

Abstract
Introduction
In order to study metastatic disease, we employed the use of two related polyomavirus middle T transgenic mouse tumor transplant models of mammary carcinoma (termed Met and Db) that display significant differences in metastatic potential.
Methods
Through suppression subtractive hybridization coupled to the microarray, we found osteopontin (OPN) to be a highly expressed gene in the tumors of the metastatic mouse model, and a lowly expressed gene in the tumors of the lowly metastatic mouse model. We further analyzed the role of OPN in this model by examining sense and antisense constructs using in vitro and in vivo methods.
Results
With in vivo metastasis assays, the antisense Met cells showed no metastatic tumor formation to the lungs of recipient mice, while wild-type Met cells, with higher levels of OPN, showed significant amounts of metastasis. The Db cells showed a significantly reduced metastasis rate in the in vivo metastasis assay as compared with the Met cells. Db cells with enforced overexpression of OPN showed elevated levels of OPN but did not demonstrate an increase in the rate of metastasis compared with the wild-type Db cells.
Conclusions
We conclude that OPN is an essential regulator of the metastatic phenotype seen in polyomavirus middle T-induced mammary tumors. Yet OPN expression alone is not sufficient to cause metastasis. These data suggest a link between metastasis and phosphatidylinositol-3-kinase-mediated transcriptional upregulation of OPN, but additional phosphatidylinositol-3-kinase-regulated genes may be essential in precipitating the metastasis phenotype in the polyomavirus middle T model.
Keywords: breast cancer, mammary gland, metastasis, migration, osteopontin
Introduction
Breast cancer is among the most common human cancers, affecting one in every eight women and accounting for an estimated 192,000 cases and more than 40,000 deaths in the United States during 2001. One of the significant predictors of breast cancer prognosis is regional and distant metastasis; yet the mechanism of metastasis and the ability to predict it are far from being fully understood.
From both a clinical and experimental perspective, a more detailed understanding of the mechanisms of metastasis is needed in order to identify better diagnostic markers and therapeutic approaches.
In order to study breast cancer, many investigators have used human derived cell lines that have yielded significant insight into the biology of breast cancer; yet these models remain an artificial in vitro system that may not reflect the in vivo biology. Over the past decade, the laboratory mouse has become the modern vehicle for human disease studies [1], and genetically engineered mice are particularly popular models for breast cancer (reviewed in [2-4]). The mouse offers an in vivo experimental system that can be manipulated and studied in great detail in order to understand the complex biology of cancer.
To study metastatic disease, we have employed the use of two related polyomavirus (PyV) middle T (mT) transgenic mouse mammary carcinoma transplant lines (termed Met and Db) that display significant differences in metastatic potential [2,5-7]. The PyV-mT system is an ideal model to study mammary carcinoma because there is rapid mammary tumor formation with 100% penetrance, because the histopathology of the PyV-mT tumors mimics that of human breast carcinoma, and because, in many cases, the human and mouse derived tumors are indistinguishable [8,9]. The PyV-mT transgene has also been used as an alternative, or a surrogate, for erbB2 in the mouse [10] as the two molecules activate similar pathways. Desai and colleagues [11] have recently shown that mammary tumors derived from PyV-mT mice and from erbB2 transgenic mice show striking similarities at the transciptome level.
Over the past few years c-erbB2 (HER2) has been shown to be a key molecule in human breast cancer [12], being overexpressed in 30–40% of human breast cancer cases [13]. PyV is capable of transforming cells by triggering signal transduction pathways that have been implicated as activated by erbB2, through interactions between its mT gene product and key cellular signaling proteins such as c-Src [14,15], Shc, and phosphatidylinositol 3-kinase (PI3-K) [16], which have all been implicated as important in human breast cancer. Specifically, with respect to PI3-K, mT interacts with the 85 kDa regulatory subunit of PI3-K to activate PI3-K [17], which has been implicated as a key signal in carcinoma invasion [18].
The Met model, derived from transgenic mice constructed with the wild-type PyV-mT line, develops rapid mammary carcinoma in all animals with 100% pulmonary metastasis [5]. In contrast, the Db model derived from animals with double site-directed mutations at amino acid residues 315 and 322 of the PyV-mT is decoupled from the PI3-K pathway. The Db model has 100% penetrance of mammary tumor but exhibits significantly fewer pulmonary metastases (9%) [7,16,19]. Similar metastatic rates were observed when Met and Db tumor lines were transplanted into syngeneic FVB mice [20]. The site-directed mutations at residues 315 and 322 interfere with the recruitment of the p85 subunit of PI3-K [16], and thereby PI3-K is not recruited and activated. This subtle difference in the mT gene significantly affects the metastatic phenotype.
Because disruption of the PI3-K pathway in this model suppresses metastasis, and because of the purported role of PI3-K in carcinoma cell invasion, we wanted to identify the key regulators that are differentially expressed between the Met tumors and the Db tumors, as well as to validate their role in metastasis. We utilized suppression subtractive hybridization (SSH) coupled to the microarray in order to initially identify targets [21,22]. SSH is a technique that generates cDNA libraries of transcripts that are differentially expressed between two populations of cells, normalizing for variable mRNA abundance by enrichment of rare transcripts [23].
Using SSH in conjunction with microarrays, we identified osteopontin (OPN) as a very highly expressed gene that was expressed more than threefold higher in the Met tumor lines compared with the Db tumor lines. Immunohistochemical analysis of the tumors confirmed the presence of OPN with a marked cytoplasmic stain in the Met tumor cells and an absence of staining in the Db tumor cells. Cell lines were developed from these tumors, and they showed differential expression of OPN in similar proportions. Furthermore, OPN expression directly correlated with the ability of the tumor-derived cell lines to migrate in response to serum. Correspondingly, antisense-mediated attenuation of OPN expression reduced the migratory capacity of Met cells (in a cell line termed Met-As), while enforced OPN expression in Db cells stimulated migration (in a cell line termed Db-S). In in vivo metastasis experiments, the wild-type Met cells showed a high rate of metastasis to the lungs of recipient mice, whereas the Met-As cells did not metastasize to the lungs of recipient mice. In contrast, the Db cells manifested a metastasis rate that was significantly lower than that of wild-type Met cells. However, the Db-S cells, with elevated OPN levels, showed no increase in the metastasis rate when compared with the wild-type Db cells.
Scanning electron microscopy (SEM) of the OPN antisense transfected Met line shows morphologic change consistent with a less metastatic phenotype, while the OPN-transfected Db cells show morphologic change consistent with a more metastatic phenotype. Given this evidence, we conclude that PyV-mT induces a metastatic phenotype, at least in part, through the PI3-K-mediated transcriptional upregulation of the matrix protein OPN, although enforced expression of OPN in the lowly metastatic Db cells does not increase their metastasis rate. We therefore conclude that OPN is essential for metastasis in the PyV-mT system, but that elevated OPN expression alone is not sufficient to induce metastasis.
Methods
Mammary tumor transplants and cell lines
Three-week-old nontransgenic FVB female mice were anesthetized with a 1:12 xylazine/1: 24 ketamine cocktail in PBS, administered at 0.012 ml/g intraperitoneally. The abdominal (#4) mammary glands were cleared of growing epithelium as previously described [24,25]. Tissue segments (1 mm3) of Met tumors and of Db tumors were transplanted into the cleared mammary fat pads. The animals were killed 28–35 days later by a double dose of anesthesia, and tumor tissue was harvested from the mammary fat pads of recipient nontransgenic FVB female mice. Mammary tumor cell lines were developed from the Met tumors and the Db tumors with a collagenase digestion technique coupled with differential centrifugation, as previously described [19,26,27]. The cells lines were then maintained and grown in 100 mm tissue culture dishes using DMEM (pH 7.4) supplemented with 10% heat-inactivated fetal bovine serum (FBS), and 1 × antibiotic/antimycotic (Invitrogen Corp., Carlsbad, CA, USA) [24,25].
cDNA subtracted libraries and arrays
RNA was isolated from the tumors by homogenizing with a Polytron generator (Brinkmann Instruments, Westbury, NY, USA), using the acid guanidinium thiocyanate–phenol–chloroform extraction method [28]. Subtracted Met libraries and Db libraries were generated using the PCR-Select cDNA Subtraction Kit, as recommended by the manufacturer (BD Biosciences Clontech, Palo Alto, CA, USA). This yielded two libraries: one containing genes expressed in Met but not in Db, and one containing genes expressed in Db but not in Met. Both subtracted libraries were subsequently subcloned into the TA-cloning vector (Invitrogen Corp.), picked and cultured in 96-well plates, PCR-amplified, and arrayed onto silanized slides as described previously [29].
