.012. [DOI] [PubMed] [Google Scholar]
- Chiti F. and Dobson C. M., Protein misfolding, functional amyloid, and human disease, Annual Review of Biochemistry, Annual Reviews, Palo Alto, 2006, pp. 333–366 [DOI] [PubMed] [Google Scholar]
- Gejyo F., Yamada T., Odani S., Nakagawa Y., Arakawa M., Kunitomo T., Kataoka H., Suzuki M., Hirasawa Y., Shirahama T., Cohen A. S., Schmid K. Biochem. Biophys. Res. Commun. 1985;129:701–706. doi: 10.1016/0006-291x(85)91948-5. [DOI] [PubMed] [Google Scholar]
- Eichner T., Radford S. E. FEBS J. 2011;278:3868–3883. doi: 10.1111/j.1742-4658.2011.08186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh C. H., Smith D. P., Kalverda A. P., Phillips S. E. V., Radford S. E. Proc. Natl. Acad. Sci. U. S. A. 2002;99:9771–9776. doi: 10.1073/pnas.152337399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichner T., Radford S. E. J. Mol. Biol. 2009;386:1312–1326. doi: 10.1016/j.jmb.2009.01.013. [DOI] [PubMed] [Google Scholar]
- Santambrogio C., Ricagno S., Colombo M., Barbiroli A., Bonomi F., Bellotti V., Bolognesi M., Grandori R. Protein Sci. 2010;19:1386–1394. doi: 10.1002/pro.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese M. F., Miranker A. D. Prion. 2009;3:1–4. doi: 10.4161/pri.3.1.8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kad N. M., Thomson N. H., Smith D. P., Smith D. A., Radford S. E. J. Mol. Biol. 2001;313:559–571. doi: 10.1006/jmbi.2001.5071. [DOI] [PubMed] [Google Scholar]
- Platt G. W., Routledge K. E., Homans S. W., Radford S. E. J. Mol. Biol. 2008;378:251–263. doi: 10.1016/j.jmb.2008.01.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosal W. S., Morten I. J., Hewitt E. W., Smith D. A., Thomson N. H., Radford S. E. J. Mol. Biol. 2005;351:850–864. doi: 10.1016/j.jmb.2005.06.040. [DOI] [PubMed] [Google Scholar]
- Myers S. L., Jones S., Jahn T. R., Morten I. J., Tennent G. A., Hewitt E. W., Radford S. E. Biochemistry. 2006;45:2311–2321. doi: 10.1021/bi052434i. [DOI] [PubMed] [Google Scholar]
- Smith D. P., Jones S., Serpell L. C., Sunde M., Radford S. E. J. Mol. Biol. 2003;330:943–954. doi: 10.1016/s0022-2836(03)00687-9. [DOI] [PubMed] [Google Scholar]
- Ladner C. L., Chen M., Smith D. P., Platt G. W., Radford S. E., Langen R. J. Biol. Chem. 2010;285:17137–17147. doi: 10.1074/jbc.M110.117234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debelouchina G. T., Platt G. W., Bayro M. J., Radford S. E., Griffin R. G. J. Am. Chem. Soc. 2010;132:17077–17079. doi: 10.1021/ja107987f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White H. E., Hodgkinson J. L., Jahn T. R., Cohen-Krausz S., Gosal W. S., Muller S., Orlova E. V., Radford S. E., Saibil H. R. J. Mol. Biol. 2009;389:48–57. doi: 10.1016/j.jmb.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft A. E. J. Am. Soc. Mass Spectrom. 2010;21:1087–1096. doi: 10.1016/j.jasms.2010.02.026. [DOI] [PubMed] [Google Scholar]
- Williams D. M., Pukala T. L. Mass Spectrom. Rev. 2013;32:169–187. doi: 10.1002/mas.21358. [DOI] [PubMed] [Google Scholar]
- Lue L. F., Kuo Y. M., Roher A. E., Brachova L., Shen Y., Sue L., Beach T., Kurth J. H., Rydel R. E., Rogers J. Am. J. Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Dickson D. W., Trojanowski J. Q., Lee V. M. Y. Exp. Neurol. 1999;158:328–337. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]
- Tomic J. L., Pensalfini A., Head E., Glabe C. G. Neurobiol. Dis. 2009;35:352–358. doi: 10.1016/j.nbd.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner B., Jappelli R., Maji S. K., Desplats P. A., Boyer L., Aigner S., Hetzer C., Loher T., Vilar M., Campionic S., Tzitzilonis C., Soragni A., Jessberger S., Mira H., Consiglio A., Pham E., Masliah E., Gage F. G., Riek R. Proc. Natl. Acad. Sci. U. S. A. 2011;108:4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reixach N., Deechongkit S., Jiang X., Kelly J. W., Buxbaum J. N. Proc. Natl. Acad. Sci. U. S. A. 2004;101:2817–2822. doi: 10.1073/pnas.0400062101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucciantini M., Giannoni E., Chiti F., Baroni F., Formigli L., Zurdo J. S., Taddei N., Ramponi G., Dobson C. M., Stefani M. