

Abstract
Aim:
To explore the potent inhibitor from one of the Traditional Chinese medicine (TCM), Epimedium sagittatum.
Methods:
We predicted the potent compound, ES03b, de novo evolution from the four Epimedium sagittatum components were verified by molecular docking, pharmacophore analysis, and analysis of quantitative structure-activity relationship (QSAR) model, which was constructed by multiple linear regression.
Results:
ES03b was chosen to undergo drug modification via de novo evolution. By analyzing the pharmacophore features, we found that the hydrophobic core in the binding site and the hydrogen bond generated at Asn663 played key roles in designing PDE5 inhibitors. ES03b generated 49 diversities (Evo01-49). Evo48 had high activity in prediction. Although the value of prediction was overestimated, Evo48 was suggested as the potent lead.
Conclusion:
In this study, we showed that the hydrophobic core in the binding site and hydrogen bond production on Asn663 played key roles to design PDE5 inhibitors. From several require validation analysis, Evo48 was suggested to be a potent inhibitor.
Keywords: phosphodiesterase5 (PDE5), Epimedium sagittatum, erectile dysfunction, pharmacophore analysis, quantitative structureactivity relationship
Introduction
Phosphodiesterase superfamily is the key enzyme for degrading the intracellular second messenger cAMP and cGMP. Phosphodiesterases (PDEs) are drug targets for treating many diseases such as heart failure, depression, asthma, inflammation, and even erectile dysfunction. Male erectile dysfunction is usually caused by low concentration of cyclic GMP. Thus, PDE5 is taken as the target of sildenafil citrate, tadalafil, and other similar drugs for the treatment of erectile dysfunction1.
Penile erection is a hemodynamic event in the smooth muscle of corpus cavernosum. This physiological response is mediated by the neurotransmitter nitric oxide (NO) that is released from both nitrergic nerves and sinusoidal endothelium. Nitric oxide stimulates the soluble guanylyl cyclase in cavernosal smooth muscle, inducing increased synthesis of cyclic GMP (cGMP) to lead relaxation of smooth muscle2. The cGMP levels in the corpus cavernosum are regulated by soluble guanylyl cyclase and cyclic nucleotide phosphodiesterases (PDEs). As mentioned above, phosphodiesterase5 (PDE5) is an enzyme for degrading cGMP. Since normal erection needs a high level concentration of cyclic GMP to decrease calcium ions in vascular smooth muscle cells of the penis, thus, the inhibitors of PDE5 were used for treating erectile dysfunction such as sildenafil citrate (Viagra) and tadalafil (Cialis)3. However, there is a cost to this treatment. The side effects of sildenafil citrate and tadalafil, owing to inhibiting PDE6 and 11, may cause headache and optical interruption4, 5, 6.
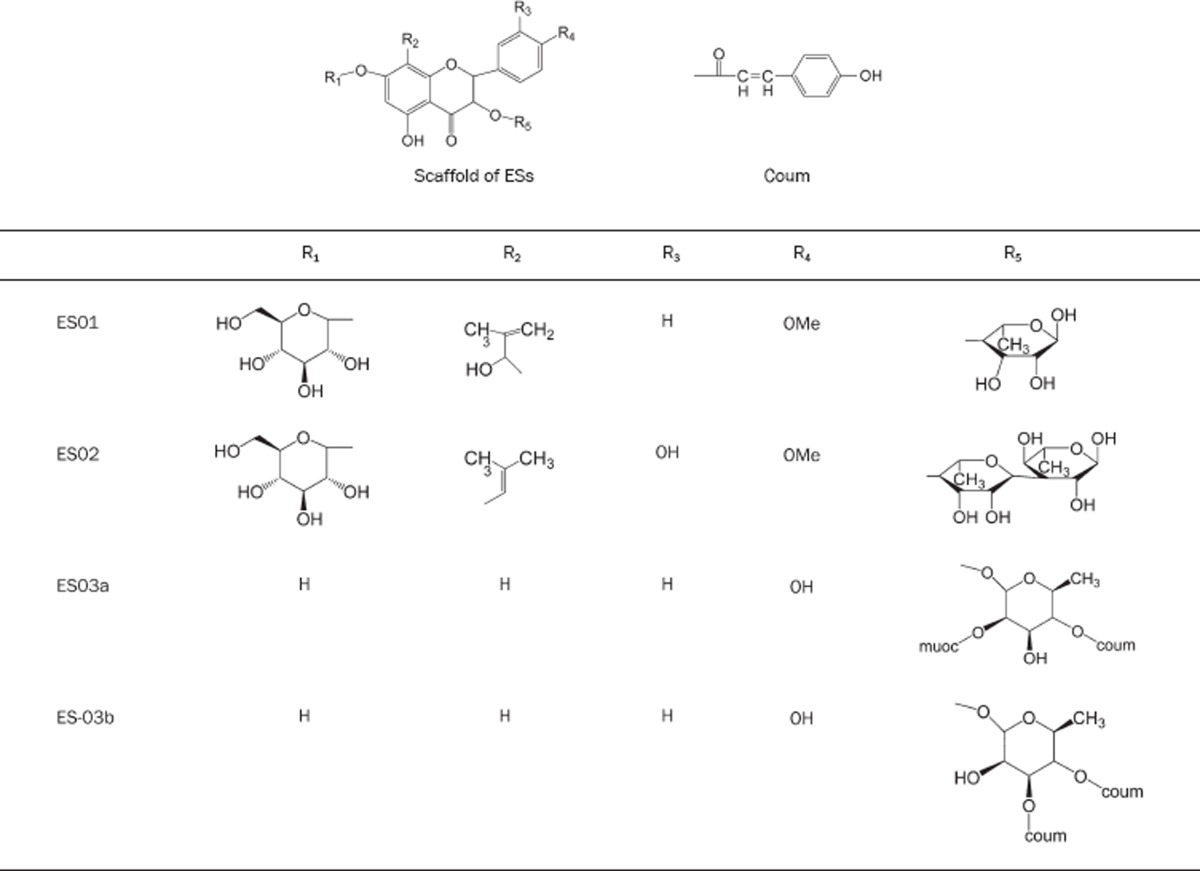
Nevertheless, certain components from natural herbs and products from Traditional Chinese Medicines (TCM) can treat erectile dysfunction7, 8. According to the reports of several studies9, 10, 11, 12, 13, ES01, ES02, ES03a, and ES03b, the components from Epimedium sagittatum, were selected to investigate the interaction with PDE5 for discovering the leading compounds to treat erectile dysfunction (Table 1).