Each SSH library was then fluorescently labeled with either Cy3-conjugated or Cy5-conjugated dCTP and hybridized to the arrays to screen for true differential expression. The slides were scanned on an Affymetrix 418 two-color fluorescent scanner (Affymetrix Inc., Santa Clara, CA, USA), each creating a 16-bit TIFF image. The images were downloaded from the scanner and analyzed using a commercial software program (ImaGene; BioDiscovery Inc., Marina Del Rey, CA, USA), which automatically detects the regions of fluorescent signal, determines the signal intensity, performs global normalization, and compiles the data into a Microsoft Excel spreadsheet for further analysis. The program determines the Cy3/Cy5 expression ratios for each data point. All ratios are log-transformed (log base 2 for simplicity).
The subcloned arrayed products were then categorized and ranked with respect to differential expression and signal intensity. Products that showed both differential expression and high signal intensity were sequenced for identity. The products were sequenced by Davis Sequencing (Davis, CA, USA).
RNA analysis and northern blots
Met and Db cells were plated at 1 × 106 cells/100 mm plate and were allowed to attach overnight. Total RNA was extracted using the TRIzol reagent (Invitrogen Corp.). Met tumors and Db tumors were harvested and the total RNA was isolated by homogenizing with a Dounce homogenizer (Kimble Kontes, Vineland, NJ, USA), using the acid guanidinium thiocyanate–phenol–chloroform extraction method [28].
For northern blot analysis, total RNA was quantitated, denatured, and electrophoresed in an agarose-formaldehyde gel, with subsequent transfer to a Hybond N nylon filter by standard methods (Amersham Biosciences Corp., Piscataway, NJ, USA). Procedures for pre-hybridization, hybridization, filter washing, and filter stripping were performed as described previously [30]. Filters were hybridized with 32P-labeled cDNA probes of mouse OPN (AF808; R&D Systems, Minneapolis, MN, USA) and 18s ribosomal RNA (as a control). The blots were scanned using a phosphorimager and normalized to 18s rRNA levels to determine the relative changes in OPN mRNA.
Histology and immunohistochemistry
Tumors were fixed overnight in formalin, were paraffin-embedded, and were sectioned at a thickness of 5 μm. Normal FVB kidney and lactating mammary glands were similarly processed as normal control tissue for OPN. Routine H&E staining was performed. Microwaving in a citrate buffer was used for antigen retrieval. For the OPN immunohistochemistry studies, anti-OPN (AF808; R&D Systems) was used at a dilution of 1: 800 and was detected using the Vector ABC kit (Vector Laboratories, Burlingame, CA, USA). The images were captured using a Kontron Model 3012 CCD camera on an Olympus BH2 microscope. They were digitized using Photoshop 6.0 with the Kontron ProgRes 'plug-in' module, were color enhanced, were balanced for contrast and were printed using a Kodak 8650 dye sublimation printer.
Production and characterization of stably transfected cell lines
To generate the constructs used for the production of stable cell lines, the full-length murine OPN cDNA was obtained by reverse-transcription PCR from Met cells using the 5' primer GGA ATT CCT TGC TTG GGT TTG CAG TCT TCT G and the 3' primer GCT CTA GAA CAT TCG TTA CAA GAT TTA TTC AC. These primers were designed to amplify the entire coding sequence, excluding the 3' termination codon and including restriction sites EcoRI and XbaI (underlined). The products were ligated in either the sense or the antisense orientation into the EcoRI and XbaI sites of the eukaryotic expression vector pCDNA3.1(+/-). The constructs were then transfected into Db and Met cell lines using standard calcium phosphate transfection methods, after which the cells were treated with G418 sulfate (Geneticin; Invitrogen Corp.) to select for the integration of the construct into the genome [31].
Control cell lines were generated by transfecting Met cells and Db cells with the empty pCDNA3.1(+/-) vector. The stably integrated lines were identified by northern blot analysis using the neomycin and OPN gene-specific probes [32]. The stable cell lines were then propagated in selective media that contained 600 μg/ml G418 sulfate. The cells were incubated in a humidified atmosphere of 95% air and 5% CO2 at 37°C. For subculturing, cells were trypsinized (0.05% trypsin and ethylenediamine tetraacetic acid [EDTA]) and were plated at 1 × 106 cells/100 mm plate.
Migration assays
For the migration assay, 24-well transwell units with 0.8 μm porosity polycarbonate filters were used (Corning, Inc., Big Flats, NY, USA). Cells were harvested by trypsinization and were counted. Then 5 × 103 cells were resuspended in 100 μl serum-free medium and placed in the upper chamber of the transwell unit. The bottom chamber contained 500 μl medium with serum as a chemoattractant [33]. The transwell units were then placed in a 37°C humidified 95% air/5% CO2 atmosphere for 18–24 hours. Cells on the upper surface were then removed by wiping with a cotton swab, while the remaining cells were fixed with 10% formalin and stained with hematoxylin. Visualization of the membrane and cells was performed with a phase contrast microscope. Migration was quantitated by determining the number of cells (averaged among four representative fields using a 20 × objective) that had migrated to the lower side of the filter.
In vivo tail vein metastasis analysis
The tail vein inoculation assay, which tests the ability of the cells to survive in the blood stream, to colonize, and to grow in the lung [34], was used to test the metastatic potential of the cells. Met cells, Met-As cells, Db cells, and Db-S cells were grown under selection (using 600 μg/ml G418 sulfate) in DMEM (pH 7.4) supplemented with 10% heat-inactivated FBS and 1 × antibiotic/antimycotic. The cells were incubated at 37°C in a humidified atmosphere containing 95% air and 5% CO2. Cells were harvested at 80–90% confluence by trypsinization (0.05% trypsin–EDTA), washed, and resuspended in 1 × PBS (pH 7.4).
For injection with each cell line, eight nude mice (Nu/Nu; Jackson Laboratories, Bar Habor, ME, USA) were used (four mice at each of two concentrations: 1 × 104 cells and 1 × 106 cells). The animals were anesthetized with 0.6% sodium pentobarbital at a dosage of 10 μl/g body weight, and cells in a total volume of 50 μl were injected into the tail vein of each mouse. Mice were monitored for a total of 6 weeks and were then sacrificed. The lungs of each mouse were analyzed by whole mount hematoxylin staining for metastatic foci.
For whole mount lung preparation, each mouse was sacrificed by anesthetic overdose. The lungs were perfused with 4% buffered formalin via cannulation of the trachea and were forcibly inflated to ensure adequate distribution of fixative. The lungs were removed and the lobes separated and placed into a Tissue-Tek uni-cassette (Sakura Finetek USA, Torrance, CA, USA) for fixation and whole mount processing. After fixation, the lungs were dehydrated with alcohol (70%, 95%, and absolute alcohol for 1 hour each step), cleared with xylene, and then hydrated for Mayer's hematoxylin staining. After 3 min of staining, the excess stain was removed with a 1% hydrochloric acid solution. The lungs were again dehydrated with alcohol, cleared in xylene, and then placed in methyl salicylate for microscopic analysis and photography.
In vivo mammary fat pad metastasis analysis
Met cells, Db cells, Met-As cells, and Db-S cells were assayed for metastatic potential by injection into the mammary fat pad of FVB mice. Each cell line was grown under G418 sulfate (600 μg/ml) selection in DMEM (pH 7.4) supplemented with 10% heat-inactivated FBS and 1 × antibiotic/antimycotic. These lines were grown at 37°C in a humidified atmosphere containing 95% air and 5% CO2, and cells were then harvested at 80–90% confluence. For the harvest, cells were trypsinized (0.05% trypsin–EDTA), washed with 1 × PBS (pH 7.4), and resuspended to a concentration of 1 × 105 cells/μl in Matrigel (BD Biosciences, Franklin Lakes, NJ, USA).
