

Abstract
Background
The transforming growth factor-β (TGF-β) signaling pathway functions to prevent tumorigenesis, and loss of sensitivity to TGF-β-mediated cell cycle arrest is nearly ubiquitous among human cancers. Our previous studies demonstrated that rapamycin potentiates TGF-β-induced cell cycle arrest in nontransformed epithelial cells and partially restores TGF-β-induced growth arrest of some human cancer cell lines. Growth arrest correlated with increased binding of p21 and p27 to cyclin-dependent kinase-2 (Cdk2), and inhibition of Cdk2 kinase activity. However, it was unclear how TGF-β caused increased binding of p21 and p27 to Cdk2.
Methods
Cell fractionation and immunofluorescence microscopy experiments were performed to examine the effect of TGF-β on the intracellular localization of Cdk2, p21, and p27. Kinase assays were performed on cytoplasmic and nuclear extracts to determine how TGF-β altered Cdk2 activity in both subcellular compartments.
Results
In breast epithelial cells treatment with TGF-β induced a decrease in nuclear Cdk2 concentrations and relocalization of Cdk2 to the cytoplasm. Cdk2 relocalization to the cytoplasm correlated with dephosphorylation of nuclear retinoblastoma tumor suppressor protein and decreased nuclear Cdk2 activity. In these epithelial cell lines, p21 and p27 were localized primarily in the cytoplasm. Decreases in nuclear Cdk2 concentrations correlated with increased binding of Cdk2 to cytoplasmic p21 and p27.
Conclusion
Cooperative growth arrest induced by treatment with TGF-β + rapamycin causes inhibition of nuclear Cdk2 activity through multiple mechanisms, including Cdk2 relocalization to the cytoplasm, increased p27 and p21 binding to Cdk2, and increased phosphorylation of nuclear Cdk2 on its inhibitory site, Tyr15.
Keywords: cyclin-dependent kinase-2, localization, p21, p27, transforming growth factor-β
Introduction
Transforming growth factor-β (TGF-β) is a ubiquitous cytokine that was originally identified as a factor able to stimulate the transformation of certain fibroblast cell lines [1]. It is now known that TGF-β is one of the most potent secreted inhibitors of cell mitogenesis and has a critical role in negatively regulating epithelial, hematopoietic, and endothelial cell proliferation. Studies in animal models of cancer indicate that TGF-β mediates tumor suppression [2,3]. Several components of the TGF-β signaling pathway, including SMADs 2 and 4 and the TGF-β receptors type I and type II (TβRI and TβRII), are downregulated or mutationally inactivated in human cancers (reviewed in [4-6]). Furthermore, epidemiological studies indicate that polymorphisms of elements of the TGF-β signaling pathway influence the risk of developing breast cancer and colon cancer [7,8]. Together, these studies indicate that TGF-β acts as a tumor suppressor, but that this TGF-β function is abrogated in cancers. Interestingly, many cancers still respond to TGF-β by transcriptional activation of TGF-β-sensitive genes, despite having lost sensitivity to the growth inhibitory effects of TGF-β. These results might indicate that the pathways through which TGF-β regulates cell cycle progression and its other biological effects are at least partly separable. If this is so, it might be possible to specifically reactivate or potentiate TGF-β-induced growth arrest in human cancers without altering other TGF-β responses.
TGF-β induces cell cycle arrest through several interdependent mechanisms, including the downregulation of c-Myc [9,10], the inhibition of Cdk4 activity [11], the inhibition of Cdk2 activity [12,13], and the inhibition of E2F-dependent transcription [14-16]. The immunosuppressant rapamycin induces the arrest of cell proliferation through mechanisms that overlap with the mechanisms of TGF-β-mediated growth inhibition, including the downregulation of c-Myc [17], the inhibition of Cdk2 [18], and the inhibition of E2F-dependent transcription [19], suggesting that TGF-β and rapamycin might cooperate to induce cell cycle arrest. Our previous studies indicated that rapamycin potentiates the TGF-β-induced growth arrest of nontransformed cells in culture, and largely restores the TGF-β-mediated cell cycle arrest of epithelial cells transformed by c-Myc and E2F1 [20]. Growth arrest induced by TGF-β + rapamycin correlates well with increased binding of p21 and p27 to Cdk2, and with inhibition of Cdk2 activity. However, it was unclear from these studies what the mechanisms were through which TGF-β and rapamycin cooperated to increase p21 and p27 binding to Cdk2 in cases where TGF-β + rapamycin did not affect total concentrations of p21 or p27.
In the present studies we show that in several epithelial cell lines p21 and p27 are localized predominantly to the cytoplasm, whereas Cdk2 is present in both the nuclear and cytoplasmic compartments. TGF-β, and to a greater extent TGF-β + rapamycin, induce a decrease in nuclear Cdk2 concentrations coincident with an increased binding of cytoplasmic Cdk2 to cytoplasmic p21 and p27. We show for the first time that TGF-β induces a change in Cdk2 subcellular localization and inhibits nuclear Cdk2 activity. The decrease in nuclear Cdk2 concentrations and activity coincides with the dephosphorylation of nuclear nuclear retinoblastoma tumor suppressor protein (Rb) and cell cycle arrest.
Materials and methods
Cell culture
HaCaT, MDA-MB-231, and NMuMG cells were cultured as described previously [20]. Primary human mammary epithelial cells (HMECs) were acquired from the Vanderbilt-Ingram Cancer Center Breast Cell Repository. HMECs were grown in Dulbecco's modified Eagle's medium:F12 medium (dilution 1:1) (Gibco-BRL, Grand Island, NY) supplemented with 1% fetal bovine serum, 10 μg/ml ascorbic acid, 2 nM β-estradiol, 35 μg/ml bovine pituitary extract, 1 ng/ml cholera toxin, 12.5 ng/ml epidermal growth factor, 0.1 mM ethanolamine, 0.1 mM phospho-ethanolamine, 1 μg/ml hydrocortisone, 1 μg/ml insulin, 0.2 mM L-glutamine, 10 nM T3, 10 μg/ml transferrin, and 15 nM sodium selenite. HaCaT, MDA-MB-231, and NMuMG cells were plated 24 hours before treatment in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 10 μg/ml insulin. HMECs were plated and treated in the medium described above.