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- Smith D. P., Radford S. E., Ashcroft A. E. Proc. Natl. Acad. Sci. U. S. A. 2010;107:6794–6798. doi: 10.1073/pnas.0913046107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. P., Woods L. A., Radford S. E., Ashcroft A. E. Biophys. J. 2011;101:1238–1247. doi: 10.1016/j.bpj.2011.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G., Ricagno S., Corazza A., Rennella E., Gumral D., Mimmi M. C., Betto E., Pucillo C. E. M., Fogolari F., Viglino P., Raimondi S., Giorgetti S., Bolognesi B., Merlini G., Stoppini M., Bolognesi M., Bellotti V. J. Mol. Biol. 2008;378:887–897. doi: 10.1016/j.jmb.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Hodkinson J. P., Jahn T. R., Radford S. E., Ashcroft A. E. J. Am. Soc. Mass Spectrom. 2009;20:278–286. doi: 10.1016/j.jasms.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Zhao M., Jiang L., Cheng P., Park J., Sawaya M. R., Pensalfinic A., Goud D., Berk A. J., Glabe C. G., Nowick J., Eisenberg D. Proc. Natl. Acad. Sci. U. S. A. 2012;109:20913–20918. doi: 10.1073/pnas.1218792109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricagno S., Colombo M., de Rosa M., Sangiovanni E., Giorgetti S., Raimondi S., Bellotti V., Bolognesi M. Biochem. Biophys. Res. Commun. 2008;377:146–150. doi: 10.1016/j.bbrc.2008.09.108. [DOI] [PubMed] [Google Scholar]
- Routledge K. E., Tartaglia G. G., Platt G. W., Vendrusco M., Radford S. E. J. Mol. Biol. 2009;389:776–786. doi: 10.1016/j.jmb.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortschansky P., Schroeckh V., Christopeit T., Zandomeneghi G., Fandrich M. Protein Sci. 2005;14:1753–1759. doi: 10.1110/ps.041266605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields M. J., Assefi N., Hodgson W., Kim E. J., Ribaudo R. K. J. Immunol. 1998;160:2297–2307. [PubMed] [Google Scholar]
- Azinas S., Colombo M., Barbiroli A., Santambrogio C., Giorgetti S., Raimondi S., Bonomi F., Grandori R., Bellotti V., Ricagno S., Bolognesi M. FEBS J. 2011;278:2349–2358. doi: 10.1111/j.1742-4658.2011.08157.x. [DOI] [PubMed] [Google Scholar]
- Okon M., Bray P., Vucelic D. Biochemistry. 1992;31:8906–8915. doi: 10.1021/bi00152a030. [DOI] [PubMed] [Google Scholar]
- Verdone G., Corazza A., Viglino P., Pettirossi F., Giorgetti S., Mangione P., Andreola A., Stoppini M., Bellotti V., Esposito G. Protein Sci. 2002;11:487–499. doi: 10.1110/ps.29002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S., Saven J. G. Protein Sci. 2006;15:200–207. doi: 10.1110/ps.051814306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza V. L., Antwi K., Baron-Rodriguez M. A., Blanco C., Vachet R. W. Biochemistry. 2010;49:1522–1532. doi: 10.1021/bi901748h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza V. L., Baron-Rodriguez M. A., Blanco C., Vachet R. W. Biochemistry. 2011;50:6711–6722. doi: 10.1021/bi2004894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba T., Hagihara Y., Higurashi T., Hasegawa K., Naiki H., Goto Y. J. Biol. Chem. 2003;278:47016–47024. doi: 10.1074/jbc.M304473200. [DOI] [PubMed] [Google Scholar]
- Schuck P. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. M., Jahn T. R., Ashcroft A. E., Radford S. E. J. Mol. Biol. 2006;364:9–19. doi: 10.1016/j.jmb.2006.08.081. [DOI] [PubMed] [Google Scholar]
- Colombo M., de Rosa M., Bellotti V., Ricagno S., Bolognesi M. FEBS J. 2012;279:1131–1143. doi: 10.1111/j.1742-4658.2012.08510.x. [DOI] [PubMed] [Google Scholar]
- Woods L. A., Platt G. W., Hellewell A. L., Hewitt E. W., Homans S. W., Ashcroft A. E., Radford S. E. Nat. Chem. Biol. 2011;7:730–739. doi: 10.1038/nchembio.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrnhoefer D. E., Bieschke J., Boeddrich A., Herbst M., Masino L., Lurz R., Engemann S., Pastore A., Wanker E. E. Nat. Struct. Mol. Biol. 2008;15:558–566. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- Hyung S. J., DeToma A. S., Brender J. R., Lee S., Vivekanandan S., Kochi A., Choi J. S., Ramamoorthy A., Ruotolo B. T., Lim M. H. Proc. Natl. Acad. Sci. U. S. A. 2013;110:3743–3748. doi: 10.1073/pnas.1220326110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessel M. M., Bernstein S., Kemper M., Teplow D. B., Bowers M. T. ACS Chem. Neurosci. 2012;3:909–918. doi: 10.1021/cn300050d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.