Ten FVB mice were used for each cell line. The mice were anesthetized and surgery was performed to expose the left #4 fat pad of each mouse. Twenty microliters of the cell–Matrigel suspension were injected into each exposed fat pad, and the mice were allowed to recover. The animals were monitored for tumor growth, and tumors were removed from the animals when they reached 1.5 cm in diameter (22–26 days postinjection). The mice were again allowed to recover and were monitored for the development of lung metastases. This tumor removal survival surgery is necessary because these tumors become very large at 3 weeks (approximately 1.5 cm), which significantly affects the health of the animals. The mice were sacrificed and lungs from the animals were harvested at 42 days postinjection. Whole mount analysis was performed on the lungs in order to identify and enumerate metastases.
Protein extraction and western blot analysis
To assess OPN protein levels, the cells/tumors were placed in a hypotonic solution (10 mM Tris) and homogenized using a Dounce homogenizer. The cell lysate was centrifuged at 15,000 × g for 10 min, and the supernatant was designated as the crude cytoplasmic fraction [31]. The intracellular protein concentration was then quantitated using the BCA assay (Pierce Chemical Company, Rockford, IL, USA). Protein preparations were subjected to SDS-PAGE on 12% gels, were transferred onto a polyvinylidenedifluoride membrane, and were incubated in a blocking solution (5% w/v Blotto). This was followed by an overnight incubation with goat antimouse OPN antibody. After being washed, membranes were incubated with horseradish peroxidase-conjugated rabbit antigoat IgG for 1 hour [31]. The enzyme bound to OPN was visualized using an enhanced chemiluminescence detection kit (ECL; AP Biotech, Piscataway, NJ, USA). The blot was then stripped and incubated with antimouse β-actin antibody as a control (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The blots were scanned using a phosphorimager and normalized to the β-actin levels to determine the relative changes in levels of OPN protein.
Scanning electron microscopy
Whole filter assemblies (0.8 μm porosity; Corning, Inc.) were fixed in modified Karnovsky's fixative consisting of 2% paraformaldehyde and 2.5% glutaraldehyde in 0.06 M Sorenson's phosphate buffer (pH 7.2–7.4). Fixation continued for 4 hours to overnight at 4°C. The filter assemblies were then rinsed three times for 2 min each in 0.1 M phosphate buffer at 4°C. Dehydration was started in 30% ethanol and then in 50% ethanol for 5 min each at 4°C. Dehydration continued in 70%, 95%, and 100% ethanol for 5 min each at room temperature. The filter assemblies were then critical point dried utilizing liquid carbon dioxide.
After drying, the filter disks were carefully cut from the filter assemblies and mounted onto SEM specimen supports using multiple thin strips of double-sided Scotch tape. One-half of each filter disk was mounted with the 'top' facing upward, and the other half was mounted with the 'bottom' facing upward. The samples were then coated with gold in a sputter coater (Ted Pella, Inc., Redding, CA, USA). The samples were examined and photographed using a Philips XL30TMP scanning electron microscope (FEICO/Philips, Hillsboro, OR, USA). Images were recorded onto 4' × 5' Polaroid 55P/N film as well as using direct digital imaging.
Results
We expected this model to provide a well-controlled system in which to identify metastasis-related genes because the Met tumors and Db tumors were propagated in the same mouse strain (FVB). The only difference between the transgenes is the presence of the two point mutations in the middle T gene in the Db mice. Our ultimate goal was to identify a key regulator that is differentially expressed in the Met tumors and in the Db tumors.
Identification of OPN using SSH and microarray analysis
Following SSH and array analysis (Fig. 1 shows a section of the SSH array), clones that displayed differential expression and high signal intensity were sequenced. We sequenced clones in batches of 10. OPN was identified three times in the first sequencing pass. The identification of a single gene three times in this initial sequencing batch was unexpected because the normalization step in the SSH procedure is designed to give equal value to both highly expressed and lowly expressed genes. In subsequent experiments, we have found that this procedure does tend to lead to clone redundancy in highly expressed genes that are differentially expressed. The OPN clones had the highest intensity signals on the array. We confirmed this in subsequent experiments using Affymetrix U74Av2 GeneChip arrays, where OPN is seen to be overexpressed in the Met tumor compared with in the Db tumors (data not shown). Since OPN displayed significant differential expression and a high level of intensity, we decided to analyze this gene in more detail before continuing the sequencing of the SSH libraries.
Figure 1.
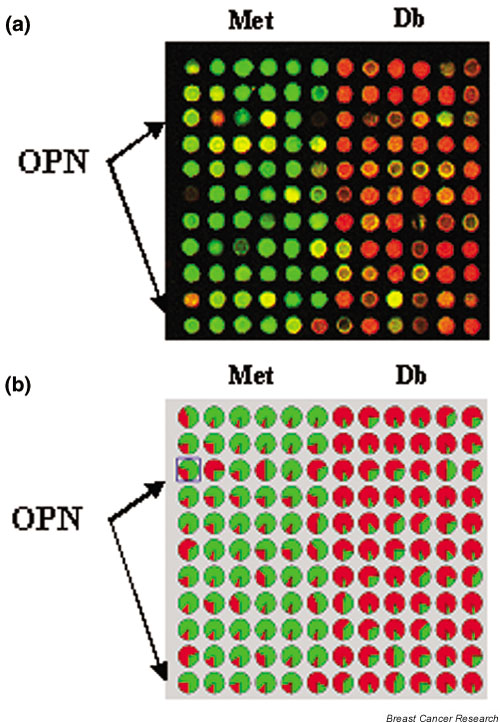
Representative region of the suppression subtractive hybridization (SSH) microarray of the Met and Db tumor lines. (a) The SSH libraries were arrayed, probed with fluorescence-labeled aliquots of each library, and scanned with a two-color fluorescent scanner. Green represents higher expression in the Met line, and red represents higher expression in the Db line. (b) Analysis of the same section of the SSH array with the GenePie function of ImaGene (BioDiscovery, Inc., Marina Del Rey, CA, USA) shows the relative percentage of signal intensity at each spot of the two fluorescent labels. Probes were selected for sequencing on the basis of high expression and differential expression. Osteopontin (OPN) was identified as being highly differentially represented in the Met subtraction library and is marked with arrows on the array in (a) and (b).
OPN transcript expression in Met and Db cell lines and tumors
Northern blot analysis was used to validate the array results in the Met tumors and the Db tumors. With respect to OPN expression, transcript levels were fourfold higher in the Met tumor versus the Db tumor. In order to study OPN at the cellular level, Met and Db cell lines were established. Northern blot analysis was also used to determine that OPN was differentially expressed in vitro in these cell lines. These analyses showed that OPN expression was 3.5-fold higher in the Met cell line versus the Db cell line. Figure 2 shows a northern blot of both the tumors and cell lines, demonstrating that the tumors and cell lines show similar differential expression of OPN.
Figure 2.
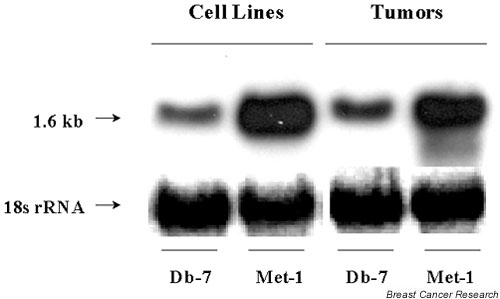
Northern blot of cell lines and tumors from Db and Met. RNA was isolated from Met and Db tumors and from Met and Db cell lines, and probed on a northern blot using 32P-labeled mouse osteopontin (OPN) cDNA. The 18s ribosomal RNA was used as a control. The 1.6 kb OPN transcript level is fourfold higher in the Met tumor than in the Db tumor, and is 3.5-fold higher in the Met cell line than in the Db cell line.