Preparation of cytoplasmic and nuclear cell extracts
Cytoplasmic and nuclear extracts were prepared with the NE-PER kit (Pierce Biotechnology, Inc, Rockford, IL) in accordance with the manufacturer's instructions. The protein concentrations of the extracts were determined with the Bradford assay (Bio-Rad Laboratories, Hercules, CA). Protein concentrations of extracts from cells subjected to different treatments were normalized. In all cases the cytoplasmic and nuclear extracts from a given cell treatment correspond to protein from an equal number of cells. HIRA (TUP1-like enhancer of split protein 1) was used as a nuclear protein marker to follow the efficiency of the fractionation procedure. HIRA is a protein previously shown to exist exclusively in the nucleus and is a substrate for Cdk2 phosphorylation [21]. In all of the extracts prepared, c-Myc and Sp1 were also present exclusively in the nuclear fractions (data not shown). c-Raf was used as a marker for the cytoplasm. Tubulin was also present exclusively in the cytoplasmic fractions, and except in the HaCaT cells, actin was also present exclusively in the cytoplasmic fractions (data not shown).
Immunoblotting and immune-complex kinase assays
Immunoblotting and immune-complex kinase assays were performed as described previously [20]. The results of kinase assays were revealed with a Fujifilm FLA-5000 PhosphoImager and quantified with Image Gauge version 3.4X software (Fujifilm Medical Systems, Inc, Alpharetta, GA). Antibodies specific for Cdk2 (sc-163), p21 (sc-6246), Raf (sc-227), Sp1 (sc-59), cyclin E (sc-481), and Cdk4 (sc-601) were purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA, USA). Antibodies specific for Rb phosphorylated on Thr821 (44-582Z) and Ser249/Thr252 (44-584Z) were from BioSource (Camarillo, CA). Antibodies recognizing total Rb (554136) and p27 (554069) were obtained from BD Transduction Laboratories (San Diego, CA). Dr Peter Adams (Fox Chase Cancer Center, Philadelphia, PA) generously provided a monoclonal antibody against HIRA. Antibodies recognizing Cdk2 phosphorylated on Thr160 (no. 2561) and Tyr15 (no. 9111) were obtained from Cell Signaling Technology, Inc (Beverly, MA).
Immunofluorescence and confocal immunofluorescence microscopy
Subconfluent cells, grown on 22 mm2 glass cover slips (VWR Scientific, Atlanta, GA), were treated for 24 h with 10 ng/ml TGF-β1. After treatment, cells were washed once with phosphate-buffered saline (PBS) and fixed with 1% paraformaldehyde/PBS for 20 min at 25°C. Cells were washed four times with PBS, permeabilized by incubation with 0.2% Triton X-100 (in PBS) for 10 min at 25°C, then washed twice more with PBS. Non-specific binding sites were blocked by incubating the cells for 2 h with 5% goat serum in PBS. Cells were then incubated with Cdk2 primary antibodies diluted in 5% serum/PBS (dilution 1:200) for 1 h at 25°C, followed by four washes with PBS. In control experiments Cdk2 staining was blocked by using the immunizing peptide (sc-163P; Santa Cruz Biotechnology, Inc, Santa Cruz, CA) at a 100-fold molar excess over the antibody.
Cells were incubated for 1 h at 25°C with goat anti-rabbit biotinylated secondary antibodies (Vector Laboratories, Inc, Burlingame, CA) diluted 1:250 in 5% serum/PBS, washed four times with PBS, and incubated with streptavidin-conjugated Cy3 (Sigma, St Louis, MO) diluted in 5% serum/PBS (dilution 1:1000) for 1 h at 25°C. Cells were washed four times with PBS, nuclei were counter-stained by incubation with 1 mM 4',6'-diamidino-2-phenylindole (DAPI; Sigma) for 5 min at 25°C, and cells were washed three times with PBS. Cover slips were mounted on 25 mm × 75 mm microslides (Fisher Scientific, Pittsburgh, PA) using AquaPolyMount (Polysciences, Warrington, PA). Actin filaments were stained using Texas Red-X phalloidin (Molecular Probes, Inc, Eugene, OR) at a dilution of 1:200. Fluorescent images were captured with a Zeiss Axiophot upright microscope and a Princeton Instruments cooled charge-coupled device digital camera. Confocal images were captured with a Zeiss LSM510 confocal microscope (Carl Zeiss MicroImaging, Inc, Thornwood, NY).
Results
TGF-β induces Cdk2 relocalization from the nucleus to the cytoplasm
TGF-β1 and rapamycin cooperate to induce growth arrest by increasing p21 and p27 binding to Cdk2, resulting in Cdk2 inhibition [20]. We hypothesized that p21 and p27 binding to Cdk2 induced by TGF-β1 + rapamycin might result from changes in the intracellular localization of p21 and p27 or Cdk2. To test this hypothesis, nontransformed mouse mammary gland epithelial NMuMG cells, MDA-MB-231 human mammary carcinoma cells, HMECs, or HaCaT human keratinocytes were treated for 24 h with normal growth medium, or medium containing 10 ng/ml TGF-β1 plus 100 nM rapamycin, and fractionated into cytoplasmic and nuclear extracts. In agreement with our previous observations [20], in parallel experiments the treatment of NMuMG, MDA-MB-231, HaCaT, and HMECs with TGF-β + rapamycin for 24 h induced a potent cell cycle arrest in 3H-thymidine incorporation and sequential cell counting assays (data not shown). The resulting extracts were analyzed by immunoblotting. c-Raf was used as a cytoplasmic marker, and HIRA [21] was used as a nuclear marker. In NMuMG cells, MDA-MB-231 cells, and HMECs, Cdk2 was distributed between the cytoplasmic and nuclear fractions, but treatment with TGF-β1 + rapamycin resulted in a selective decrease in nuclear Cdk2 concentrations (Fig. 1a). Similar results were obtained with Balb/MK mouse keratinocytes (data not shown).
Figure 1.
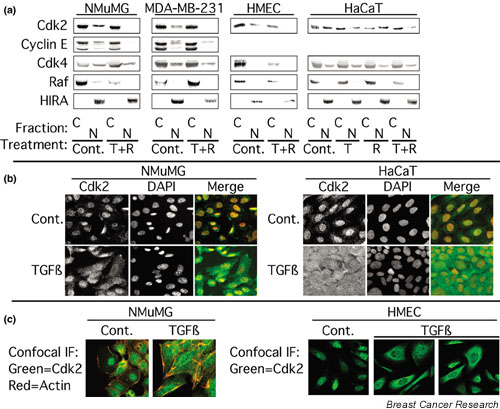
TGF-β alters Cdk2 localization. (a) NMuMG, MDA-MB-231, primary human mammary epithelial (HMEC), or HaCaT cells were treated for 24 h with normal growth medium (Cont.), 10 ng/ml TGF-β1 (T), 100 nM rapamycin (R), or a combination of both (T + R). The cells were subjected to subcellular fractionation into nuclear (N) and cytoplasmic (C) extracts as described in Materials and Methods and the resulting fractions were analyzed by immunoblotting with the indicated antibodies. HIRA served as a marker for the nucleus, and Raf was used as a marker for the cytoplasm. (b) NMuMG cells (left panel) or HaCaT cells (right panel) were treated for 24 h with 10 ng/ml TGF-β1 and the cells were stained with antibodies specific for Cdk2 (shown in green), and with 4',6'-diamidino-2-phenylindole (DAPI; shown in red) as a nuclear stain, as described in Materials and Methods. In the merged images, Cdk2 and DAPI staining are overlaid. (c) NMuMG cells (left panel) treated as in (b) were stained for Cdk2 (green) and actin (red) and were imaged by confocal microscopy. HMECs (right panel) treated as in (b) were stained for Cdk2 (green) and were imaged by confocal microscopy.
