

Abstract
Introduction
The oncoprotective role of food-derived polyphenol antioxidants has been described but the implicated mechanisms are not yet clear. In addition to polyphenols, phenolic acids, found at high concentrations in a number of plants, possess antioxidant action. The main phenolic acids found in foods are derivatives of 4-hydroxybenzoic acid and 4-hydroxycinnamic acid.
Methods
This work concentrates on the antiproliferative action of caffeic acid, syringic acid, sinapic acid, protocatechuic acid, ferulic acid and 3,4-dihydroxy-phenylacetic acid (PAA) on T47D human breast cancer cells, testing their antioxidant activity and a number of possible mechanisms involved (interaction with membrane and intracellular receptors, nitric oxide production).
Results
The tested compounds showed a time-dependent and dose-dependent inhibitory effect on cell growth with the following potency: caffeic acid > ferulic acid = protocatechuic acid = PAA > sinapic acid = syringic acid. Caffeic acid and PAA were chosen for further analysis. The antioxidative activity of these phenolic acids in T47D cells does not coincide with their inhibitory effect on tumoral proliferation. No interaction was found with steroid and adrenergic receptors. PAA induced an inhibition of nitric oxide synthase, while caffeic acid competes for binding and results in an inhibition of aryl hydrocarbon receptor-induced CYP1A1 enzyme. Both agents induce apoptosis via the Fas/FasL system.
Conclusions
Phenolic acids exert a direct antiproliferative action, evident at low concentrations, comparable with those found in biological fluids after ingestion of foods rich in phenolic acids. Furthermore, the direct interaction with the aryl hydrocarbon receptor, the nitric oxide synthase inhibition and their pro-apoptotic effect provide some insights into their biological mode of action.
Keywords: apoptosis, aryl hydrocarbon receptor, breast cancer cells (T47D), cell proliferation, phenolic acids
Introduction
The oncoptrotective properties of exogenous antioxidants have been documented in a number of epidemiological, intervention and in vitro studies (for a recent review see [1]). However, the mechanisms implicated are far from being clarified. Antioxidant effects, steroid receptor binding, direct interaction with intracellular elements and signaling systems and, recently, aryl hydrocarbon receptor (AhR) binding and modification of subsequent signaling pathways [2-15] have been proposed as possible mechanisms for the mediation of the oncoprotective effect of these agents. Exogenous antioxidants are exclusively produced by plants; they are divided into water-soluble antioxidants (e.g. vitamin C) and lipid-soluble antioxidants (e.g. vitamin A, vitamin E, β-carotene). In addition, a rich, heterogeneous class of substances, antioxidant (poly)phenols, characterized by the presence of one or multiple phenolic rings in their molecular structure, is also present in plant sources. This phenolic ring can be present either in the oxidized form (quinone) or in the reduced form (phenol). Exogenous antioxidants belong to distinct classes; for example, simple phenolic acids (e.g. caffeic acid), phytoalexins (stilbenoids, e.g. resveratrol) or flavonoids (catechins, quercetin) [16-18]. They further polymerize and form high molecular weight substances like tannins.
The majority of studies dealing with antioxidants focuses on the action of polyphenolic substances. Nevertheless, in a number of foods, in addition to polyphenols, simple phenolic acid antioxidants may occur, especially derivatives of 4-hydroxybenzoic acid and 4-hydroxycinnamic acid [19-21]. Few studies exist on the possible role of phenolic acids in cancer prevention and antigenotoxicity [22]. The present work concentrates on the antiproliferative action of caffeic acid, syringic acid, sinapic acid, protocatechuic acid, ferulic acid and 3,4-dihydroxyphenylacetic acid (PAA) (Fig. 1) on the human breast cancer T47D cell line, at concentrations more or less similar to those expected from normal consumption of foods. Our results indicate that phenolic acids produce growth inhibition of cancer cells, in vitro, indicating an additional protective effect on hormone-dependent breast tumors.
Figure 1.
The phenolic acids used in the present study.
Materials and methods
Cell lines and culture conditions
The hormone-sensitive breast cancer cell line T47D was purchased from the European Collection of Cell Cultures (Salisbury, UK). Cells were cultured in DMEM medium, supplemented with 10% fetal bovine serum (FBS), in a humidified atmosphere of 5% CO2 in air, at 37°C. Culture media and serum were from Gibco BRL (Life Technologies, Paisley, UK). FBS was assayed, prior to use, for the presence of polyphenol oxidase (seruloplasmin) and transferrin, by conventional nephelometric techniques, using a QM300 nephelometer, and commercial kits both by Kallestad/Pasteur (Paris, France). No measurable levels of either substance were found in all FBS batches tested.
Cell viability and growth assay
Cells were plated in a 24-well ELISA plate, at an initial density of 2 × 104 cells, with 1.0 ml medium per well. One day after seeding (designated as day 0) the medium was replaced, the different substances were introduced and the cells were grown for 5 days. Caffeic acid (97% purity), ferulic acid (99% purity) and protocatechuic acid (99% purity) were purchased from Aldrich Chemical Co. (Milwaukee, WI, USA). Sinapic acid (98% purity), syringic acid (98% purity) and PAA were from Sigma Chemical Co. (St Louis, MO, USA). Cell growth and viability were measured by the tetrazolium salt [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT)] assay [23]. All experiments were performed at least three times, in triplicate. Initial experiments comparing the MTT assay with the absolute cell number did not show significant differences, indicating that the MTT assay can be used for the determination of cell number, even in the presence of antioxidants, and/or H2O2.
H2O2 treatment
Cells were seeded in 24-well plates at an initial density of 150,000 cells/well. After 24 hours, the medium was replaced, FBS was omitted and the different phenolic acids (10-8 M) were introduced. Twenty-four hours later the culture medium was discarded, and fresh medium containing different concentrations of H2O2 (0.05–5.0 mM) was provided. After 3 hours at 37°C, cells were washed in PBS and their viability was determined by the MTT method, as already described. Although preliminary experiments did not show any interference of the serum with H2O2, the serum was eliminated from all the experiments. Cell viability was not influenced for the short periods of the experiment by the absence of serum.
Nitric oxide synthase assay
Nitric oxide synthase (NOS) activity was assayed by the transformation of radioactive arginine to citrulline [24,25]. Briefly, cells were detached from dishes by trypsin-EDTA, washed with PBS, harvested in PBS–1 mM EDTA, and homogenized with repeated pipetting with 250 μl homogenization buffer (250 mM Tris–EDTA, 10 mM EDTA, 10 mM EGTA). Nuclei and unbroken cells were separated by centrifugation at 12,000 × g for 15 min and discarded, while the supernatant was used for the assay of NOS. The concentration of proteins was adjusted at 10 μg/ml.
A reaction mixture (sufficient for 10 data points) was prepared with 250 μl of 50 mM Tris–HCl (pH 7.4) containing 6 μM tetrahydrobiopterin, 2 μM flavin adenine dinucleotide and 2 μM flavin adenine mononucleotide, 50 μl of 10 mM NADPH, 10 μl [3H]arginine (Amersham, Little Chalfont, UK), 50 μl of 6 mM CaCl2 and 40 μl distilled water. Forty microliters of this reaction mixture were mixed with 10 μl protein extract and incubated for 1 hour at 37°C. During this incubation time [3H]arginine is converted by NOS to [3H]citrulline. The reaction was stopped with 400 μl ice-cold 50 mM HEPES (pH 5.5)–5 mM EDTA. Nonreacted arginine was eliminated by resin absorption (AG 50 Wx*; BioRad Laboratories, Hercules, CA, USA). The eluate was mixed with scintillation fluid (SigmaFluor; Sigma Chemical Co.) and the radioactivity was measured in a liquid scintillation counter (Tricarb; Packard Instrument Co., Meriden, CT, USA), with 60% efficiency for tritium.