Immunohistochemical staining for OPN in Met tumors and Db tumors
Ten Met tumors and 10 Db tumors from different transplant generations were histologically and immunohistochemically analyzed. H&E staining of the tumor sections showed that the Met tumors formed more glands and had lower mitotic indices and smaller nuclei than the Db tumors. Immunohistochemistry staining with OPN antibody confirmed that OPN was differentially expressed at the protein level and originated from the tumor cells (Fig. 3).
Figure 3.
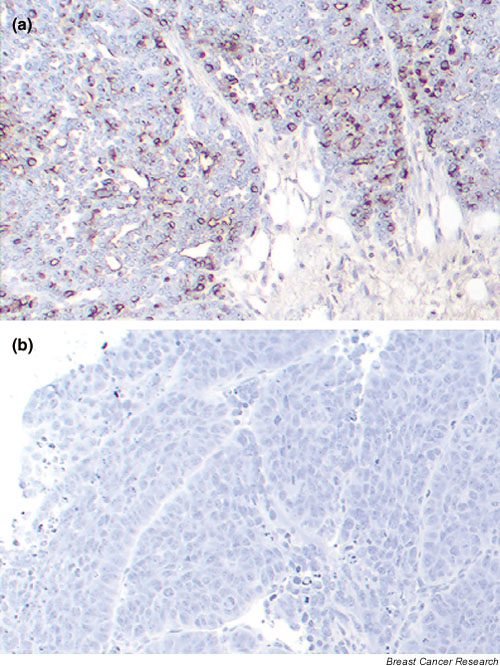
Immunohistochemical analysis of osteopontin (OPN) in the Met and Db tumor tissues. Sections from 10 Met tumors and from 10 Db tumors were stained using immunohistochemistry with antimouse OPN antibody. Representative sections are shown. (a) The Met tumor shows diffuse cytoplasmic OPN staining in the tumor cells, particularly in the periphery of the tumor. (b) The Db tumor cells generally did not stain for OPN.
As a group, Met tumors showed diffuse cytoplasmic OPN staining with more intense staining in the peripheral areas of the tumor. The OPN staining was weak or absent in the lumina of the tumor glands. The Db tumors, as a group, showed OPN staining in less than 1% of the tumor cells, usually located either close to the stroma or close to necroses. In both groups, extracellular OPN was present in necrotic tumor areas.
For comparison, we performed immunohistochemical staining of two mammary glands from normal lactating FVB mice. This showed that OPN was localized in the lumen and at the luminal membrane of alveolar cells and ductal cells (data not shown). Because of its differential expression at both the RNA level and the protein level, OPN was chosen to test the hypothesis that this gene, identified by array analysis, plays a significant role with regard to cell migration/metastasis in the PyV-mT model.
Met cells have a higher migratory potential than Db cells
Because of the known role of OPN in increased cell migration [35], we tested the idea that Met cells are more capable of migration than are Db cells. Migration assays were used to determine the migratory potential of these cells. Figure 4a shows a typical 40 × magnification field of the transwell unit membrane after removal of the nonmigratory cells. Met cells demonstrated a migratory capacity approximately 19 times that of Db cells (P = 0.001; Fig. 4c) in response to serum.
Figure 4.
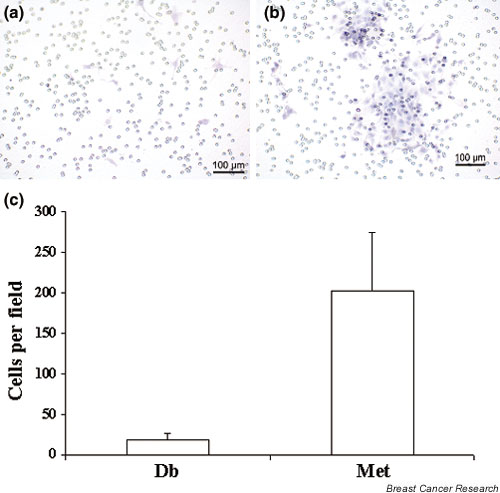
Migration of Db cell lines and Met cell lines in response to serum. Cells were placed in serum-free medium in the upper chamber of transwell units with 8 μm porosity polycarbonate filter membranes. Medium with 10% FBS was used as a chemoattractant in the lower chamber. Cells were incubated for 18–24 hours, and the cells on the upper surface were removed and the cells on the lower surface were fixed and stained. Cells were visualized with a phase contrast microscope and were photographed. (a) A representative field (40 × magnification) showing very few Db cells migrated in response to the serum. (b) A representative field (40 × magnification) of Met cells showing a much higher number of migratory cells. (c) Migration was quantitated by assessing the mean number of cells that had migrated through the membrane in four representative 20 × fields. Met cells demonstrated a migratory capacity 19 times higher than Db cells (P = 0.001).
Inhibition of the OPN gene results in the reduction of migration
Given this cell line model that showed differential expression of OPN and differential migration potential, we tested the hypothesis that OPN is essential to the establishment and maintenance of the metastatic phenotype in our model. In order to test this hypothesis, we generated Met sublines and Db sublines having suppressed or enforced expression of OPN, respectively. These were generated by stable transfection of the indicated cell lines with expression constructs containing the full-length OPN cDNA in either the sense or the antisense orientation, and were referred to as Met-As and Db-S, respectively. Vector-only controls were also generated for each cell line. Immunoblot analysis of detergent lysates demonstrated that OPN antisense expression in Met markedly reduced basal OPN expression to barely detectable levels, similar to that observed in the Db vector control (Fig. 5a). Conversely, OPN expression was successfully elevated in Db-S. Relative OPN expression was determined by densitometry, indicating that OPN was reduced eightfold and was increased 3.5-fold in Met-As and in Db-S, respectively, compared with their vector control counterparts (Fig. 5b,5c).
Figure 5.
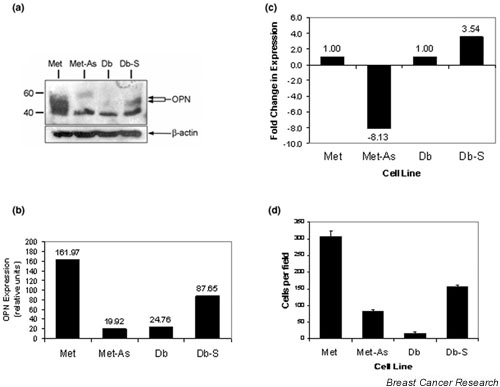
Analysis of osteopontin (OPN) expression and cell migration in Met-As and Db-S stable cell lines. (a) Detergent lysates were prepared from the indicated cell lines. Proteins were size-fractionated by 7.5% SDS-PAGE and transferred to a polyvinylidenedifluoride membrane and analyzed for OPN expression with antimouse antibody (AF808; R&D Systems, Minneapolis, MN, USA). OPN migrated as a doublet, presumably due to isoforms generated by different levels of glycosylation (arrows). Molecular mass standards are indicated on the left-hand side. The lowermost band present in all four lanes is due to nonspecific binding. (b) Relative OPN expression and (c) the fold change in OPN expression in Met-As cells and Db-S cells compared with their relevant vector-transfected control cell lines were determined in the blot in (a) using the TotalLab version 2.00 software package (NonLinear Dynamics, Ltd, Durham, NC, USA). (d) Migration assays were performed on the Db-S cells, Met-As cells, Db control cells, and Met control cells. Met-As cells showed a threefold decrease (P < 0.001) in the number of cells that migrated in comparison with the Met control cells. Db-S cells showed a 10-fold increase (P = 0.001) in migration when compared with the Db control cells.
Migration assays were performed to determine the native migration potential of all the lines (Fig. 5d). Five unique clones from each line were tested for their migration capability. As a group, the Met-As clones showed decreased migration, while the Db-S clones showed an increase in its migration capacity. A representative clone from each line was used for more definitive analysis. Met-As showed a threefold decrease in the number of cells that migrated, as compared with the Met control (P < 0.001). Db-S cells showed a more than 10-fold increase in the number of migratory cells as compared with the Db control (P = 0.001).