Under the same conditions, TGF-β1 + rapamycin had no effect on Cdk4 (Fig. 1a) or Cdc2 localization (data not shown). In NMuMG cells and MDA-MB-231 cells, cyclin E localization paralleled Cdk2 localization, suggesting that effects of treatment on intracellular localization were specific for Cdk2 complexes. In HaCaT cells, TGF-β1 caused a small decrease in nuclear Cdk2 concentrations, rapamycin had no effect, but TGF-β1 + rapamycin induced a larger decrease in nuclear Cdk2 concentrations than TGF-β1 alone (Fig. 1a). Because we showed previously that TGF-β1 and rapamycin cooperate to induce cell cycle arrest [20], these results are consistent with the idea that decreases induced in nuclear Cdk2 concentrations by TGF-β1 + rapamycin constitute a novel mechanism by which TGF-β1 mediates cell cycle arrest.
To confirm the results of the cell fractionation experiments we performed immunofluorescence microscopy studies. As shown in Fig. 1b (left panel), Cdk2 was largely nuclear in control NMuMG cells, but TGF-β1 induced a diffuse localization of Cdk2 throughout the entire cell volume. No staining was observed when the Cdk2 primary antibody was omitted, or blocked with an excess of immunizing peptide (data not shown). TGF-β1 + rapamycin induced changes in Cdk2 localization similar to those induced by TGF-β1 alone (data not shown); however, cells treated with TGF-β1 + rapamycin were more rounded than TGF-β1-treated cells, making it difficult to distinguish between cytoplasmic and nuclear Cdk2 staining. In HaCaT cells (Fig. 1b, right panel) Cdk2 was predominantly nuclear, but treatment with TGF-β1 induced a diffuse localization of Cdk2 throughout the entire cell volume, as observed in NMuMG cells. Similar results were obtained in immunofluorescence microscopy experiments employing HMECs (data not shown). The results in Fig. 1b were confirmed by confocal immunofluorescence microscopy (Fig. 1c and data not shown). These experiments demonstrate that Cdk2 was localized to the nucleus of untreated NMuMG cells, but that TGF-β1 induced the relocalization of Cdk2 throughout the entire cell volume, as demarcated by cortical actin staining. In HMECs (Fig. 1c, right panel) TGF-β1 treatment induced a more cytoplasmic localization of Cdk2 with partial exclusion from the nucleus. Taken together, our results indicate that TGF-β1, and to a greater extent TGF-β1 + rapamycin, induced a partial relocalization of Cdk2 from the nucleus to the cytoplasm.
Decreased nuclear Cdk2 concentrations correlate with dephosphorylation of nuclear Rb
Cdk2 has several nuclear substrates whose phosphorylation is thought to play a role in cell cycle progression [21-25]. We hypothesized that TGF-β1 + rapamycin-induced decreases in nuclear Cdk2 concentrations might result in cell cycle arrest through decreased phosphorylation of nuclear Cdk2 substrates. In particular, Rb is an important Cdk2 substrate, and decreased phosphorylation of nuclear Rb should inhibit E2F-dependent transcription and induce cell cycle arrest. Thus, cytoplasmic Cdk2 relocalization induced by TGF-β1 + rapamycin could explain the ability of TGF-β1 + rapamycin to inhibit E2F-dependent transcription and induce growth arrest.
To test the hypothesis that Cdk2 relocalization induced by TGF-β1 + rapamycin results in Rb dephosphorylation, cytoplasmic and nuclear extracts were analyzed by immunoblotting with phospho-specific and total Rb antibodies. In untreated NMuMG cells, Rb was predominantly nuclear and existed in two forms, a slower-migrating hyperphosphorylated form and a faster-migrating hypophosphorylated form (Fig. 2). In NMuMG cells treated with TGF-β1 + rapamycin, Rb existed almost exclusively in the hypophosphorylated form. To examine changes in the phosphorylation status of individual Rb phosphorylation sites we employed phosphorylation-site-specific antibodies against phospho-Rb(Thr821) and phospho-Rb(Ser249/ Thr252). Thr821 is phosphorylated in vitro preferentially by cyclin E/cyclin A–Cdk2 complexes and phosphorylation at this site inhibits E2F binding [26]. Ser249/Thr252 are phosphorylated in vitro preferentially by cyclin D1–Cdk4 complexes, and the biological function of these phosphorylation sites is unknown. In NMuMG cells, Rb phosphorylated on Thr821 was observed primarily in the cytoplasm and phosphorylation levels did not change significantly with TGF-β1 + rapamycin treatment. Interestingly, Rb phosphorylated on Ser249/Thr252 existed in both the nucleus and cytoplasm of untreated NMuMG cells, but was not observed in either compartment of NMuMG cells treated with TGF-β1 + rapamycin.
Figure 2.
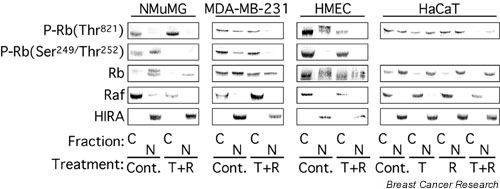
TGF-β and rapamycin together induce the dephosphorylation of nuclear Rb. The cytoplasmic (C) and nuclear (N) extracts prepared in Fig. 1a were analyzed by immunoblotting with antibodies specific for Rb phosphorylated on Thr821 (P-Rb(Thr821)), Rb phosphorylated on Ser249/Thr252 (P-Rb(Ser249/Thr252)), and total Rb. Where two bands of total Rb are apparent, the upper band represents hyperphosphorylated Rb and the lower band hypophosphorylated Rb. Cont., control; T + R, 10 ng/ml TGF-β1 plus 100 nM rapamycin; HMEC, primary human mammary epithelial cells.