Assay for AhR binding and CYP1A1 activity
Cells were plated in 24-well culture dishes (with an initial density of 150 × 103 cells/well). When the cell culture reached approximately 70–80% confluency, AhR binding was performed in serum-free RPMI medium in a total volume of 0.4 ml. [3H]TCDD (34.7 Ci/mmol; ChemSyn Laboratories, Lenexa, Kansas, USA) was used in a final concentration of 5 nM, diluted in dimethyl sulfoxide, while a 200-fold molar excess of unlabeled TCDD (Wellington Laboratories, Greyhound, Birkenhead, UK) was used for nonspecific binding estimation. Phenolic acids were used with final concentrations ranging from 10-12 to 10-6 M. The medium was removed after 2 hours of incubation at 37°C in a 5% CO2 atmosphere, and cells were washed once with ice-cold PBS containing 10% FBS and afterwards once with cold PBS. Cells were then detached from dishes with 0.1 ml trypsin–EDTA, and filtered on Whatman GF/B glass fiber filters (1.0 μm), prewetted in cold PBS (4°C). The filters were then washed with cold (-80°C) acetone, allowed to dry and the radioactivity was measured in a 3 ml scintillation cocktail (SigmaFluor; Sigma Chemical Co.) using a liquid scintillation counter (Tricarb; Packard Instrument Co.), with 60% efficiency for tritium. All measurements were performed in triplicate [26,27].
The activity of CYP1A1, an enzyme induced by AhR activation, was assayed by the O-dealkylation of ethoxyresorufin (EROD) [26] (Molecular Probes, Leiden, The Netherlands). Cells were cultured in a black, clear bottom, 96-well plate. When the cells reach 50–60% confluency 5 nM TCDD were added, diluted in dimethyl sulfoxide. Caffeic acid and PAA (in ethanol) were added at the indicated concentrations. Blank, control and assay wells received the same amount of dimethyl sulfoxide and ethanol. After 24 hours of incubation at 37°C in an atmosphere of 95% air and 5% CO2, the medium was removed and the plates frozen sequentially at -20°C, in dry ice and at -80°C. Afterwards, cells were thawed at room temperature for 10 min, and BSA (diluted in 50 mM Tris, pH 7.2) was added at a final concentration of 1.33 μg/ml. Ethoxyresorufin (diluted in 1.5% methanol in 50 mM Tris, pH 7.2) was added at a final concentration of 5 μM. The plates were placed on a plate shaker at 37°C for 15 min. The EROD reaction was started by adding 1.67 mM NADPH in 25 μl of 50 mM Tris (pH 7.2). The reaction was carried out at room temperature for 7 min and stopped by adding 150 μl ice-cold methanol. The plates were allowed to sit, at room temperature, for 20–30 min prior to measuring. Fluoerescence was measured at 530 nm excitation wavelength and 590 nm emission wavelength with a Microplate Fluorescence Reader FLX800 (Bio-Tech Instruments Inc, Winooski, Vermont, USA). Results were calculated against a standard curve of resorufin concentration ranging from 0 to 500 nM (Molecular Probes), diluted in methanol [27].
Apoptosis assay
Cells treated with 10-7 M phenolic acids for 5 days were transferred from the culturing wells to a staining tube and washed with 4 ml PBS, containing 1% BSA, at 4°C. After medium removal (200 × g for 10 min at 4°C), and washing of cells with cold PBS, 3 ml cold absolute ethanol were added, incubated at 4°C for 1 hour, washed twice in cold PBS, and provided with 1 ml of 50 μg/ml propidium iodide in 3.8 mM sodium citrate, and 50 μl of 10 μg/ml RNase A solution. Cells were incubated for 3 hours at 4°C, and analyzed by flow cytometry, using a Beckton-Dickinson FACSArray apparatus (Beckton-Dickinson, Franklin Lakes, NJ, USA) and analyzed with the CELLQuest (Beckton Dickinson) and ModFit LT (Verify Software, Topsham, MN, USA) software.
For the double staining, using annexin V and propidium iodide, cells treated with phenolic acids were transferred from the culturing wells to a staining tube and washed with 4 ml PBS, containing 1% BSA, at 4°C. After medium removal (200 × g for 10 min at 4°C), 100 μl of 2 μg/ml annexin V–FITC was added, in a staining buffer (10 mM Hepes, 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, pH 7.4), and incubated for 10 min in the dark. Propidium iodide (1 μg/tube) was then added and cells were analyzed within 20 min by flow cytometry [28].
Semiquantitative western blot analysis of apoptotic proteins
At the end of each experiment, T47D cells were washed twice with PBS, removed by scraping and centrifuged at 430 × g. Cell lysis was completed at 4°C by shaking the pellet vigorously for 30 min reconstituted in a lysis buffer composed of 50 mM Tris–HCl (pH 8), 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP40 and freshly added protein inhibitors 10 μg/ml phenylmethylsulfonyl fluoride and 1 μg/ml aprotinin. Solid cellular debris was removed by centrifugation at 12,000 × g for 15 min. The cytoplasmic fractions were collected and stored at -80°C.
Protein concentration was measured by the Bio-Rad Protein Assay Kit II (BioRad Laboratories) following the instructions of the manufacturer. Samples of cytoplasmic protein fractions, containing 20 μg protein, were solubilized with SDS-PAGE sample buffer and electrophoresed through a 12% SDS gel. The resulting protein bands were transferred to nitrocellulose membranes, using an LKB electroblot apparatus (LKB, Bromma, Sweden). Standard western blotting procedures were employed. Band intensities were quantified by PC-based Image Analysis (Image Analysis Inc., Ontario, Canada). The antibodies used were: as primary antibody, anti-human Bcl-2 monoclonal antibody (clone 124, 1:200; DAKO A/S, Glostrup, Denmark), the rabbit polyclonal anti-serums against Bax, Bak, Bcl-xs/l and Bad (1:100; Santa Cruz Biotechnology, Inc., CA, USA), the anti-Fas (1:2500) and anti-FasL (1:1000; Transduction Laboratories, Lexington, KY, USA); and as secondary antibody, goat peroxidase-conjugated anti-mouse IgG (1:10,000; Chemicon International Inc., CA, USA) or anti-rabbit IgG (1:4000; Immunotech, Marseille, France). For purposes of normalization the blots were also stained with a monoclonal anti-actin antibody in a dilution of 1:400 (Amersham).
RT-PCR assays
NOS and CYP1A1 transcripts were measured by semiquantitative multiplex RT-PCR versus the constitutive gene of actin. Cells were cultured in six-well plates 24 hours prior to the addition of phenolic acids. Samples were taken after 2, 6, 12 and 24 hours of treatment. Total RNA was extracted with TRIzol® reagent (Invitrogen, Carisbad, CA, USA) according to the manufacturer's protocol, with an additional step of 70% ethanol wash.
For the RT reaction 1 μg RNA was used. First, DNA was eliminated with DNase I amplification grade treatment (Invitrogen) for 20 min at 25°C, followed by heat inactivation for 10 min at 65°C. Then cDNA synthesis was performed using SuperScript™ II RNA H- reverse trascriptase (Invitrogen), 5 μM poly d(T) (Amersham Pharmacia Biotech) and 1 μl ribonuclease inhibitor rRNasin® (Promega, Madison, WI, USA), in a total volume of 20 μl, for 1 hour at 42°C, which was stopped after incubation for 5 min at 95°C.