Overall, we observed a direct relationship between OPN protein expression and cell migration. The overexpression of OPN correlates with an increased migration potential of cells, and the inhibition of OPN correlates with a decrease in migration.
In vivo tail vein metastasis assays
To assess the importance of OPN with respect to metastasis in the mouse, we performed in vivo tail vein injections of wild-type Met cells, Met-As cells, Db cells, and Db-S cells into nude mice. In both the low concentration (1 × 104) sets and the high concentration (1 × 106) sets of cell injections, the Met cells showed tumor metastasis to the lung while the Met-As cells showed no tumor metastasis. The Db control cells are known to embolically colonize the lungs in tail vein injection experiments, and subsequent analysis of the Db cells and Db-S cells showed metastasis rates and tumor volumes similar to those of Met cells, despite low metastatic frequency in orthograft transplantations [20].
In summary, 100% of the animals in the Met group, the Db group, and the Db-S group showed metastasis. This metastasis analysis was assessed by gross, whole mount, and histologic analysis. Figure 6a,6b shows images of the whole mounts of lungs from a representative Met wild-type animal and from a representative Met-As animal. Figure 6c is a photograph of the lung cavity of a mouse injected with 1 × 106 Met cells. In all of the animals injected with 1 × 106 Met cells, the lungs were nearly filled with tumor and it was impossible to count individual foci. In these cases, we estimated the amount of lung volume occupied by the tumor.
Figure 6.
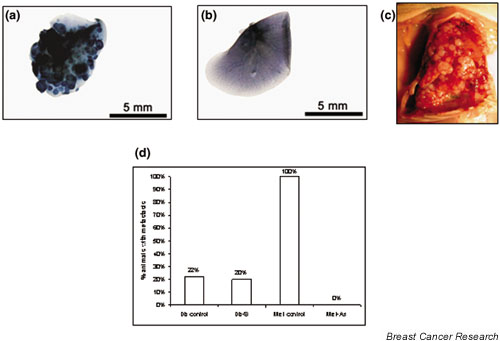
In vivo metastasis analysis via tail vein injection assays and mammary fat pad injection assays. Using the tail vein, nude mice were inoculated with Met wild-type cells and Met-As cells. The lungs of these mice were dissected at 6 weeks and were subjected to whole mount hemotoxylin staining. (a) Lungs of mice injected with 1 × 106 Met wild-type cells were nearly completely infiltrated with metastatic foci (dark regions). (b) Lungs of mice injected with 1 × 106 Met-As cells were free of metastatic foci. (c) Lung cavities of mice injected with 1 × 106 Met wild-type cells were filled with grossly visible metastatic foci. (d) Met cells and Met-As cells were injected in the mammary fat pad of FVB mice. Whole mount analysis of the lungs was performed 42 days after injection. One hundred percent of the Met animals had metastasis (n = 6), while metastasis was not seen in any of the Met-As animals (n = 10). In contrast, in the Db group, two out of nine animals (22%) had metastasis, and the Db-S group had a similar rate of metastasis (two out of 10 animals [20%]).
In vivo fat pad metastasis analysis
Because the tail vein experiments do not recapitulate all the necessary steps that a cell needs to perform in order to metastasize from an ectopic site, we performed mammary fat pad injections with Met cells, Db cells, Met-As cells, and Db-S cells in order to mimic the entire metastasis cascade.
All the groups had approximately the same growth rate in terms of tumor size (data not shown), and tumors were removed at days 22–26 from each animal. The animals were then followed for an additional 21 days and sacrificed at day 42 in order to examine the lungs. Three mice injected with Met cells expired at tumor harvest due to significant blood loss from the large vascular tumors. One mouse in the Db group expired after the surgery due to unknown causes. These four mice were necropsied and no metastasis was found. These were not included in the final analysis.
Results from whole mount analysis of the lungs are depicted in Fig. 6d. The Met group had the highest rate of metastasis, the Db group and the Db-S group had a low metastasis rate, while the Met-As group had no metastasis. In the Met group, the number of metastatic foci per animal ranged from one to eight, except for one animal that expired 18 days after the survival surgery, whose lungs were completely filled with tumor. The animals in the Db group and the Db-S group with metastasis each had a single focus of metastatic tumor.
Sense and antisense constructs alter the morphology of the cells
Finally, we wanted to test the hypothesis that the change in migratory potential in these cells is due to the effect of OPN on the cytological architecture of the cell. Using SEM, we examined the morphology of the various cell lines (Fig. 7). The surface membrane of the Met cells was extremely rough with numerous and highly branched filopodia, whereas the Db cells were much smoother and lacked branched filopodia. In contrast, the Met-As cells had a smoother cell surface with fewer branched filopodia when compared with the Met wild-type cells, resembling the Db wild-type cells. The Db-S cells had a rougher exterior and had a minor increase in the number of branching filopodia compared with the Db wild-type cells.
Figure 7.
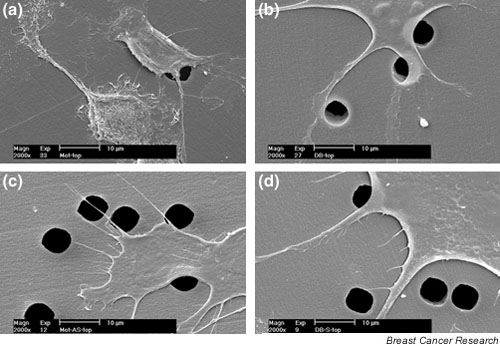
Alterations in cell morphology by induction and inhibition of osteopontin (OPN). Scanning electron microscopy was used to visualize differences in cell morphology between the transfected cell lines. (a) Micrograph of control Met cells, (b) micrograph of control Db cells, (c) micrograph of Met-As cells, and (d) micrograph of Db-S cells. These images indicate a correlation between lower OPN expression and a decrease in branched filopodia and roughness of the cell membrane.
Discussion
The goal of this study was to identify and functionally validate key molecules that may be responsible for the metastatic phenotype seen in the PyV-mT transgenic model. We show that OPN is transcriptionally over-expressed in the metastatic line with mT/PI3-K interaction, compared with the lowly metastatic line without mT/PI3-K interaction. We further demonstrate the differential expression of the OPN protein by western blot and immunohistochemistry. In vitro and in vivo studies demonstrate that OPN is required for the metastatic phenotype in the PyV-mT. This is demonstrated by the Met-As cells, in which OPN expression is nearly completely abolished. The suppression of OPN was seen to eliminate the ability of the PyV-mT cells to migrate in vitro, to colonize the lungs in tail vein injections, and to metastasize to the lungs in the mammary fat pad assay. In contrast, the studies in the Db cells demonstrate that overexpression of OPN alone is not sufficient to effect a metastatic phenotype, as evidenced by the lack of correlation between increased OPN levels and the metastasis rate in the Db-S cells. This suggests that other PI3-K-mediated genes are important and may cooperate with OPN in producing the metastasis phenotype in the PyV-mT system.
OPN has been shown to be important in the developing mammary gland and in cancer. It is a necessary component for mammary gland development and lactation, and is particularly important in the differentiation of the terminal alveolar structure [36-38]. With regard to its role in cancer, previous studies have shown OPN to be a secreted, integrin-binding glycophosphoprotein whose plasma concentrations are elevated in metastatic breast cancer and are associated with increased tumor burden and decreased survival [39]. In addition, it has been shown, by immunohistochemistry and in situ hybridization, that OPN expression is associated with decreased disease survival and decreased overall survival in node-negative breast cancer [40].