In untreated MDA-MB-231 cells, both unphosphorylated Rb and Rb phosphorylated on Thr821 and Ser249/Thr252 were observed in both the nuclear and cytoplasmic compartments. Strikingly, treatment with TGF-β1 + rapamycin specifically decreased the phosphorylation of Rb at Thr821 and Ser249/Thr252 in the nuclear compartment but not in the cytoplasm. In HMECs, TGF-β1 + rapamycin induced the dephosphorylation of nuclear, but not cytoplasmic, Rb at Thr821. In contrast, Rb phosphorylated on Ser249/Thr252 was observed primarily in the cytoplasm, and phosphorylation was minimally decreased by treatment with TGF-β1 + rapamycin. In HaCaT cells, Rb was primarily nuclear, whereas Rb phosphorylated on Thr821 was distributed evenly between the nucleus and the cytoplasm. Rb phosphorylation on Ser249/Thr252 was not detected in HaCaT cells. Importantly, TGF-β1 and rapamycin cooperated to induce Rb dephosphorylation on Thr821 in both the nucleus and the cytoplasm and resulted in a mobility shift consistent with Rb dephosphorylation. Taken together, these results indicate that TGF-β1 + rapamycin blocked the phosphorylation of nuclear Rb in all cases, but only blocked the phosphorylation of cytoplasmic Rb in specific instances.
p21 and p27 are predominantly cytoplasmic in mammary epithelial cells and human keratinocytes
Because TGF-β1 + rapamycin induced the cytoplasmic relocalization of Cdk2 (Fig. 1), and our previous studies [20] demonstrated that TGF-β1 and rapamycin cooperate to induce p21 and p27 binding to Cdk2, we examined the intracellular localization of p21 and p27. Immunoblot analysis of the cytoplasmic and nuclear extracts demonstrated that in NMuMG cells, MDA-MB-231 cells, and HMECs, p27 was localized primarily to the cytoplasm of both untreated cells and cells treated with TGF-β1 + rapamycin (Fig. 3). The low concentrations of nuclear p27 in NMuMG and HMECs was further decreased by treatment with TGF-β + rapamycin. p27 expression was not detected in HaCaT cells. p21 expression was primarily cytoplasmic in the four cell lines examined in both untreated cells and cells treated with TGF-β1 + rapamycin. TGF-β1 + rapamycin significantly increased total p21 concentrations in both MDA-MB-231 and HaCaT cells without affecting p21 intracellular localization. Taken together, the results in Figs 1 and 3 indicate that TGF-β1 induces increased p21 and p27 concentrations in the cytoplasm that correlate temporally with Cdk2 relocalization to this compartment.
Figure 3.
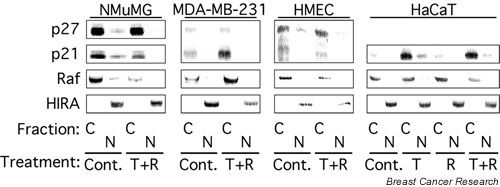
p21 and p27 are localized primarily to the cytoplasm of NMuMG, MDA-MB-231, HMEC, and HaCaT cells. The cytoplasmic (C) and nuclear (N) subcellular fractions prepared in Fig. 1a were analyzed by immunoblot with antibodies specific for p27 or p21. HIRA served as a marker for the nucleus and Raf was used as a marker for the cytoplasm. Cont., control; T + R, 10 ng/ml TGF-β1 plus 100 nM rapamycin; HMEC, primary human mammary epithelial cells.
Cytoplasmic relocalization of Cdk2 coincides with increased Cdk2 binding to cytoplasmic p21 and p27
The hypothesis that the cytoplasmic localization of Cdk2 results in increased binding of Cdk2 to cytoplasmic p21 and p27 was tested by preparing cytoplasmic and nuclear extracts from treated and untreated MDA-MB-231 (Fig. 4a) or NMuMG (Fig. 4b) cells. Treated cells were subjected to subcellular fractionation and the fractions were analyzed in Cdk2 coimmunoprecipitation experiments. As expected, loss of Cdk2 from the nuclear fraction of MDA-MB-231 cells after treatment with TGF-β + rapamycin resulted in increased binding of Cdk2 to cytoplasmic p21 (Fig. 4a). p27 binding to Cdk2 was not detected in MDA-MB-231 cell extracts. In NMuMG cells, treatment with TGF-β1 resulted in decreased Cdk2 concentrations in the nucleus that correlated with an increase in Cdk2 binding to p27 in the cytoplasm (Fig. 4b). Interestingly, rapamycin alone had little effect on Cdk2 localization or Cdk2 binding to p27; however, rapamycin potentiated a TGF-β1-induced decrease in nuclear concentrations of Cdk2 that coincided with increased binding of Cdk2 to primarily cytoplasmic, but also nuclear, p27. These results are consistent with our previous report that TGF-β1 and rapamycin cooperate to induce p27 binding to Cdk2 [20], but extend these observations by demonstrating that binding of Cdk2 to cytoplasmic p27 and p21 coincides with decreased nuclear Cdk2 concentrations.
Figure 4.
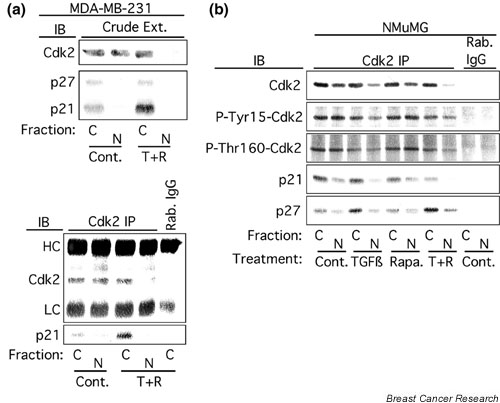
Cdk2 relocalization to the cytoplasm correlates with increased binding to cytoplasmic p21 and p27. (a) Crude MDA-MB-231 cytoplasmic (C) or nuclear (N) extracts (Crude ext.) from cells treated as in Fig. 1 were subjected to immunoblot (IB) analysis with antibodies specific for Cdk2, p27, or p21 (top panel), or immunoprecipitated with a Cdk2 antibody or antibody from preimmune rabbit IgG (Rab. IgG), and the immunoprecipitates (Cdk2 IP) were probed with antibodies specific for Cdk2, p27, or p21 (bottom panel). (b) NMuMG cytoplasmic or nuclear extracts from cells treated as in Fig. 1 were subjected to immunoprecipitation with Cdk2 antibody or preimmune rabbit IgG, and the immunoprecipitates were probed with antibodies specific for Cdk2, Cdk2 phosphorylated on Tyr15 (P-Tyr15–Cdk2), Cdk2 phosphorylated on Thr160 (P-Thr160-Cdk2), p27, or p21. Cont., control; Rapa., 100 nM rapamycin; T + R, 10 ng/ml transforming growth factor-β1 plus 100 nM rapamycin. LC, immunoglobulin light chain; HC, immunoglobulin heavy chain.