Multiplex PCR reactions were performed using 1 μl cDNA product, the DNA primers, 200 μM each dNTP (Invitrogen) and 1 U DyNAzyme II polymerase (Finzyme, Ozyme, France) in a total volume of 25 μl for 35 cycles, with a 30 s extension period. The specific set of DNA primers (MWG Biotech, Ebersberg, Germany), all in a concentration of 500 nM, and annealing temperatures used were: endothelial nitric oxide synthase (eNOS) (forward, 5'-AATCCTGTATGGCTCCGAGA-3' and reverse, 5'-GGGACACCACGTCATACTCA-3') at 58.3°C, inducible nitric oxide synthase (iNOS) (forward, 5'-ACAGGAGGGGTTAAAGCTGC-3' and reverse, 5'-TTGTCTCCAAGGGACCAGG-3') at 59.1°C, and CYP1A1 (forward, 5'-CTCTTAGGTGCTTGAGAGCCC-3' and reverse, 5'-CATCAGCA TCTATGTGGCCC-3') at 60°C. Actin primers (forward, 5'-GGTGGCTTTTAGGATGGCAAG-3' and reverse, 5'-ACTGGAACGGTGAAGGTGACA-3') were added to the PCR mix at a concentration of 100 nM and 250 nM, for eNOS and iNOS or for CYP1A1, respectively. PCR products were electrophorized in 3% agarose gel in 0.5 × TBE buffer at 100 mV, in a horizontal apparatus (Horizon® 11–14; Life Technologies, Gibco BRL, Goettingen, Germany) and band intensities were measured using the Molecular Analyst Software (BioRad).
Statistical analysis
Statistical analysis was performed by parametric methods, with the aid of the microcomputer programs Origin V 5.0 (Microcal Software, Northampton, MA, USA) and Systat V 10.0 (SPSS, Chicago, IL, USA).
Results
Phenolic acids affect the proliferation of T47D cells
All tested phenolic acids showed a time-dependent (Fig. 2a) and a dose-dependent inhibition of cell growth (Fig. 2b). The maximum effect was observed on the 5th day of incubation (two proliferation cycles). Caffeic acid was the most potent inhibitor of cell growth [inhibitory concentration 50% (IC50) = 2.17 × 10-9 M]. The number of cells remained unchanged after two or three proliferation cycles (20%, as compared with the control). This effect was also obvious in the time-course curves.
Figure 2.
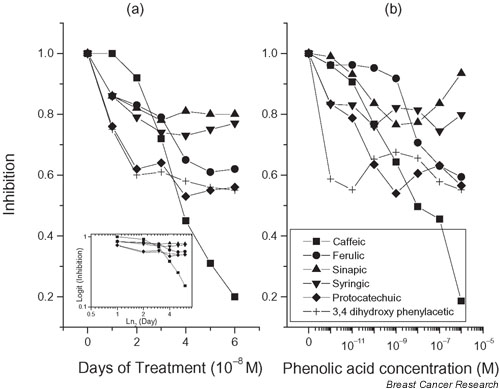
Time effect and dose effect of phenolic acids on the proliferation of T47D breast cancer cells. (a) Cells (2 × 104) were seeded in 24-well culture plates, and incubated for the indicated number of days with 10-8 M of different phenolic acids. In another series of experiments, cells were cultured in the absence of phenolic acids. The figure presents the ratio of the cell number in the presence/absence of the different phenolic acids. A typical experiment is presented, which was performed three more times with similar results. Inset: Data presented in the figure were analyzed by the application of the exponential decay equation 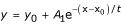 , in which y is the observed ratio of the cell number in the presence/absence of the phenolic acid, y0 is the maximal observed decrease of cell proliferation (plateau), A1 is 1 - y0, x0 is the initial time of observation (= 0), and x is the time (in days) of the y observation (abscissa of the y observation). The equation after transformation is rearranged as ln [(y - y0) / (1 - y0)] = -xt, the left side of which is the logit of the earlier quantity. From this equation one can obtain the calculation of t, which is an estimate of the half-life (t1/2) of the effect of the applied agent. In the case of the monotonous curves (sinapic acid, syringic acid, protocatechuic acid, and 3,4-dihydroxy-phenylacetic acid), this t1/2 was varying between 0.9 and 1.3 days. In the case of ferulic acid, in which the observed curve follows that of sinapic acid and ferulic acid for short incubation times and jumps to that of protocatechuic acid and 3,4-dihydroxy-phenylacetic acid for longer incubation times, the calculated t1/2 was 5.7 days. Finally, for caffeic acid, in which a sigmoidal curve was observed, data were not well fitted to the curve and the calculated t1/2 was 4.1 days. (b) Dose effect of phenolic acids on T47D cell proliferation. Cells (2 × 104) were incubated for 5 days in the absence or the presence of the indicated concentrations of phenolic acids, ranging from 10-12 to 10-6 M. The figure presents the ratio of the number of cells in the presence/absence of the indicated phenolic acids. A typical experiment is presented, repeated three more times with similar results.
, in which y is the observed ratio of the cell number in the presence/absence of the phenolic acid, y0 is the maximal observed decrease of cell proliferation (plateau), A1 is 1 - y0, x0 is the initial time of observation (= 0), and x is the time (in days) of the y observation (abscissa of the y observation). The equation after transformation is rearranged as ln [(y - y0) / (1 - y0)] = -xt, the left side of which is the logit of the earlier quantity. From this equation one can obtain the calculation of t, which is an estimate of the half-life (t1/2) of the effect of the applied agent. In the case of the monotonous curves (sinapic acid, syringic acid, protocatechuic acid, and 3,4-dihydroxy-phenylacetic acid), this t1/2 was varying between 0.9 and 1.3 days. In the case of ferulic acid, in which the observed curve follows that of sinapic acid and ferulic acid for short incubation times and jumps to that of protocatechuic acid and 3,4-dihydroxy-phenylacetic acid for longer incubation times, the calculated t1/2 was 5.7 days. Finally, for caffeic acid, in which a sigmoidal curve was observed, data were not well fitted to the curve and the calculated t1/2 was 4.1 days. (b) Dose effect of phenolic acids on T47D cell proliferation. Cells (2 × 104) were incubated for 5 days in the absence or the presence of the indicated concentrations of phenolic acids, ranging from 10-12 to 10-6 M. The figure presents the ratio of the number of cells in the presence/absence of the indicated phenolic acids. A typical experiment is presented, repeated three more times with similar results.
A second group of the phenolic acids is composed of ferulic acid, protocatechuic acid and PAA. All three compounds inhibited cell growth by 40%, showing one-half of the potency of caffeic acid. Of these, PAA was the most potent inhibitor, with IC50 < 10-12 M, followed by protocatechuic acid (IC50 = 2 × 10-11 M) and ferulic acid (IC50 = 2.3 × 10-9 M). Finally, sinapic acid and syringic acid were only partial inhibitors of cell growth, decreasing cell proliferation by 20%, with IC50 values ranging from 7 × 10-11 M (sinapic acid) to < 10-12 M (syringic acid).
Considering the time effect, three different groups of compounds are depicted. Caffeic acid has a proper time effect with a half-life of 3.2 days. Sinapic acid, syringic acid, protocatechuic acid and PAA have the same time effect with a half-life of 1.1 days, but with a different final effect, as described for the dose effect. Finally, ferulic acid shows a biphasic effect, with a short time effect comparable with that of sinapic acid and syringic acid, and a longer lasting effect similar to these of protocatechuic acid and PAA. This is better seen in the inset of Fig. 2a, which depicts the logit of the inhibition of cell growth on the ordinate, and the log2 of the incubation time on the abcissa.
In conclusion, all tested phenolic acids produced a significant inhibition of T47D cell proliferation, with IC50 values ranging from the nanomolar to the picomolar range. The concentration of 10-7 M was thus chosen for the following experiments, corresponding in all cases to the maximum effective concentration.
We then focused on two out of the six substances: caffeic acid (i.e. the major effective antiproliferative compound; Fig. 2) and PAA (the precursor molecule for the synthesis of the other more complex phenolic molecules) [29].