Rudland and colleagues [41] recently showed that OPN is tightly linked with decreased survival in stage I and stage II breast cancer. Urquidi and colleagues [42] have demonstrated overexpression of OPN in sublines of the MDA-MB-435 breast tumor cell line that were selected for a high metastatic potential as compared with the non-metastatic sublines. Studies in humans and human derived cell lines demonstrate that OPN is associated with both breast carcinoma and with metastatic breast carcinoma, and confirm that the breast cancer cells are a significant source of OPN expression [43]. In nonhuman studies, Oates and colleagues [44] have shown that increasing the expression of OPN is sufficient to produce a metastatic phenotype in a previously benign cell line (rat mammary tumor line). Behrend and colleagues [45] have shown in nonmammary systems that attenuation of OPN with anti-sense methods can reduce metastasis in ras-transformed NIH 3T3 cells. Gardner and colleagues [46] have shown the same effect in transformed Rat1 fibroblasts.
These studies, in humans and in animal models, demonstrate an association of OPN with breast/mammary carcinoma, and specifically with metastatic disease. To observe a more direct association of OPN and metastatic potential, we used our model system, with its metastatic line and a nonmetastatic line, to manipulate OPN expression by overexpressing it in the nonmetastatic line and by abrogating its expression in the metastatic line, and observing whether the presence or absence of OPN can affect the phenotype with regard to metastasis. Cell migration was used for this initial analysis, as it is one of the hallmarks of invasive behavior [47]. In the Met lines, using antisense methodology, we were able to significantly decrease the OPN protein expression to nearly undetectable levels. This decrease in OPN protein expression was associated with a threefold decrease in the number of cells that migrated, as compared with the vector-transfected Met control. In in vivo tail vein and mammary fat pad injection experiments, this decrease in OPN expression was associated with an elimination of metastatic ability. We viewed these results as indicating that OPN is essential for the metastasis phenotype in the Met lines.
In turn, with the overexpression of the OPN protein in the Db-S cells (3.5-fold higher than the control Db cells and 6.5-fold higher than the Met-As cells), there was a significant increase in the migratory potential of these cells. Specifically, the migration of the Db-S cells was increased at least 10-fold over the vector-transfected wild-type Db cells, twofold over the Met-As cells, and was shown to be about one-half the rate of the Met wild-type cells. We therefore postulated that this level of OPN overexpression would have further consequences in the in vivo experiments.
The observation that there was no increase in the metastasis rate in the fat pad experiments in the Db-S line, despite increased OPN expression and the increased migration of the cells in vitro, was surprising. The Db-S cells had a similar rate of growth assessed by tumor size when compared with the wild-type lines and Met-As. In addition, the primary tumors remained positive for the expression of OPN, as demonstrated by immunohistochemical analysis. These observations suggest that in the Db system OPN may play a role in the migration capacity of the cells, but other genes are necessary to recapitulate the entire metastatic cascade. Because the Db system is deficient in terms of the activation of PI3-K, these cooperative genes are most probably PI3-K dependent.
The tail vein metastasis assays were uninformative in the Db system. As such, they are illustrative of the limitations of this assay. The wild-type Db cells colonize the lung with the same frequency as the wild-type Met cells, even though the Db cells demonstrated a low migration capacity and have a low metastasis rate in mammary fat pad assays. The tail vein assays test the ability of the cells to survive the circulatory system, to attach to a particular site, to proliferate, and in some circumstances, to invade. The assays do not test the ability of the cell to invade and enter the circulation system from a distant site (as in the mammary fat pad or subcutaneous implantation). This assay therefore needs to be viewed with caution in assessing the ability of cells to metastasize.
Extracellular OPN and intracellular OPN were found by immunohistochemistry. The extracellular OPN was observed in the tumor necroses in both tumor lines, and may be derived from former tumor cells or from the serum. OPN is known to bind to components of the extracellular matrix. The intracellular localization in the Met tumor cells was diffusely cytoplasmic. In the normal lactating mammary gland, OPN is predominantly found in the lumen of ducts and alveoli. Western blotting with OPN antibody on cell extracts and concentrated media (10-fold and 100-fold concentrated) shows that OPN levels in the cell extracts are similar to the levels in our previous blots, and that OPN levels in the concentrated media fractions are undetectable (data not shown).
These data support our immunohistochemistry observations that OPN may be mislocalized and preferentially sequestered in the cytoplasm. We suggest that the potential mislocalization of OPN and potential reduction in secreted OPN in these cells may lead to protein complexes, which alter signaling pathways and increase the migratory behavior of the cells. It has been demonstrated that OPN induces increased urokinase production [48], leading to the induction of a number of proteases (pro-MMP-1, pro-MMP-2, pro-MMP-3, pro-MMP-9, pro-MMP-14) [49,50] that can digest various components of the extracellular matrix as well as being able to activate several growth factors (e.g. scatter factor/hepatocyte growth factor, transforming growth factor beta, basic fibroblast growth factor) that may be involved in cell migration and invasion. In addition, SEM of these cells suggests that OPN has an impact on the reorganization and the remodeling of the cytoskeleton. Likewise, Zohar and colleagues have shown that OPN may bind with an ezrin–radixin–moesin complex (band 4.1 proteins) in cooperation with CD44, in order to stimulate cytoskeleton remodeling, signaling, and migration [51].
Our studies suggest that, in the PyV-mT model, an active PI3-K pathway is associated with or induces OPN expression. These observations are not just specific to the transplanted tumor lines analyzed in this study. Maglione and colleagues [52] have shown OPN protein expression in nontransplanted mammary tumors derived from PyV-mT transgenic mice, and we have shown that nontransplanted mammary tumors derived from Db transgenic mice do not stain positive for OPN protein (unpublished results). This suggests that the recruitment of PI3-K by PyV-mT has a direct effect on OPN expression.
Zhang and colleagues [53] recently showed that the PI3-K/Akt axis activates OPN, which increases cell motility and anchorage independence. In addition, Kansra and colleagues [54] showed that PI3-K is essential for the activation of the mitogen-activated protein kinase (MAPK) cascade (ERK and p38). Interestingly, it has been demonstrated that p44/42 ERK/MAPK and p38 MAPK activity increases OPN expression. Specifically, p38 and ERK directly increase OPN transcription [55,56]. Therefore, in our Met model, due to the activation of PI3-K, OPN gene expression may be directly activated via PI3-K, or it may be activated further downstream or via cross-talk through the MAPK pathway. The direct binding of OPN to CD44v3, the integrin pathway, or other downstream pathways may then lead to the metastatic cascade.
The observation that the reconstitution of OPN activity in the Db cells does not restore the metastatic phenotype suggests that OPN itself is not sufficient for metastasis and that other PyV-mT-triggered genes or pathways are necessary. Hutchinson and colleagues [7] demonstrated that restoration of Akt activity in Db mice induces rapid tumor formation, but does not reinstate the metastatic capacity seen in the PyV-mT model with the activated PI3-K/Akt axis. We interpret these results, as well as our results, to indicate that additional PI3-K-related genes are required to functionally interact with OPN to produce the fully metastatic phenotype. The restoration of OPN alone in the Db cells is therefore insufficient to mediate metastasis because one or more cooperating PI3-K-associated components are absent. However, in the Met-As cells, where the PI3-K binding component of PyV-mT is not altered, the loss of metastatic ability can be directly attributed to the suppression of OPN expression. The requirement of these cooperating molecules in addition to OPN may help explain the recent data by Wang-Rodriguez and colleagues [57], which demonstrate that there was not a strong correlation between OPN-positive breast cancer and metastatic disease.
Conclusion
From our studies, it is apparent that OPN plays an important role in the metastasis of PyV-mT mammary tumors. The results of these experiments demonstrate that OPN is essential for the metastatic phenotype in PyV-mT, but that it alone is not sufficient to produce the metastatic phenotype. This hypothesis suggests that, in OPN-positive tumors, therapy targeting OPN may be feasible for inhibiting the metastasis of breast cancer–either by acting at the receptor level to block OPN binding, or by inhibiting the activity of the PI3-K pathway or the MAPK pathway.
Competing interests
None declared.
Abbreviations
DMEM = Dulbecco's modified Eagle medium; EDTA = ethylenediamine tetraacetic acid; FBS = fetal bovine serum; H&E = hematoxylin and eosin; MAPK = mitogen-activated protein kinase; mT = middle T; OPN = osteopontin; PBS = phosphate-buffered saline; PCR = polymerase chain reaction; PI3-K = phosphatidylinositol-3-kinase; PyV = polyomavirus; SEM = scanning electron microscopy; SSH = suppression subtractive hybridization.