We hypothesized that the phosphorylation state of Cdk2 at activating or inhibitory sites might differ between the nucleus and the cytoplasm. To test this hypothesis we examined Cdk2 phosphorylation at the inhibitory phosphorylation site, Tyr15, and on the activating phosphorylation site, Thr160, using phosphorylation state-specific antibodies (Fig. 4b). Although concentrations of nuclear Cdk2 were quite low in the NMuMG cells treated with TGF-β1 + rapamycin, concentrations of phospho-Tyr15 Cdk2 were decreased only slightly, indicating that the stoichiometry of phosphorylation of nuclear Cdk2 on the inhibitory site, Tyr15, was increased by treatment with TGF-β1 + rapamycin. Similarly, the amount of p27 binding to nuclear Cdk2 was also increased in NMuMG cells treated with TGF-β1 + rapamycin. Taken together, these results suggest that TGF-β1 and rapamycin cooperate to inhibit the activity of nuclear Cdk2 through several mechanisms including decreasing the total concentration of nuclear Cdk2, increasing the stoichiometry of phosphorylation of nuclear Cdk2 on the inhibitory site, Tyr15, and increasing the stoichiometry of binding of p27 to nuclear Cdk2.
TGF-β and rapamycin cooperate to inhibit nuclear, but not cytoplasmic, Cdk2 activity
To verify that TGF-β1 and rapamycin cooperate to inhibit nuclear Cdk2 activity specifically, we prepared nuclear and cytoplasmic extracts from NMuMG cells treated as in Fig. 4. The extracts were immunoprecipitated with antibodies specific for Cdk2 and subjected to immune-complex kinase assays with histone H1 as the substrate. Results were analyzed by phosphorimaging and immunoblot analysis of the kinase reactions (Fig. 5). Interestingly, treatment with TGF-β1 preferentially inhibited cytoplasmic Cdk2 activity while having little effect on nuclear Cdk2 activity. Rapamycin weakly inhibited cytoplasmic Cdk2 activity and increased nuclear Cdk2 activity. Strikingly, treatment with TGF-β1 + rapamycin inhibited cytoplasmic Cdk2 activity to a similar extent to that of TGF-β1 alone, but inhibited nuclear Cdk2 activity much more potently than TGF-β alone.
Figure 5.
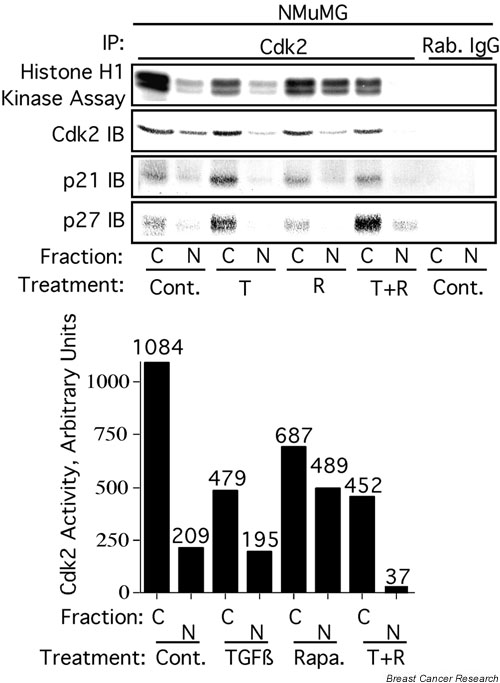
Transforming growth factor-β (TGF-β) and rapamycin cooperate to inhibit nuclear Cdk2 activity specifically. NMuMG cytoplasmic (C) or nuclear (N) extracts from cells treated as in Fig. 1 were subjected to immunoprecipitation (IP) with anti-Cdk2 antibody or antibody from preimmune serum (Rab. IgG) as in Fig. 4. The immunoprecipitates were subjected to immune-complex kinase assays with histone H1 as the substrate. The kinase reactions/immunoprecipitates were analyzed by phosphorimaging (histone H1 kinase assay) and by immunoblotting (IB) with antibodies specific for Cdk2, p21, or p27. The bar graph in the lower panel is a quantification of the histone H1 kinase assays. Cont., control; T, 10 ng/ml TGF-β1; R, 100 nM rapamycin.
It is interesting that neither TGF-β nor rapamycin alone exhibited a strong inhibitory effect on soluble nuclear Cdk2 activity, whereas together they strongly inhibited soluble nuclear Cdk2 activity. Comparison of Figs 4b and 5 indicates that this cooperative behavior results from a combination of effects. First, rapamycin potentiates the TGF-β-induced decrease in soluble nuclear Cdk2 concentrations. Rapamycin alone has little effect on Cdk2 localization. Second, although the concentration of soluble nuclear Cdk2 is significantly lower in cells treated with TGF-β + rapamycin relative to TGF-β-treated cells, the concentration of Tyr15-phosphorylated Cdk2 is similar in both treatments. Rapamycin alone has no effect on the degree of Cdk2 phosphorylation on Tyr15. This suggests that rapamycin increases the stoichiometry of soluble nuclear Cdk2 phosphorylation on its inhibitory site, Tyr15, in the context of TGF-β treatment. It is unclear how rapamycin alone stimulates soluble nuclear Cdk2 activity, but it might result from increased phosphorylation of Cdk2 on its activating site, Thr160. Third, although the concentration of soluble nuclear Cdk2 is significantly lower in cells treated with TGF-β + rapamycin relative to TGF-β-treated cells, the amount of p27 that coimmunoprecipitates with Cdk2 is actually higher. Rapamycin alone has no effect on the binding of p27 to soluble nuclear Cdk2. This suggests that rapamycin increases the stoichiometry of binding of p27 to soluble nuclear Cdk2 in the context of TGF-β treatment.
Discussion
It has long been thought that the cyclin-dependent kinases Cdk2 and Cdk4/Cdk6 have essential and unique roles in regulating cell division. This presumption was weakened by the observation that cyclin D1 deficiency is rescued by a knockin of cyclin E [27]. More recently it was shown that mice lacking cyclin E [28] or Cdk2 [29,30] are viable. Additionally, several cancer cell lines proliferate normally in the absence of Cdk2 [31]. These observations call into question the relevance of Cdk2 inhibition by TGF-β + rapamycin. However, it should be pointed out that several cancer cell lines are arrested in the cell cycle by the overexpression of dominant-negative Cdk2 or p27 [31]. Furthermore, cells lacking cyclin E are relatively resistant to oncogenic transformation [28], and cells lacking Cdk2 exhibit increased contact inhibition and delayed entry into S phase [30]. It was proposed [28] that a low cyclin-dependent kinase activity might be required for normal cell proliferation, but that a higher cyclin-dependent kinase activity might be required for oncogenic transformation. Thus, even if Cdk2 is not absolutely required for cell proliferation it could serve as a central target of growth inhibitory, tumor suppressive pathways involved in preventing uncontrolled cell proliferation.