Effect of phenolic acids in the cell cycle and apoptosis
Figure 3a presents the effect of 48 hours of incubation with 10-7 M caffeic acid and PAA on the cell cycle. As shown, apparent apoptosis is obvious after PAA incubation, a result found equally when cells were stained with annexin V and propidium iodide (Fig. 3c). The analysis of cell cycle phases, presented in Fig. 3b, indicates that the number of nonapoptotic cells in the G0/G1 phase is significantly decreased after caffeic acid incubation. In contrast, both phenolic acids decrease the number of cells in the G2/M phase, and increase the S phase of T47D cells.
Figure 3.
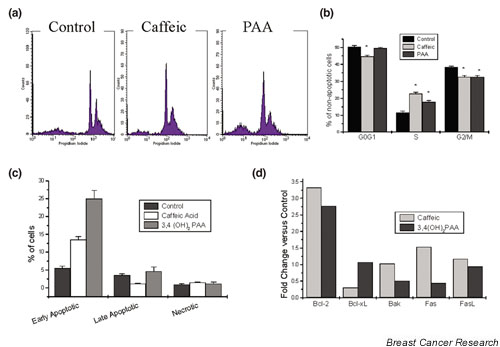
Effect of caffeic acid and 3,4-dihydroxy-phenylacetic acid (PAA) on the cell cycle and apoptosis after long (two cell cycles) incubation times. Cells were incubated with the indicated phenolic acids for 5 days. The medium, containing 10-7 M of the agents, was changed every other day. At the end of the incubation period, cells were removed from plates by scraping, stained with annexin V and/or propidium iodide (PI), as described in Materials and methods, and analyzed by flow cytometry. Early apoptotic changes were identified as the cell population having a normal (diploid) DNA content and annexin V staining. Late apoptotic cells were those presenting a hypodiploid DNA content identified by PI staining. After permeabilization of cells, staining with PI and flow cytometry revealed cell cycle phases. Necrotic cells were identified by forward and side scatter. (a) Typical flow cytometric analysis of the cell cycle. Cells were stained with PI. (b) Cumulative cell cycle phases of nonapoptotic cells (mean ± standard error of the mean of three measurements). (c) Cumulative effects of phenolic acids on apoptosis (mean ± standard error of the mean of three experiments). Cells were stained with annexin V and PI. (d) Effect of phenolic acids on apoptosis-related proteins. Semiquantitative western blot analysis on cell homogenates from T47D cell cultures treated with 10-7 M various phenolic acids for 5 days. At the end of the incubation period, cells were removed from plates by scraping and apoptosis-related proteins were measured with western blot analysis, as described in Materials and methods. Quantification results are depicted, expressed as the percentage of nontreated control values. Data are mean ± standard error of the mean of three independent experiments.
Apoptosis can be distinguished at early stages by the exposure of phosphatidylserine moieties on cell membranes, identified by annexin V binding, while late apoptosis is characterized by the appearance of DNA fragmentation. Figure 3c depicts early and late apoptosis in T47D cells, produced 5 days after the application of caffeic acid and PAA. Both phenolic acids induced apoptosis after 5 days of incubation. Necrotic cells were constantly low, indicating that these substances are not cytotoxic, at least at the concentrations used. It is interesting to note that, even at long incubation times (5 days), the main finding is early apoptotic changes. In addition, the effect of PAA was more prominent than that of caffeic acid.
Analysis of apoptotic-related proteins is depicted in Fig. 3d. Both phenolic acids (caffeic and PAA) induced significantly the anti-apoptotic protein Bcl-2 (3.3-fold and 2.8-fold, respectively). In addition, the pro-apoptotic FasL protein was induced by caffeic acid (1.6-fold). In contrast, the same phenolic acid decreased significantly the levels of the anti-apoptotic Bcl-xl protein. PAA, on the contrary, decreased significantly the levels of the pro-apoptotic proteins Bak and Fas, indicating different signaling pathways leading to apoptosis.
Phenolic acids have been reported to have an intrinsic free radical scavenging and antioxidant activity. In many in vitro systems, PAA was reported to be the strongest antioxidant, followed by caffeic acid [30]. In order to explore the possibility that phenolic acids might exert their antiproliferative action on T47D cells acting as antioxidants, we have incubated these cells with phenolic acids (10-7 M), and exposed them, after 24 hours, to varying concentrations of H2O2. As shown in Fig. 4, PAA produced a significant shift to the effective dose 50% value of H2O2. In contrast, caffeic acid, which exhibited the stronger antiproliferative effect on this cell system, did not show any notable antioxidant activity.
Figure 4.

Effect of phenolic acids on the inhibition of H2O2 cytotoxicity. (a) Cells (15 × 104) were incubated for 24 hours with the different phenolic acids (10-7 M). The medium was then discarded and replaced with a fresh one, in which fetal bovine serum was omitted, containing the indicated concentrations of H2O2. Incubation was followed for 3 hours, and then the viability of cells was determined by the MTT method. Results are presented of an experiment (in triplicate) performed four times with similar results. (b) The difference of effective concentration 50% values of H2O2 obtained in the presence of phenolic acids, as compared with control experiments. Bars represent the standard error of the mean of four different experiments performed in triplicate. IC50, inhibitory concentration 50%; PAA, 3,4-dihydroxy-phenylacetic acid.
Mechanism of action of phenolic acids in breast cancer cells
It appears that wine flavonoids and stilbens show an interaction with steroid hormone receptors in T47D cells [31]. We therefore tested phenolic acids for a similar interaction and also for a possible interaction with adrenergic receptors, reported to be implicated in prostate cancer cell growth arrest [32]. Finally, we examined the interaction of phenolic acids with the NOS system, also known to be involved in the cellular action of wine antioxidants [33]. In contrast to wine polyphenols [31], however, no interaction of either phenolic acid with estrogen, progesterone or adrenergic receptors was found (data not shown).
Previous reports from our group show that a number of polyphenolic antioxidants interact with the NOS-producing system [33]. Our data, presented in Fig. 5, indicate that, in cellular homogenates of T47D cells, PAA directly decreased NOS activity by 40%, while caffeic acid did not show any notable inhibition. In contrast, RT-PCR assays of the two isoforms of NOS (eNOS and iNOS) after variable incubation times showed that PAA transiently increased iNOS transcription (within 2 hours), followed by a 50% decrease. Meanwhile, eNOS transcription was reduced by 50%. A normalization occurs thereafter, followed by a linear decrease at longer incubation times.
Figure 5.
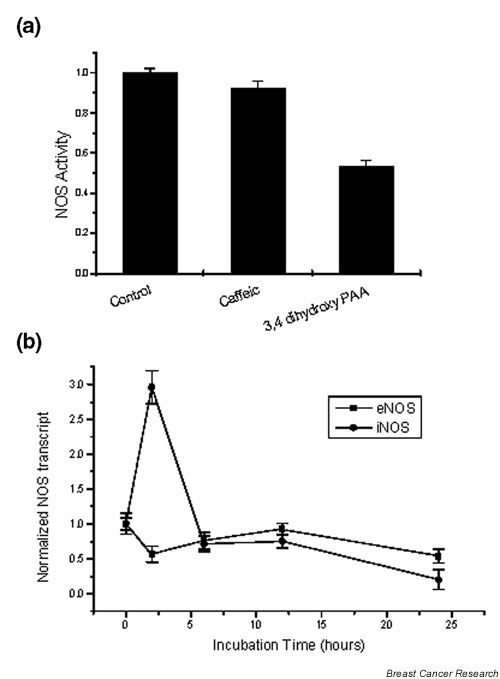
(a) Effect of 24 hours of incubation with phenolic acids (10-7M) on the activity of nitric oxide synthase (NOS) in the cytoplasm of T47D cells. (b) Time course of the effect of 3,4-dihydroxy-phenylacetic acid (PAA) (10-7 M) on the transcript of endothelial nitric oxide synthase (eNOS) and inducible nitric oxide synthase (iNOS). RT-PCR data for either enzyme were normalized for actin, and divided by the corresponding control values (control = 1 in all cases).