Acknowledgments
Acknowledgements
The authors would like to express their appreciation to Dr William Muller for creating these transgenic mouse models. They would also like to thank Sze Chun Chan for his work on the immunohistochemical studies, and Paul Hoffman, Noelle Niemand, and Ruria Namba for their expert technical assistance. This work was supported by Grant R01-CA89140-01 from the National Cancer Institute, by Grant 6KB-0074 from the California Breast Cancer Research Program, by the Deutsche Forschungsgemeinschaft (BA 1433/4-1; AR, individual grant), and by the and National Centers for Research Resources-U42RR14905 (to EM, AB, and RC).
References
- Paigen K. A miracle enough: the power of mice. Nat Med. 1995;1:215–220. doi: 10.1038/nm0395-215. [DOI] [PubMed] [Google Scholar]
- Cardiff RD, Bern HA, Faulkin LJ, Daniel CW, Smith GH, Young LJ, Medina D, Gardner MB, Wellings SR, Shyamala G, Guzman RC, Rajkumar L, Yang J, Thordarson G, Nandi S, MacLeod CL, Oshima RG, Man AK, Sawai ET, Gregg JP, Cheung AT, Lau DH. Contributions of mouse biology to breast cancer research. Comp Med. 2002;52:12–31. [PubMed] [Google Scholar]
- Hutchinson JN, Muller WJ. Transgenic mouse models of human breast cancer. Oncogene. 2000;19:6130–6137. doi: 10.1038/sj.onc.1203970. [DOI] [PubMed] [Google Scholar]
- Muller WJ, Neville MC. Introduction: signaling in mammary development and tumorigenesis. J Mammary Gland Biol Neoplasia. 2001;6:1–5. doi: 10.1023/A:1009540813521. [DOI] [PubMed] [Google Scholar]
- Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel PM, Hardy WR, Muller WJ. Mammary gland neoplasia: insights from transgenic mouse models. Bioessays. 2000;22:554–563. doi: 10.1002/(SICI)1521-1878(200006)22:6<554::AID-BIES8>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Hutchinson J, Jin J, Cardiff RD, Woodgett JR, Muller WJ. Activation of Akt (protein kinase B) in mammary epithelium provides a critical cell survival signal required for tumor progression. Mol Cell Biol. 2001;21:2203–2212. doi: 10.1128/MCB.21.6.2203-2212.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardiff RD. Validity of mouse mammary tumour models for human breast cancer: comparative pathology. Microsc Res Tech. 2001;52:224–230. doi: 10.1002/1097-0029(20010115)52:2<224::AID-JEMT1007>3.3.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Cardiff RD, Wagner U, Henninghausen L. Mammary cancer in humans and mice: a tutorial for comparative pathology. Vet Pathol. 2001;38:357–358. doi: 10.1354/vp.38-4-357. [DOI] [PubMed] [Google Scholar]
- Guy CT, Cardiff RD, Muller WJ. Activated neu induces rapid tumor progression. J Biol Chem. 1996;271:7673–7678. doi: 10.1074/jbc.271.16.9567. [DOI] [PubMed] [Google Scholar]
- Desai KV, Xiao N, Wang W, Gangi L, Greene J, Powell JI, Dickson R, Furth P, Hunter K, Kucherlapati R, Simon R, Liu ET, Green JE. Initiating oncogenic event determines gene-expression patterns of human breast cancer models. Proc Natl Acad Sci USA. 2002;99:6967–6972. doi: 10.1073/pnas.102172399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WJ, Ho J, Siegel PM. Oncogenic activation of Neu/ErbB-2 in a transgenic mouse model for breast cancer. Biochem Soc Symp. 1998;63:149–157. [PubMed] [Google Scholar]
- Barnes DM, Bartkova J, Camplejohn RS, Gullick WJ, Smith PJ, Millis RR. Overexpression of the c-erbB-2 oncoprotein: why does this occur more frequently in ductal carcinoma in situ than in invasive mammary carcinoma and is this of prognostic significance? Eur J Cancer. 1992;28:644–648. doi: 10.1016/s0959-8049(05)80117-0. [DOI] [PubMed] [Google Scholar]
- Oostra BA, Harvey R, Ely BK, Markham AF, Smith AE. Transforming activity of polyoma virus middle-T antigen probed by site-directed mutagenesis. Nature. 1983;304:456–459. doi: 10.1038/304456a0. [DOI] [PubMed] [Google Scholar]
- Courtneidge SA, Smith AE. Polyoma virus transforming protein associates with the product of the c-src cellular gene. Nature. 1983;303:435–439. doi: 10.1038/303435a0. [DOI] [PubMed] [Google Scholar]
- Webster MA, Hutchinson JN, Rauh MJ, Muthuswamy SK, Anton M, Tortorice CG, Cardiff RD, Graham FL, Hassell JA, Muller WJ. Requirement for both Shc and phosphatidylinositol 3' kinase signaling pathways in polyomavirus middle T-mediated mammary tumorigenesis. Mol Cell Biol. 1998;18:2344–2359. doi: 10.1128/mcb.18.4.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman M, Kaplan DR, Schaffhausen B, Cantley L, Roberts TM. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature. 1985;315:239–242. doi: 10.1038/315239a0. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Rabinovitz I, Wang HH, Toker A, Mercurio AM. Activation of phosphoinositide 3-OH kinase by the alpha6beta4 integrin promotes carcinoma invasion. Cell. 1997;91:949–960. doi: 10.1016/s0092-8674(00)80486-9. [DOI] [PubMed] [Google Scholar]
- Cheung ATW, Young LJT, Chen PCY, Chao CY, Ndoye A, Barry PA, Muller WJ, Cardiff RD. Microcirculation and metastasis in a new mouse mammary tumor model system. Int J Oncol. 1997;11:69–77. [PubMed] [Google Scholar]
- Borowsky AD, Namba R, Niemand N, Tepper CG, McGoldrick ET, Cardiff RD, Gregg JP. In American Association for Cancer Research Annual Meeting: 2003; Toronto, Canada. Philadelphia, PA: AACR; 2003. Tumors from two related mouse models of mammary carcinoma show differential metastasis rate and regulation of Akt signal effectors. [Google Scholar]
- Welford SM, Gregg J, Chen E, Garrison D, Sorensen PH, Denny CT, Nelson SF. Detection of differentially expressed genes in primary tumor tissues using representational differences analysis coupled to microarray hybridization. Nucleic Acids Res. 1998;26:3059–3065. doi: 10.1093/nar/26.12.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang GP, Ross DT, Kuang WW, Brown PO, Weigel RJ. Combining SSH and cDNA microarrays for rapid identification of differentially expressed genes. Nucleic Acids Res. 1999;27:1517–1523. doi: 10.1093/nar/27.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diatchenko L, Lau YF, Campbell AP, Chenchik A, Moqadam F, Huang B, Lukyanov S, Lukyanov K, Gurskaya N, Sverdlov ED, Siebert PD. Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc Natl Acad Sci USA. 1996;93:6025–6030. doi: 10.1073/pnas.93.12.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeOme KB, Faulkin LJJ, Bern HA, Blair PB. Development of mammary tumors from hyperplastic alveolar nodules transplanted into gland-free mammary fat pads of female C3H mice. Cancer Res. 1959;19:515–525. [PubMed] [Google Scholar]
- Young LJT. The cleared mammary fat pad and the transplantation of mammary gland morphological structures and cells. In: Ip MMaABB, editor. Methods in Mammary Gland Biology and Breast Cancer Research. New York: Kluwer Academic/Plenum Publishers; 2000. pp. 67–74. [Google Scholar]