We propose that in addition to these considerations concerning Cdk2 kinase activity, the role of Cdk2 in cell cycle progression must also take into account the biological effects of regulated changes in the subunit composition of Cdk2 complexes and changes in the intracellular localization of Cdk2 and the components of Cdk2 complexes. We have previously demonstrated that TGF-β and rapamycin cooperate to inhibit Cdk2 activity and that this parallels changes in the subunit composition of Cdk2 complexes [20]. Here we present data indicating that Cdk2 function might also be regulated by its intracellular localization and that Cdk2 intracellular localization is linked with changes in the subunit structure of Cdk2 complexes. In short, these results suggest that TGF-β and rapamycin together regulate Cdk2 function in mammary epithelial cells through a novel mode involving changes in Cdk2 intracellular localization, and that in some cell types p21 and p27 might act not only to inhibit Cdk2 directly but also to control Cdk2 intracellular localization and access to substrates.
Cdk2 is often assumed to reside primarily in the nucleus; however, several reports indicate that Cdk2 localization is dynamically regulated. Serum deprivation induces Cdk2 localization to the cytoplasm, and it was concluded that serum-induced Cdk2 relocalization from the cytoplasm to the nucleus overcomes the primary barrier to Cdk2 activation and the exit of cells from quiescence [32]. Similarly, the ability of human cytomegalovirus infection to drive the proliferation of serum-deprived or contact-inhibited cells correlates with Cdk2 translocation from the cytoplasm into the nucleus [33]. More recently it was shown that vitamin D induces Cdk2 relocalization to the cytoplasm [34].
Cdk2 localization to the cytoplasm represents an effective mechanism for inducing growth arrest because several proteins that are intimately involved in cell proliferation are localized predominantly to the nucleus and are substrates for Cdk2-dependent phosphorylation. These proteins include HIRA [21], p220NPAT [24], BRCA1 [22], and nucleophosmin [25]. Cyclin A–Cdk2 complexes promote DNA replication in part through the phosphorylation of nuclear Cdc6, resulting in Cdc6 export from the nucleus and subsequent degradation [23]. Further, the pocket proteins Rb, p107, and p130 exist at least partly in the nucleus, and Cdk2 regulates E2F-dependent transcription through the phosphorylation of the pocket proteins. Despite the importance of Cdk2 localization in regulating cell proliferation, little is known about the mechanisms controlling the subcellular targeting of Cdk2.
Previously we showed [20] that cell cycle arrest induced by TGF-β + rapamycin results from Cdk2 inhibition due to increased p21 and p27 binding to Cdk2. The observations presented here extend these observations by showing that, in addition to directly binding and inhibiting Cdk2, cytoplasmically localized p21 and p27 might also act as targeting subunits to sequester Cdk2 away from nuclear targets such as Rb. The data in this study suggest that the mechanism behind Cdk2 relocalization probably involves p21 and p27 because decreases in nuclear Cdk2 concentrations correlate with increased binding of Cdk2 to cytoplasmic p21 and p27. Further, rapamycin potentiates TGF-β1-induced Cdk2 translocation in parallel with the potentiation of Cdk2 binding to cytoplasmic p21 and p27. The observation that TGF-β1 and rapamycin together induce p27 binding to Cdk2 in both the nucleus and the cytoplasm suggests that increased Cdk2 binding to p27 is independent of intracellular localization and suggests that Cdk2 relocalization to the cytoplasm results from increased binding of Cdk2 to the predominantly cytoplasmic p27.
Immunofluorescence microscopy studies indicate that some Cdk2 is still present in the nucleus after TGF-β treatment, but nuclear extracts from TGF-β-treated cells contain very little Cdk2. To address this apparent discrepancy, cell pellets remaining after the nuclear extraction procedure were extracted by boiling in Laemmli SDS sample buffer and the resulting extracts were subjected to immunoblot analysis. These experiments indicated that in both untreated and TGF-β-treated cells Cdk2 exists in the nucleus in an insoluble form that can be extracted by SDS (data not shown). Thus, two pools of nuclear Cdk2 exist, a soluble nuclear fraction and an insoluble nuclear fraction. Our results indicate that the soluble nuclear Cdk2 fraction is the most relevant target of TGF-β and rapamycin with respect to Rb dephosphorylation and cell cycle arrest induced by TGF-β and rapamycin.
Like Cdk2, p21 and p27 are generally considered to be nuclear proteins; however, several reports indicate that p21 and p27 distribution between the nucleus and the cytoplasm is regulated. p27 intracellular localization is controlled by several phosphorylation sites, including Ser10 [35-37], Thr157 [38-40], and Thr198 [41,42]. Phosphorylation of Ser10 induces p27 export to the cytoplasm and stabilizes the protein, and the phosphorylation status of this residue does not alter the intrinsic ability of p27 to inhibit Cdk2 activity [35]. Thr157 phosphorylation induces the cytoplasmic localization of p27; however, Thr157, unlike Ser10, is not conserved in murine p27. Therefore Thr157 phosphorylation cannot explain the cytoplasmic localization of p27 observed in NMuMG cells. Phosphorylation of p27 on Thr198 promotes p27 binding to 14-3-3 proteins and localization to the cytoplasm [41,42].
On the basis of the assumption that Cdk2 is predominantly nuclear, it has been presumed that cytoplasmic p21 and p27 are not involved in regulating the cell cycle. In fact it has been proposed that p21 and p27 have different functions in the cytoplasm and might act as cytoplasmic oncoproteins [43,44], in part by inhibiting apoptosis. A recent report [45] suggests that p27 in the cytoplasm interacts with F-actin and regulates cell motility. Although p21 and p27 probably have different functions in the cytoplasm as opposed to the nucleus, our results indicate that cytoplasmic p21 and p27 bind Cdk2 and might in fact serve to control Cdk2 localization in a regulated manner. These results are consistent with those in a recent report [46] that identified an isoform of p27, p27(Kip1R), that was retained in the cytoplasm but inhibited cell proliferation in a manner similar to the more extensively studied p27 isoform, p27(Kip1). Thus, categorizing p21 and p27 as tumor suppressors when localized to the nucleus, and oncogenic when localized to the cytoplasm, is probably an oversimplification. Further studies are warranted to determine the precise cellular functions of cytoplasmically localized p21, p27, and Cdk2.