Another receptor–effector system that recently gained increased attention is the AhR system. The interaction of several antioxidants with the AhR has already been established. Resveratrol, a stilbene found in red wine, appears to be a pure AhR competitive antagonist [34]. It appears that quercetin and kaempferol regulate CYP1A1 gene expression through binding to the AhR [8,9]. We therefore tested the possible interaction of caffeic acid and PAA with this receptor system. As shown in Fig. 6a, only caffeic acid displaced radiolabeled TCDD from the AhR, with an IC50 value of 158 nM (Fig. 6b) comparable with that of the prototype ligand. This effect is 100 times higher compared with the cell growth inhibition by caffeic acid (2.17 nM).
Figure 6.
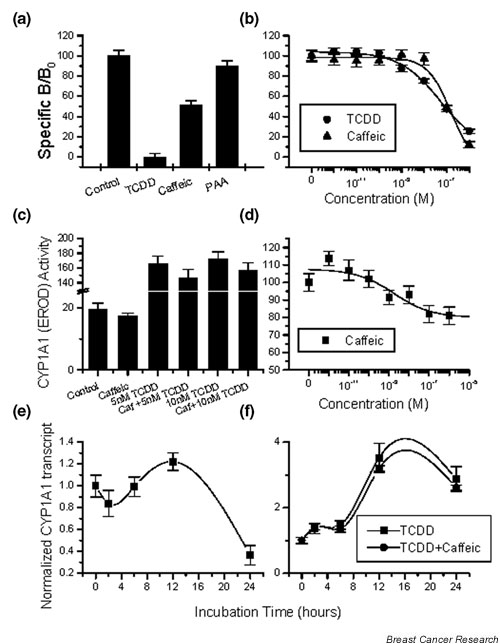
Interaction of phenolic acids with the aryl hydrocarbon receptor (AhR). (a) Interaction of 10-7 M phenolic acids on the AhR against TCDD. (b) Competition of caffeic acid on aryl hydrocarbon binding. Homologous displacement with TCDD given for comparison. (c) Inhibition of basal and TCDD-stimulated CYP1A1 (O-dealkylation of ethoxyresorufin [EROD]) activity by 10-7 M caffeic acid. (d) Dose effect of caffeic acid on the (EROD) activity of CYP1A1, after 24 hours of incubation. The time course of (e) the basal and (f) the TCDD-stimulated CYP1A1 transcript, after application of 10-7 M caffeic acid. RT-PCR data were normalized for actin, and divided by the corresponding control values (control = 1 in all cases). PAA, 3,4-dihydroxy-phenylacetic acid.
Activation of the AhR leads to a nuclear translocation, an association with specific transcription factors (aryl hydrocarbon nuclear translocator protein [ARNT]) and a modification of CYP1A1 expression. In order to identify whether the association of caffeic acid with the AhR is agonistic or antagonistic, we have assayed both the activity of CYP1A1 with the EROD method [26], and the CYP1A1 transcript with RT-PCR. As depicted in Fig. 6c, the interaction of caffeic acid with the AhR resulted in an inhibition of basal and TCDD-stimulated activity of CYP1A1. The observed IC50 value for this inhibition was 10.8 nM (Fig. 6d). This inhibitory effect was partial (maximum inhibition, 25%). In order to discriminate between a direct action of caffeic acid on the enzyme and a modification of transcription, we performed a time course of the CYP1A1 transcript with RT-PCR. Basal levels of CYP1A1 transcript were inhibited by 70% after 24 hours of incubation (Fig. 6e,6f). In parallel, a weak effect of TCDD-stimulated CYP1A1 transcription was observed.
Discussion
A great number of reports have in recent years dealt with antioxidants and their action on cancer cell proliferation (for a recent review see [1]). The great majority of these studies have been targeted towards polyphenolic antioxidants, active in chronic degenerative diseases, including cardiovascular diseases and cancer. In contrast, phenolic acids were overlooked in spite of the fact that these substances are found in appreciable concentrations in an important number of vegetable foods [19-22] (for a recent review see [29]). The present study investigated the antiproliferative action of these simple phenolics on cell proliferation of the hormone-sensitive T47D breast cancer cell line. The choice of the phenolic acids was based on their concentration in a number of plant foods, and on the fact that some of these phenolic acids (e.g. PAA) are precursors of more complex (poly)phenolic molecules [29].
Our data showed that all six phenolic acids tested possess a dose-dependent and time-dependent inhibitory antiproliferative effect on T47D cells. Nevertheless, a differential effect for each phenolic acid was found, with IC50 values varying from the nanomolar to the picomolar range. The time course of phenolic acids varies equally. Indeed, caffeic acid exerts its action later than other phenolic acids, suggesting a different mode of action. Ferulic acid, structurally related to caffeic acid, shows a bimodal effect, with a short time component and a long time component. Finally, all other phenolic acids show a half-maximal effect, achieved after 2 days. Comparing the structures of the different phenolic acids, presented in Fig. 1, our data suggest that the two hydroxyl groups on the phenolic ring and the three carbon side chains are both essential for the antiproliferative activity. The shortening of the side chain produces a loss of the antiproliferative activity, which is more apparent in both the methylation of one or both -OH group(s) and the p-OH substitution found in syringic acid and sinapic acid. In addition, shortening of the side chain confers an increased IC50 value, indicating a possible increase of the transmembrane transit of the compounds or, alternatively, an increased interaction with an unknown membrane constituent.
To examine the possible mode of action of simple phenolic acids on T47D cell growth, we have concentrated on two out of the six phenolic acids: caffeic acid, which is the most potent, and PAA, which is a precursor for the synthesis of other more complex molecules [29]. Furthermore, we have tested low concentrations of these phenolic acids comparable with those found in the body after consumption of foods. In contrast to polyphenols [31], we have not detected any interaction of these two compounds with either estrogen, progesterone or adrenergic receptors in T47D cells. Cell cycle analysis revealed that phenolic acids increase the number of nonapoptotic cells in the S phase and decrease the G2/M phase of the cell cycle. In addition, both substances induced apoptosis within 5 days. PAA appears to be a more potent inducer of apoptosis than caffeic acid (Fig. 3).
Phenolic acids were reported to possess a major antioxidant activity in different systems [30]. We have therefore investigated whether the antiproliferative and pro-apoptotic effects of caffeic acid and PAA were correlated with their inhibition of H2O2 toxicity, an indirect measurement of their antioxidant activity. A differential effect between caffeic acid and PAA was found (Fig. 4a,4b). Indeed, caffeic acid did not show any notable shift of the H2O2-induced toxicity (IC50 = 0.28 mM, as compared with IC50 = 0.51 mM of control cells). In contrast, PAA produced a major (two logarithms) shift of the H2O2 curve (IC50 = 67.01 mM). It is interesting to note that PAA decreased the activity of NOS, and concomitantly decreased the possible production of endogenous reactive nitrogen species.
The role of nitric oxide (NO) in cancer is ubiquitous (for a discussion see [35]). NO was reported to inhibit cell proliferation, to induce differentiation and to decrease the metastatic spread of different tumor cell lines [36], although this effect seems to be related to the type and the origin of the cancer cell studied [37], and to the oxidative status of the cells [38,39]. Polyphenols have been reported to affect NO production [40] and some of its biological effects [41,42]. Although the role of the NO/NOS system in breast cancer is controversial (for a review of the current bibliography see, for example, [43]), inhibition of NOS activity has been considered a possible target for anticancer treatment. In MCF7 breast cancer cells, inhibitors of NO synthesis (NG-nitro-L-arginin methyl ester) and NO scavengers induced apoptosis [44], via a p53-associated pathway, while in T47D cells suppression of NO production triggers an induction of apoptosis via a FKHRL1 (FOXO3a) kinase pathway, independent of phosphoinositide 3-kinase-Akt and caspase 3 activation [45]. In this respect, the decrease in NOS activity by PAA could explain its pro-apoptotic effect (Fig. 5). It is further interesting to note that, in addition to the inhibition of enzyme activity, PAA decreases NOS transcripts after long (> 12 hours) incubation times. At shorter incubation times, however, a substantial increase of iNOS was found, indicating differential regulation of transcription.