- Bourguignon LY, Gunja-Smith Z, Iida N, Zhu HB, Young LJ, Muller WJ, Cardiff RD. CD44v(3,8-10) is involved in cytoskeleton-mediated tumor cell migration and matrix metalloproteinase (MMP-9) association in metastatic breast cancer cells. J Cell Physiol. 1998;176:206–215. doi: 10.1002/(SICI)1097-4652(199807)176:1<206::AID-JCP22>3.3.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Young LJT, Cardiff RD, McGrath CM. Tissue Culture Association Manual. Gaithersburg, MD: Tissue Culture Association; 1976. Primary epithelial cell dome culture of mouse mammary tumors; pp. 161–167. [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Jessen KA, Satre MA. Induction of mouse retinol binding protein gene expression by cyclic AMP in Hepa 1-6 cells. Arch Biochem Biophys. 1998;357:126–130. doi: 10.1006/abbi.1998.0821. [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current Protocols in Molecular Biology. New York: Wiley Press; 1991. [Google Scholar]
- Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- Merzak A, Koocheckpour S, Pilkington GJ. CD44 mediates human glioma cell adhesion and invasion in vitro. Cancer Res. 1994;54:3988–3992. [PubMed] [Google Scholar]
- Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- Soga N, Connolly JO, Chellaiah M, Kawamura J, Hruska KA. Rac regulates vascular endothelial growth factor stimulated motility. Cell Commun Adhes. 2001;8:1–13. doi: 10.3109/15419060109080703. [DOI] [PubMed] [Google Scholar]
- Rittling SR, Novick KE. Osteopontin expression in mammary gland development and tumorigenesis. Cell Growth Differ. 1997;8:1061–1069. [PubMed] [Google Scholar]
- Nemir M, Bhattacharyya D, Li X, Singh K, Mukherjee AB, Mukherjee BB. Targeted inhibition of osteopontin expression in the mammary gland causes abnormal morphogenesis and lactation deficiency. J Biol Chem. 2000;275:969–976. doi: 10.1074/jbc.275.2.969. [DOI] [PubMed] [Google Scholar]
- Baik MG, Lee MJ, Choi YJ. Gene expression during involution of mammary gland [review] Int J Mol Med. 1998;2:39–44. [PubMed] [Google Scholar]
- Singhal H, Bautista DS, Tonkin KS, O'Malley FP, Tuck AB, Chambers AF, Harris JF. Elevated plasma osteopontin in metastatic breast cancer associated with increased tumor burden and decreased survival. Clin Cancer Res. 1997;3:605–611. [PubMed] [Google Scholar]
- Tuck AB, O'Malley FP, Singhal H, Harris JF, Tonkin KS, Kerkvliet N, Saad Z, Doig GS, Chambers AF. Osteopontin expression in a group of lymph node negative breast cancer patients. Int J Cancer. 1998;79:502–508. doi: 10.1002/(SICI)1097-0215(19981023)79:5<502::AID-IJC10>3.3.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Rudland PS, Platt-Higgins A, El-Tanani M, De Silva Rudland S, Barraclough R, Winstanley JH, Howitt R, West CR. Prognostic significance of the metastasis-associated protein osteopontin in human breast cancer. Cancer Res. 2002;62:3417–3427. [PubMed] [Google Scholar]
- Urquidi V, Sloan D, Kawai K, Agarwal D, Woodman AC, Tarin D, Goodison S. Contrasting expression of thrombospondin-1 and osteopontin correlates with absence or presence of metastatic phenotype in an isogenic model of spontaneous human breast cancer metastasis. Clin Cancer Res. 2002;8:61–74. [PubMed] [Google Scholar]
- Sharp JA, Sung V, Slavin J, Thompson EW, Henderson MA. Tumor cells are the source of osteopontin and bone sialopro-tein expression in human breast cancer. Lab Invest. 1999;79:869–877. [PubMed] [Google Scholar]
- Oates AJ, Barraclough R, Rudland PS. The identification of metastasis-related gene products in a rodent mammary tumour model. Biochem Soc Trans. 1996;24:353S. doi: 10.1042/bst024353s. [DOI] [PubMed] [Google Scholar]
- Behrend EI, Craig AM, Wilson SM, Denhardt DT, Chambers AF. Reduced malignancy of ras-transformed NIH 3T3 cells expressing antisense osteopontin RNA. Cancer Res. 1994;54:832–837. [PubMed] [Google Scholar]
- Gardner HA, Berse B, Senger DR. Specific reduction in osteopontin synthesis by antisense RNA inhibits the tumorigenicity of transformed Rat1 fibroblasts. Oncogene. 1994;9:2321–2326. [PubMed] [Google Scholar]
- Sliva D, Rizzo MT, English D. Phosphatidylinositol 3-kinase and NF-kappaB regulate motility of invasive MDA-MB-231 human breast cancer cells by the secretion of urokinase-type plasminogen activator. J Biol Chem. 2002;277:3150–3157. doi: 10.1074/jbc.M109579200. [DOI] [PubMed] [Google Scholar]
- Tuck AB, Arsenault DM, O'Malley FP, Hota C, Ling MC, Wilson SM, Chambers AF. Osteopontin induces increased invasiveness and plasminogen activator expression of human mammary epithelial cells. Oncogene. 1999;18:4237–4246. doi: 10.1038/sj.onc.1202799. [DOI] [PubMed] [Google Scholar]
- Baricos WH, Cortez SL, el-Dahr SS, Schnaper HW. ECM degradation by cultured human mesangial cells is mediated by a PA/plasmin/MMP-2 cascade. Kidney Int. 1995;47:1039–1047. doi: 10.1038/ki.1995.150. [DOI] [PubMed] [Google Scholar]
- Mazzieri R, Masiero L, Zanetta L, Monea S, Onisto M, Garbisa S, Mignatti P. Control of type IV collagenase activity by components of the urokinase–plasmin system: a regulatory mechanism with cell-bound reactants. Embo J. 1997;16:2319–2332. doi: 10.1093/emboj/16.9.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zohar R, Suzuki N, Suzuki K, Arora P, Glogauer M, McCulloch CA, Sodek J. Intracellular osteopontin is an integral component of the CD44–ERM complex involved in cell migration. J Cell Physiol. 2000;184:118–130. doi: 10.1002/(SICI)1097-4652(200007)184:1<118::AID-JCP13>3.3.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Maglione JE, Moghanaki D, Young LJ, Manner CK, Ellies LG, Joseph SO, Nicholson B, Cardiff RD, MacLeod CL. Transgenic Polyoma middle-T mice model premalignant mammary disease. Cancer Res. 2001;61:8298–8305. [PubMed] [Google Scholar]
- Zhang G, He B, Weber GF. Growth factor signaling induces metastasis genes in transformed cells: molecular connection between Akt kinase and osteopontin in breast cancer. Mol Cell Biol. 2003;23:6507–6519. doi: 10.1128/MCB.23.18.6507-6519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansra V, Groves C, Gutierrez-Ramos JC, Polakiewicz RD. Phosphatidylinositol 3-kinase-dependent extracellular calcium influx is essential for CX(3)CR1-mediated activation of the mitogen-activated protein kinase cascade. J Biol Chem. 2001;276:31831–31838. doi: 10.1074/jbc.M009374200. [DOI] [PubMed] [Google Scholar]
- Sodhi CP, Batlle D, Sahai A. Osteopontin mediates hypoxia-induced proliferation of cultured mesangial cells: role of PKC and p38 MAPK. Kidney Int. 2000;58:691–700. doi: 10.1046/j.1523-1755.2000.00215.x. [DOI] [PubMed] [Google Scholar]
- You J, Reilly GC, Zhen X, Yellowley CE, Chen Q, Donahue HJ, Jacobs CR. Osteopontin gene regulation by oscillatory fluid flow via intracellular calcium mobilization and activation of mitogen-activated protein kinase in MC3T3-E1 osteoblasts. J Biol Chem. 2001;276:13365–13371. doi: 10.1074/jbc.M009846200. [DOI] [PubMed] [Google Scholar]
- Wang-Rodriguez J, Urquidi V, Rivard A, Goodison S. Elevated osteopontin and thrombospondin expression identifies malignant human breast carcinoma but is not indicative of metastatic status. Breast Cancer Res. 2003;5:R136–R143. doi: 10.1186/bcr620. [DOI] [PMC free article] [PubMed] [Google Scholar]