Conclusion
Our results demonstrate for the first time that TGF-β1 regulates Cdk2 intracellular localization in mammary epithelial cells and human keratinocyes and that this process is involved in potent and specific inhibition of nuclear Cdk2 activity that results in the dephosphorylation of nuclear Rb. Regulation of Cdk2 localization by TGF-β1 and TGF-β1 + rapamycin correlates with increased Cdk2 binding to cytoplasmic p21 and p27, in the absence of changes in total p27 concentrations. These results suggest that p21 and p27 not only serve as Cdk2 inhibitory proteins but might also serve as Cdk2 intracellular targeting subunits. Studies are currently under way to determine the precise mechanisms by which TGF-β1 regulates the intracellular localization and association of Cdk2 with p21 and p27.
Competing interests
None declared.
Abbreviations
Cdk2 = cyclin-dependent kinase-2; Cdk4 = cyclin-dependent kinase-4; DAPI = 4',6'-diamidino-2-phenylindole; HMECs = primary human mammary epithelial cells; PBS = phosphate-buffered saline; Rb = retinoblastoma tumor suppressor protein; TβRI and TβRII = TGF-β receptors type I and type II; TGF-β = transforming growth factor-β.
Acknowledgments
Acknowledgements
We thank Dr Peter Adams (Fox Chase Cancer Center, Philadelphia, PA) for providing the anti-HIRA antibody, and Dr Harold Moses and Dr Jennifer Pietenpol for reading the manuscript critically. This work was supported by NIH Grant R01-CA93651 [BL], Vanderbilt-Ingram Comprehensive Cancer Center support grant CA68485, the Frances Williams Preston Laboratories of the TJ Martell Foundation, and the Robert J Kleberg, Jr, and Helen C. Kleberg Foundation. The Vanderbilt University Cell Imaging Core Resource is supported by NIH grants CA68485 and DK20593.
References
- Keski-Oja J, Leof EB, Lyons RM, Coffey RJ, Jr, Moses HL. Transforming growth factors and control of neoplastic cell growth. J Cell Biochem. 1987;33:95–107. doi: 10.1002/jcb.240330204. [DOI] [PubMed] [Google Scholar]
- Pierce DF, Jr, Gorska AE, Chytil A, Meise KS, Page DL, Coffey RJ, Jr, Moses HL. Mammary tumor suppression by transforming growth factor β1 transgene expression. Proc Natl Acad Sci USA. 1995;92:4254–4258. doi: 10.1073/pnas.92.10.4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottinger EP, Jakubczak JL, Haines DC, Bagnall K, Wakefield LM. Transgenic mice overexpressing a dominant-negative mutant type II transforming growth factor beta receptor show enhanced tumorigenesis in the mammary gland and lung in response to the carcinogen 7,12-dimethylbenz-[a]-anthracene. Cancer Res. 1997;57:5564–5570. [PubMed] [Google Scholar]
- Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- Kelly DL, Rizzino A. Growth regulatory factors and carcinogenesis: the roles played by transforming growth factor beta, its receptors and signaling pathways. AntiCancer Res. 1999;19:4791–4807. [PubMed] [Google Scholar]
- Kretzschmar M. Transforming growth factor-beta and breast cancer: Transforming growth factor-beta/SMAD signaling defects and cancer. Breast Cancer Res. 2000;2:107–115. doi: 10.1186/bcr42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv E, Cauley J, Morin PA, Saiz R, Browner WS. Association between the T29→C polymorphism in the transforming growth factor β1 gene and breast cancer among elderly white women: the study of osteoporotic fractures. JAMA. 2001;285:2859–2863. doi: 10.1001/jama.285.22.2859. [DOI] [PubMed] [Google Scholar]
- Pasche B, Kolachana P, Nafa K, Satagopan J, Chen YG, Lo RS, Brener D, Yang D, Kirstein L, Oddoux C, Ostrer H, Vineis P, Varesco L, Jhanwar S, Luzzatto L, Massague J, Offit K. TβR-I(6A) is a candidate tumor susceptibility allele. Cancer Res. 1999;59:5678–5682. [PubMed] [Google Scholar]
- Pietenpol JA, Holt JT, Stein RW, Moses HL. Transforming growth factor β1 suppression of c-myc gene transcription: role in inhibition of keratinocyte proliferation. Proc Natl Acad Sci USA. 1990;87:3758–3762. doi: 10.1073/pnas.87.10.3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrow MG, Kawabata M, Aakre M, Moses HL. Overexpression of the c-Myc oncoprotein blocks the growth-inhibitory response but is required for the mitogenic effects of transforming growth factor β1. Proc Natl Acad Sci USA. 1995;92:3239–3243. doi: 10.1073/pnas.92.8.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-β-induced cell cycle arrest. Nature. 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- Cipriano SC, Chen YQ. Insensitivity to growth inhibition by TGF-β1 correlates with a lack of inhibition of the CDK2 activity in prostate carcinoma cells. Oncogene. 1998;17:1549–1556. doi: 10.1038/sj.onc.1202069. [DOI] [PubMed] [Google Scholar]
- Schwarz JK, Bassing CH, Kovesdi I, Datto MB, Blazing M, George S, Wang XF, Nevins JR. Expression of the E2F1 transcription factor overcomes type β transforming growth factor-mediated growth suppression. Proc Natl Acad Sci USA. 1995;92:483–487. doi: 10.1073/pnas.92.2.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JM, Hu PP, Shen X, Yu Y, Wang XF. E2F4-RB and E2F4-p107 complexes suppress gene expression by transforming growth factor β through E2F binding sites. Proc Natl Acad Sci USA. 1997;94:4948–4953. doi: 10.1073/pnas.94.10.4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iavarone A, Massague J. E2F and histone deacetylase mediate transforming growth factor beta repression of cdc25A during keratinocyte cell cycle arrest. Mol Cell Biol. 1999;19:916–922. doi: 10.1128/mcb.19.1.916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Stoneley M, Willis AE. Translational induction of the c-myc oncogene via activation of the FRAP/TOR signalling pathway. Oncogene. 1998;17:769–780. doi: 10.1038/sj.onc.1201990. [DOI] [PubMed] [Google Scholar]
- Morice WG, Wiederrecht G, Brunn GJ, Siekierka JJ, Abraham RT. Rapamycin inhibition of interleukin-2-dependent p33cdk2 and p34cdc2 kinase activation in T lymphocytes. J Biol Chem. 1993;268:22737–22745. [PubMed] [Google Scholar]