Caffeic acid conversely seems to modify cell proliferation through interaction with the xenobiotic receptor (AhR)–CYP system. The aryl hydrocarbon or xenobiotic receptor (AhR) is a ligand-activated nuclear transcription factor that binds structurally diverse environmental contaminants (for a recent review see [46]). Upon ligand binding, the AhR translocates to the nucleus and heterodimerizes with the ARNT [47,48]. The AhR–ARNT heterodimer binds to dioxin (or xenobiotic) responsive elements of genes encoding xenobiotic-metabolizing enzymes such as CYP1A1, CYP1A2, CYP1B1, glutathione S-transferase, UDP-glucuronosyltransferase1A6, and NAD(P)H quinone oxidoreductase-1 [49-51]. It is interesting to note that an additional antioxidant-responsive element has been discovered at the regulatory region of the latter three enzymes (reviewed in [52]). ARNT, on the contrary, appears to be a common dimerization partner for many nuclear transcription factor proteins [53-58], most probably playing the role of crosstalk integrator between diverse signaling pathways.
In addition to the induction of xenobiotic-metabolizing enzymes, the AhR may have other pleiotropic actions. Indeed, the AhR is involved in cell cycle regulation (through interaction with c-myc, p53 Rb and other cycle regulating elements), in induction of phase II metabolizing enzymes, in antioxidant response, and in induction of pro-apoptotic or anti-apoptotic genes (reviewed in [52]). Direct and/or indirect interactions of the AhR and NF-κB transcription factors have been reported (reviewed in [59]). In this respect, modification of NF-κB levels may modify the concentration of a number of apoptotic-related factors. Our results suggest that caffeic acid may bind to the AhR, being an inhibitor of its action, thus decreasing the transcription and activity of CYP1A1, both in basal and TCDD-treated cells. This implies either a direct effect on the enzyme molecule or a competition for the AhR with the (at present unknown) endogenous ligand of the AhR [52]. This latter hypothesis seems more probable as, in our experimental conditions, the same inhibitory pattern was found in either case (20% decrease). To our knowledge, this is the first report indicating an interaction of phenolic acids with the AhR.
It was recently shown that the effect of TCDD is exerted via binding to AhR. AhR–TCDD complex in turn induces CYP1A1 [15], resulting in a significant increase in the DNA binding activity of NF-κB and apolipoprotein 1, and a sustained activation of these two transcription factors. It is of note that this activation was blocked by antioxidants [15]. On the contrary, activation of the Fas receptor induces the phosphorylation of NF-κB transcription factor, resulting in induction of apoptosis in a number of various cell types [60,61]. Considering the role of NF-κB in cancer cell apoptosis [62], it is tentative to hypothesize that caffeic acid may act by inhibiting this pathway. This hypothesis is further supported by the stimulation effect of caffeic acid on pro-apoptotic Fas receptor.
In an effort to uncover other pathways of apoptosis, involved in the pro-apoptotic actions of phenolic acids on T47D cells, we have also tested their effects on the members of the other major family of apoptosis-related factors, the Bcl-2 proteins. Bcl-2 proteins are strongly expressed in human breast cancer cells, including the T47D cells [63]. Surprisingly, both phenolic acids elevated the protein content of the apoptosis-preventing Bcl-2 protein. It is possible that a Bcl-2-related mechanism is activated to short-term counteract the stress signals generated by the apoptosis-inducing factor FasL in order to rescue the cells from programmed death. Another possibility is that Bcl-2 related anti-apoptotic proteins, at the outer mitochondrial membrane, increased to counteract the pro-oxidant effects of phenolic acids locally [64].
Conclusions
The present work suggests that phenolic acids exert a direct antiproliferative action. This action is evident at low concentrations, comparable with those found in biological fluids after ingestion of foods rich in phenolic acids. Furthermore, the direct interaction with the AhR (caffeic acid), the interaction with the NOS system (PAA) and the pro-apoptotic effect of phenolic acids provide insights about their mode of action.
Competing interests
None declared.
Abbreviations
AhR = aryl hydrocarbon receptor; ARNT = aryl hydrocarbon nuclear translocator protein; BSA = bovine serum albumin; DMEM = Dulbecco's modified Eagle's medium; ELISA = enzyme-linked immunosorbent assay; eNOS = endothelial nitric oxide synthase; EROD = ethoxyresorufin-O-de-ethylase; FBS = fetal bovine serum; IC50= inhibitory concentration 50%; iNOS = inducible nitric oxide synthase; MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide; NF = nuclear factor; NO = nitric oxide; NOS = nitric oxide synthase; PAA = 3,4-dihy-droxy-phenylacetic acid; PBS = phosphate-buffered saline; PCR = polymerase chain reaction; RT = reverse transcriptase.
References
- Kampa M, Nistikaki A, Hatzoglou A, Kouimtzoglou H, Boskou D, Vercauteren J, Gravanis A, Castanas E. Phenolic acids and polyphenols are potent inhibitors of cancer cell proliferation: assessment and mechanisms of action. In: El-Hadrami I, Daayf F, editor. In Polyphenols 2002: Recent Advances in Polyphenols Research. Marrakech, Morocco: Groupe Polyphenols; 2002. pp. 62–93. [Google Scholar]
- Bonnesen C, Eggleston IM, Hayes JD. Dietary indoles and isothiocyanates that are generated from cruciferous vegetables can both stimulate apoptosis and confer protection against DNA damage in human colon cell lines. Cancer Res. 2001;61:6120–6130. [PubMed] [Google Scholar]
- Reiners JJ, Jr, Lee JY, Clift RE, Dudley DT, Myrand SP. PD98059 is an equipotent antagonist of the aryl hydrocarbon receptor and inhibitor of mitogen-activated protein kinase kinase. Mol Pharmacol. 1998;53:438–445. doi: 10.1124/mol.53.3.438. [DOI] [PubMed] [Google Scholar]
- Reiners JJ, Jr, Clift R, Mathieu P. Suppression of cell cycle progression by flavonoids: dependence on the aryl hydrocarbon receptor. Carcinogenesis. 1999;20:1561–1566. doi: 10.1093/carcin/20.8.1561. [DOI] [PubMed] [Google Scholar]
- Wang WL, Porter W, Burghardt R, Safe SH. Mechanism of inhibition of MDA-MB-468 breast cancer cell growth by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Carcinogenesis. 1997;18:925–933. doi: 10.1093/carcin/18.5.925. [DOI] [PubMed] [Google Scholar]
- Zacharewski TR, Bondy KL, McDonell P, Wu ZF. Antiestrogenic effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on 17 beta-estradiol-induced pS2 expression. Cancer Res. 1994;54:2707–2713. [PubMed] [Google Scholar]
- Brockdorff BL, Skouv J, Reiter BE, Lykkesfeldt AE. Increased expression of cytochrome p450 1A1 and 1B1 genes in anti-estrogen-resistant human breast cancer cell lines. Int J Cancer. 2000;88:902–906. doi: 10.1002/1097-0215(20001215)88:6<902::AID-IJC10>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Ciolino HP, Daschner PJ, Yeh GC. Resveratrol inhibits transcription of CYP1A1 in vitro by preventing activation of the aryl hydrocarbon receptor. Cancer Res. 1998;58:5707–5712. [PubMed] [Google Scholar]
- Ciolino HP, Yeh GC. The flavonoid galangin is an inhibitor of CYP1A1 activity and an agonist/antagonist of the aryl hydrocarbon receptor. Br J Cancer. 1999;79:1340–1346. doi: 10.1038/sj.bjc.6690216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinck PH, Forkert PG, Riddick DS, Okey AB, Michnovicz JJ, Bradlow HL. Ah receptor binding properties of indole carbinols and induction of hepatic estradiol hydroxylation. Biochem Pharmacol. 1993;45:1129–1136. doi: 10.1016/0006-2952(93)90258-X. [DOI] [PubMed] [Google Scholar]
- Le Ferrec E, Lagadic-Gossmann D, Rauch C, Bardiau C, Maheo K, Massiere F, Le Vee M, Guillouzo A, Morel F. Transcriptional induction of CYP1A1 by oltipraz in human Caco-2 cells is aryl hydrocarbon receptor- and calcium-dependent. J Biol Chem. 2002;277:24780–24787. doi: 10.1074/jbc.M111319200. [DOI] [PubMed] [Google Scholar]
- Lee JE, Safe S. Involvement of a post-transcriptional mechanism in the inhibition of CYP1A1 expression by resveratrol in breast cancer cells. Biochem Pharmacol. 2001;62:1113–1124. doi: 10.1016/S0006-2952(01)00763-8. [DOI] [PubMed] [Google Scholar]
- Liu RM, Vasiliou V, Zhu H, Duh JL, Tabor MW, Puga A, Nebert DW, Sainsbury M, Shertzer HG. Regulation of [Ah] gene battery enzymes and glutathione levels by 5,10-dihydro-indeno[1,2-b]indole in mouse hepatoma cell lines. Carcinogenesis. 1994;15:2347–2352. doi: 10.1093/carcin/15.10.2347. [DOI] [PubMed] [Google Scholar]
- McDougal A, Wormke M, Calvin J, Safe S. Tamoxifen-induced antitumorigenic/antiestrogenic action synergized by a selective aryl hydrocarbon receptor modulator. Cancer Res. 2001;61:3902–3907. [PubMed] [Google Scholar]
- Puga A, Barnes SJ, Chang C, Zhu H, Nephew KP, Khan SA, Shertzer HG. Activation of transcription factors activator protein-1 and nuclear factor-kappaB by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem Pharmacol. 2000;59:997–1005. doi: 10.1016/S0006-2952(99)00406-2. [DOI] [PubMed] [Google Scholar]
- Aherne S, O'Brien N. Dietary flavonols: chemistry, food content and metabolism. Nutrition. 2002;18:75–81. doi: 10.1016/S0899-9007(01)00695-5. [DOI] [PubMed] [Google Scholar]
- Hollman P, Katan M. Dietary flavonoids: intake, health effects and bioavailability. Food Chem Technol. 1999;37:937–942. doi: 10.1016/S0278-6915(99)00079-4. [DOI] [PubMed] [Google Scholar]
- Rice-Evans C, Miller N, Paganga G. Structure–antioxidant activity relationships of flavonoids and phenolic acids. Free Radic Biol Med. 1996;20:933–956. doi: 10.1016/0891-5849(95)02227-9. [DOI] [PubMed] [Google Scholar]
- Bianco A, Ucella N. Biophenolic components of olives. Food Res Int. 2000;33:475–485. doi: 10.1016/S0963-9969(00)00072-7. [DOI] [Google Scholar]
- Boskou D. Olive oil. In: Simopoulos AP, Visioli F, editor. In Mediterranean diets. Basel: Karger; 2000. pp. 56–74. [Google Scholar]
- Herrmann K. Occurence and contents of hydroxycinnamic and hydroxybenzoic acid compounds in foods. Crit Rev Food Sci Nutr. 1989;28:315–347. doi: 10.1080/10408398909527504. [DOI] [PubMed] [Google Scholar]
- Clifford MN. Chlorogenic acids and other cinnamates. J Sci Food Agric. 2000;80:1033–1043. doi: 10.1002/(SICI)1097-0010(20000515)80:7<1033::AID-JSFA595>3.3.CO;2-K. [DOI] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1973;65:53–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Marletta MA. Nitric oxide synthase structure and mechanism. J Biol Chem. 1993;268:12231–12234. [PubMed] [Google Scholar]
- Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- Kennedy S, Lorenzen A, James C, Collins B. Ethoxyresorufin-O-deethylase and porphyrin analysis chicken embryo hepatocyte cultures with a fluorescence multiwell plate reader. Anal Biochem. 1993;211:102–112. doi: 10.1006/abio.1993.1239. [DOI] [PubMed] [Google Scholar]
- Burke M, Thompson S, Weaver R, Wolf C, Mayer R. Cytochrome P450 specifities of alkoxyresorufin O-dealkylation in human and rat liver. Biochem Pharmacol. 1994;48:923–936. doi: 10.1016/0006-2952(94)90363-8. [DOI] [PubMed] [Google Scholar]
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis: flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Anexin-V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-I. [DOI] [PubMed] [Google Scholar]
- Boskou D, Visioli F. Biophenols in olive oil and olives. In: Pilar Vaquero M, Garcia-Arias T, Carbajal A, editor. Bioavailability of Micronutrients and Minor Dietary Compounds Metabolic and Technological Aspects. Trivandrum, India: Research Signpost; 2003. pp. 161–169. [Google Scholar]
- Papadopoulos G, Boskou D. Antioxidant effect of natural phenols on olive oil. J Am Oil Chem Soc. 1991;68:669–671. [Google Scholar]
- Damianaki A, Bakogeorgou E, Kampa M, Notas G, Hatzoglou A, Panagiotou S, Gemetzi C, Kouroumalis E, Martin P-M, Castanas E. Potent inhibitory action of red wine polyphenols on human breast cancer cells. J Cell Biochem. 2000;78:429–441. doi: 10.1002/1097-4644(20000901)78:3<429::AID-JCB8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Kampa M, Hatzoglou A, Thermos K, Castanas E. Direct interaction of opioids with alpha 1 adrenergic receptors in the human prostate cancer cell line LNCaP. Action on cell proliferation. HSBMB Newslett. 1999;46:30–35. [Google Scholar]
- Kampa M, Hatzoglou A, Notas G, Damianaki A, Bakogeorgou E, Gemetzi C, Kouroumalis E, Martin P-M, Castanas E. Wine antioxidant polyphenols inhibit the proliferation of human prostate cancer cell lines. Nutr Cancer. 2000;37:105–115. doi: 10.1207/S15327914NC372_16. [DOI] [PubMed] [Google Scholar]
- Casper RF, Quesne M, Rogers IM, Shirota T, Jolivet A, Milgrom E, Savouret JF. Resveratrol has antagonist activity on the aryl hydrocarbon receptor: implications for prevention of dioxin toxicity. Mol Pharmacol. 1999;56:784–790. [PubMed] [Google Scholar]
- Wink DA, Vodovotz Y, Laval J, Laval F, Dewhirst MW, Mitchell JB. The multifaceted roles of nitric oxide in cancer. Carcinogenesis. 1998;19:711–721. doi: 10.1093/carcin/19.5.711. [DOI] [PubMed] [Google Scholar]
- Bani D, Masini E, Bello MG, Bigazzi M, Sacchi TB. Relaxin activates the L-arginine-nitric oxide pathway in human breast cancer cells. Cancer Res. 1995;55:5272–5275. [PubMed] [Google Scholar]
- Adami A, Crivellente F, De Prati AC, Cavalieri E, Cuzzolin L, Tommasi M, Suzuki H, Benoni G. Biotransformation and cytotoxic properties of NO-donors on MCF7 and U251 cell lines. Life Sci. 1998;63:2097–2105. doi: 10.1016/S0024-3205(99)80006-X. [DOI] [PubMed] [Google Scholar]