- Brennan P, Babbage JW, Thomas G, Cantrell D. p70s6k integrates phosphatidylinositol 3-kinase and rapamycin-regulated signals for E2F regulation in T lymphocytes. Mol Cell Biol. 1999;19:4729–4738. doi: 10.1128/mcb.19.7.4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law BK, Chytil A, Dumont N, Hamilton EG, Waltner-Law ME, Aakre ME, Covington C, Moses HL. Rapamycin potentiates transforming growth factor beta-induced growth arrest in nontransformed, oncogene-transformed, and human cancer cells. Mol Cell Biol. 2002;22:8184–8198. doi: 10.1128/MCB.22.23.8184-8198.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall C, Nelson DM, Ye X, Baker K, DeCaprio JA, Seeholzer S, Lipinski M, Adams PD. HIRA, the human homologue of yeast Hir1p and Hir2p, is a novel cyclin-cdk2 substrate whose expression blocks S-phase progression. Mol Cell Biol. 2001;21:1854–1865. doi: 10.1128/MCB.21.5.1854-1865.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffner H, Jiang W, Craig AG, Hunter T, Verma IM. BRCA1 is phosphorylated at serine 1497 in vivo at a cyclin-dependent kinase 2 phosphorylation site. Mol Cell Biol. 1999;19:4843–4854. doi: 10.1128/mcb.19.7.4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Wells NJ, Hunter T. Multistep regulation of DNA replication by Cdk phosphorylation of HsCdc6. Proc Natl Acad Sci USA. 1999;96:6193–6198. doi: 10.1073/pnas.96.11.6193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Kennedy BK, Lawrence BD, Barbie DA, Matera AG, Fletcher JA, Harlow E. NPAT links cyclin E-Cdk2 to the regulation of replication-dependent histone gene transcription. Genes Dev. 2000;14:2283–2297. doi: 10.1101/gad.827700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuyama Y, Horn HF, Kawamura K, Tarapore P, Fukasawa K. Specific phosphorylation of nucleophosmin on Thr199 by cyclin-dependent kinase 2-cyclin E and its role in centrosome duplication. J Biol Chem. 2001;276:21529–21537. doi: 10.1074/jbc.M100014200. [DOI] [PubMed] [Google Scholar]
- Knudsen ES, Wang JY. Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol Cell Biol. 1997;17:5771–5783. doi: 10.1128/mcb.17.10.5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Whoriskey W, Park MY, Bronson RT, Medema RH, Li T, Weinberg RA, Sicinski P. Rescue of cyclin D1 deficiency by knockin cyclin E. Cell. 1999;97:767–777. doi: 10.1016/s0092-8674(00)80788-6. [DOI] [PubMed] [Google Scholar]
- Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S, Rideout WM, Bronson RT, Gardner H, Sicinski P. Cyclin E ablation in the mouse. Cell. 2003;114:431–443. doi: 10.1016/s0092-8674(03)00645-7. [DOI] [PubMed] [Google Scholar]
- Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M, Barbacid M. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet. 2003;35:25–31. doi: 10.1038/ng1232. [DOI] [PubMed] [Google Scholar]
- Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P. Cdk2 knockout mice are viable. Curr Biol. 2003;13:1775–1785. doi: 10.1016/j.cub.2003.09.024. [DOI] [PubMed] [Google Scholar]
- Tetsu O, McCormick F. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell. 2003;3:233–245. doi: 10.1016/s1535-6108(03)00053-9. [DOI] [PubMed] [Google Scholar]
- Bresnahan WA, Boldogh I, Ma T, Albrecht T, Thompson EA. Cyclin E/Cdk2 activity is controlled by different mechanisms in the G0 and G1 phases of the cell cycle. Cell Growth Differ. 1996;7:1283–1290. [PubMed] [Google Scholar]
- Bresnahan WA, Thompson EA, Albrecht T. Human cytomegalovirus infection results in altered Cdk2 subcellular localization. J Gen Virol. 1997;78:1993–1997. doi: 10.1099/0022-1317-78-8-1993. [DOI] [PubMed] [Google Scholar]
- Yang ES, Burnstein KL. Vitamin D inhibits G1 to S phase progression in LNCaP prostate cancer cells through p27Kip1 stabilization and Cdk2 mislocalization to the cytoplasm. J Biol Chem. 2003;78:46862–46868. doi: 10.1074/jbc.M306340200. [DOI] [PubMed] [Google Scholar]
- Ishida N, Kitagawa M, Hatakeyama S, Nakayama K. Phosphorylation at serine 10, a major phosphorylation site of p27Kip1, increases its protein stability. J Biol Chem. 2000;275:25146–25154. doi: 10.1074/jbc.M001144200. [DOI] [PubMed] [Google Scholar]
- Rodier G, Montagnoli A, Di Marcotullio L, Coulombe P, Draetta GF, Pagano M, Meloche S. p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis. EMBO J. 2001;20:6672–6682. doi: 10.1093/emboj/20.23.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm M, Yoshimoto T, Crook MF, Nallamshetty S, True A, Nabel GJ, Nabel EG. A growth factor-dependent nuclear kinase phosphorylates p27Kip1 and regulates cell cycle progression. EMBO J. 2002;21:3390–3401. doi: 10.1093/emboj/cdf343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, Arteaga CL. PKB/Akt mediates cell-cycle progression by phosphorylation of p27Kip1 at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8:1145–1152. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, Franssen E, Slingerland JM. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–1160. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- Viglietto G, Motti ML, Bruni P, Melillo RM, D'Alessio A, Califano D, Vinci F, Chiappetta G, Tsichlis P, Bellacosa A, Fusco A, Santoro M. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27Kip1 by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med. 2002;8:1136–1144. doi: 10.1038/nm762. [DOI] [PubMed] [Google Scholar]
- Fujita N, Sato S, Tsuruo T. Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2003;278:49254–49260. doi: 10.1074/jbc.M306614200. [DOI] [PubMed] [Google Scholar]
- Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2002;277:28706–28713. doi: 10.1074/jbc.M203668200. [DOI] [PubMed] [Google Scholar]
- Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol. 2003;13:65–70. doi: 10.1016/S0962-8924(02)00043-0. [DOI] [PubMed] [Google Scholar]
- Reed SI. Cell cycling? Check your brakes. Nat Cell Biol. 2002;4:E199–E201. doi: 10.1038/ncb0802-e199. [DOI] [PubMed] [Google Scholar]
- McAllister SS, Becker-Hapak M, Pintucci G, Pagano M, Dowdy SF. Novel p27kip1 C-terminal scatter domain mediates Rac-dependent cell migration independent of cell cycle arrest functions. Mol Cell Biol. 2003;23:216–228. doi: 10.1128/MCB.23.1.216-228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano K, Zeng Y, Hirano M, Nishimura J, Kanaide H. Sequence requirement for nuclear localization and growth inhibition of p27Kip1R, a degradation-resistant isoform of p27Kip1. J Cell Biochem. 2003;89:191–202. doi: 10.1002/jcb.10499. [DOI] [PubMed] [Google Scholar]