- Farias-Eisner R, Chaudhuri G, Aeberhard E, Fukuto JM. The chemistry and tumoricidal activity of nitric oxide/hydrogen peroxide and the implications to cell resistance/susceptibility. J Biol Chem. 1996;271:6144–6151. doi: 10.1074/jbc.271.11.6144. [DOI] [PubMed] [Google Scholar]
- Darley-Usmar V, Wiseman H, Halliwell B. Nitric oxide and oxygen radicals: a question of balance. FEBS Lett. 1995;369:131–135. doi: 10.1016/0014-5793(95)00764-Z. [DOI] [PubMed] [Google Scholar]
- Visioli F, Bellosta S, Galli C. Oleuropein, the bitter principle of olives, enhances nitric oxide production by mouse macrophages. Life Sci. 1998;62:541–546. doi: 10.1016/S0024-3205(97)01150-8. [DOI] [PubMed] [Google Scholar]
- Ahmad N, Srivastava RC, Agarwal R, Mukhtar H. Nitric oxide synthase and skin tumor promotion. Biochem Biophys Res Commun. 1997;232:328–331. doi: 10.1006/bbrc.1997.6275. [DOI] [PubMed] [Google Scholar]
- Ho YS, Lee HM, Mou TC, Wang YJ, Lin JK. Suppression of nitric oxide-induced apoptosis by N-acetyl-L-cysteine through modulation of glutathione, bcl-2, and bax protein levels. Mol Carcinogen. 1997;19:101–113. doi: 10.1002/(SICI)1098-2744(199707)19:2<101::AID-MC5>3.3.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Thomsen LL, Miles DW. Role of nitric oxide in tumour progression: lessons from human tumours. Cancer Metastasis Rev. 1998;17:107–118. doi: 10.1023/A:1005912906436. [DOI] [PubMed] [Google Scholar]
- Mortensen K, Skouv J, Hougaard DM, Larsson LI. Endogenous endothelial cell nitric-oxide synthase modulates apoptosis in cultured breast cancer cells and is transcriptionally regulated by p53. J Biol Chem. 1999;274:37679–37684. doi: 10.1074/jbc.274.53.37679. [DOI] [PubMed] [Google Scholar]
- Radisavljevic Z. Nitric oxide suppression triggers apoptosis through the FKHRL1 (FOXO3A)/ROCK kinase pathway in human breast carcinoma cells. Cancer. 2003;97:1358–1363. doi: 10.1002/cncr.10081. [DOI] [PubMed] [Google Scholar]
- Safe S. Molecular biology of the Ah receptor and its role in carcinogenesis. Toxicol Lett. 2001;120:1–7. doi: 10.1016/S0378-4274(01)00301-0. [DOI] [PubMed] [Google Scholar]
- Reyes H, Reisz-Porszasz S, Hankinson O. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science. 1992;256:1193–1195. doi: 10.1126/science.256.5060.1193. [DOI] [PubMed] [Google Scholar]
- Hoffman EC, Reyes H, Chu F-F, Sander F, Conley LH, Brooks BA, Hankinson O. Cloning of a factor required for activity of the Ah (dioxin) receptor. Science. 1991;252:954–958. doi: 10.1126/science.1852076. [DOI] [PubMed] [Google Scholar]
- Safe S, Wormke M, Samudio I. Mechanisms of inhibitory aryl hydrocarbon receptor–estrogen receptor crosstalk in human breast cancer cells. J Mam Gland Biol Neoplasia. 2000;5:295–306. doi: 10.1023/A:1009550912337. [DOI] [PubMed] [Google Scholar]
- Kobayash IA, Numayamatsuruta K, Sogawa K, Fujii-Kuriyama Y. CBP/p300 functions as a possible transcriptional coactivator of Ah receptor nuclear translocator (Arnt) J Biochem. 1999;397:250–257. doi: 10.1093/oxfordjournals.jbchem.a021812. [DOI] [PubMed] [Google Scholar]
- Kumar MB, Tarpey RW, Perdew GH. Differential recruitment of coactivator RIP140 by Ah and estrogen receptors: absence of a role for LXXLL motifs. J Biol Chem. 1999;274:22155–22164. doi: 10.1074/jbc.274.32.22155. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol. 2000;59:65–85. doi: 10.1016/S0006-2952(99)00310-X. [DOI] [PubMed] [Google Scholar]
- Ema M, Morita M, Ikawa S, Tanaka M, Matsuda Y, Gotoh O, Saijoh Y, Fujii H, Hamada H, Kikuchi Y, Fujii-Kuriyama Y. Two new members of the murine Sim gene family are transcriptional repressors and show different expression patterns duting mouse embryogenesis. Mol Cell Biol. 1996;16:5865–5875. doi: 10.1128/mcb.16.10.5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ema M, Taya S, Yokotan IN, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1a regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci USA. 1997;94:4273–4278. doi: 10.1073/pnas.94.9.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffett P, Reece M, Pelltier J. The murine Sim-2 gene product inhibits transcription by active repression and functional interference. Mol Cell Biol. 1997;17:4933–4947. doi: 10.1128/mcb.17.9.4933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst MR, Fan CM, Tessier-Lavigne M, Hankinson O. Two murine homologs of the Drosophila single-minded protein that inteact with the mouse aryl hydrocarbon receptor nuclear translocator protein. J Biol Chem. 1997;272:4451–4457. doi: 10.1074/jbc.272.7.4451. [DOI] [PubMed] [Google Scholar]
- McGuire J, Coumailleau P, Whitelaw ML, Gustafsson JA, Poellinger L. The basic helix–loop–helix/PAS factor Sim is associated with hsp90. Implications for regulation by interaction with partner factors. J Biol Chem. 1995;270:31353–31357. doi: 10.1074/jbc.270.52.31353. [DOI] [PubMed] [Google Scholar]
- Hogenesch JB, Chan WK, Jackiw VH, Brown RC, Gu YZ, Pray-Grant M, Perdew GH, Bradfield CA. Characterization of a subset of the basic–loop–helix–PAS superfamily that interacts with components of the dioxin signaling pathway. J Biol Chem. 1997;272:8581–8593. doi: 10.1074/jbc.272.13.8581. [DOI] [PubMed] [Google Scholar]
- Tian Y, Rabson AB, Gallo MA. Ah receptor and NF-kappaB interactions: mechanisms and physiological implications. Chem Biol Interact. 2002;141:97–115. doi: 10.1016/S0009-2797(02)00068-6. [DOI] [PubMed] [Google Scholar]
- Lu B, Wang L, Medan D, Toledo D, Huang C, Chen F, Shi X, Rojanasakul Y. Regulation of Fas (CD95)-induced apoptosis by nuclear factor-kappaB and tumor necrosis factor-alpha in macrophages. Am J Physiol Cell Physiol. 2002;283:C831–C838. doi: 10.1152/ajpcell.00045.2002. [DOI] [PubMed] [Google Scholar]
- Afford SC, Ahmed-Choudhury J, Randhawa S, Russell C, Youster J, Crosby HA, Eliopoulos A, Hubscher SG, Young LS, Adams DH. CD40 activation-induced, Fas-dependent apoptosis and NF-kappaB/AP-1 signaling in human intrahepatic biliary epithelial cells. Faseb J. 2001;15:2345–2354. doi: 10.1096/fj.01-0088com. [DOI] [PubMed] [Google Scholar]
- Kim DW, Gazourian L, Quadri SA, Romieu-Mourez R, Sherr DH, Sonenshein GE. The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene. 2000;19:5498–5506. doi: 10.1038/sj.onc.1203945. [DOI] [PubMed] [Google Scholar]
- Kumar R, Vadlamudi R, Adam L. Apoptosis is mammary gland and cancer. Endocr Relat Cancer. 2000;7:257–269. doi: 10.1677/erc.0.0070257. [DOI] [PubMed] [Google Scholar]
- Loo G. Redox-sensitive mechanisms of phytochemical-mediated inhibition of cancer cell proliferation. J Nutr Biochem. 2003;14:64–73. doi: 10.1016/S0955-2863(02)00251-6. [DOI] [PubMed] [Google Scholar]