Table 1. The structures of Epimedium sagittatum components.
In the past few years, we had focused on computer-aided drug design (CADD) in several researches14, 15, 16, 17, 18, 19, 20, 21. In our experience, before the de novo design, a suitable scoring function should be selected to evaluate the protein-ligand interaction. CADD includes protein based method and ligand based method. Both of them were used for improve reliability. The major benefit of CADD in drug discovery is that it costs much less than any biomedical test of inhibition in cells just like in the references from APS22, 23, 24, 25, 26, 27, 28.
The ligand based method is built on regression analysis for molecular structure and properties against activities, while the protein based method focuses on the docking procedure in which the structure of the protein would be docked with many kinds of inhibitors and the binding energies would be calculated29, 30. When the patterns of results in the two methods agree with each other, it indicates a reliable outcome.
In this study, sildenafil citrate (Viagra) and tadalafil (Cialis) were selected as the control set in this protocol (Figure 1)31. We expected to discover some compounds from nature products that would have higher activities than that of Viagra and Cialis.
Figure 1.

The control group included (A) cGMP (B) Sildenafil citrate, and (C)Tadalafil.
Materials and methods
Dataset
All the molecular simulated performances were based on Discovery Studio modeling 2.0 (Accelrys Inc, San Diego, USA). Three-dimensional (3D) molecule model of PDE5-sildenafil citrate complex was downloaded from Protein Data Bank website (PDB ID: 1UDT)32. Components of Epimedium sagittatum (ES01, ES02, ES03a, ES03b) (Figure 1) were drew by ChemDraw Ultra 10.0 (Cambridgesoft Inc, USA) and transformed to 3D molecule models by Chem3D Ultra 10.0 (Cambridgesoft Inc, USA). The forcefield of the Chemistry at Harvard Macromolecular mechanics (CHARMm) would be applied to 3D molecule models of PDE5- sildenafil citrate complex and Epimedium sagittatum components. CHARMm uses a flexible and comprehensive empirical energy function that is a summation of many individual energy terms. The energy function is based on separable internal coordinate terms and pairwise nonbond interaction terms. Then, hydrogens would be added appropriately and partial charges of this system would be estimated and assigned. This step is considered to be a rough approximation of the solvent effect and should not be combined with explicit solvent in simulations.
Docking and scoring function prediction
The docking scores and binding energy were calculated from Accelrys Discovery studio V2.0. The protocol was as follows: Receptor-Ligand Interactions\Dock Ligands (LigandFit), Simulation\Energy Minimization, and Receptor-Ligand Interactions\Calculate Binding Energies; all steps were executed with the Chemistry at Harvard Macromolecular mechanics (CHARMm) forcefield. We set the control group, which included cGMP, sildenafil citrate and tadalafil with PDE5 for calculating their docking scores and binding energies. We set the maximum poses retained as 10, RMS threshold for diversity as 1.5, and score threshold for diversity as 20.0. The minimization algorithm was set. The minimization energy tolerance was zero and the gradient tolerance was 0.001 by default.
The same protocols was used to calculate docking scores and binding energies for components of Epimedium sagittatum (ES01, ES02, ES03a, ES03b) with PDE5, and compare with docking scores of the control group.
Ligand de novo evolution
The component which has the highest score would be selected into the next protocol: Receptor-Ligand Interactions\De Novo Evolution. The drug design program from pharmacophore methodology could change side groups from the setting data which based on target component for getting higher binding affinity to PDE5. The de novo evolution results were calculated docking scores for checking their docking scores.
Pharmacophore analysis
The protocols: pharmacophore\Interaction Generation, Common Feature Pharmacophore Generation, and Ligand Pharmacophore Mapping on Accelrys Discovery studio 2.0 were employed. The receptor and ligands were analyzed the active site for hydrogen bond donors, acceptors, and hydrophobes. The results of the calculation were interaction maps31, 33, 34, 35, 36.
Multiple linear regression (MLR) analysis
We calculated the properties of the molecules from Xia's study37(Table S1). The properties contained AlogP, molecular energy, molecular weight, molecular solubility, number of rotatable bonds, number of rings, number of aromatic rings, number of hydrogen bond acceptors, number of hydrogen bond donors, and molecular polar surface area (Table S2). Multiple linear regression analysis was used to build the QSAR model. Those properties were associated to drug activities by the QSAR model38, 39, 40.
Table S1. The structures of molecules for building QSAR model.
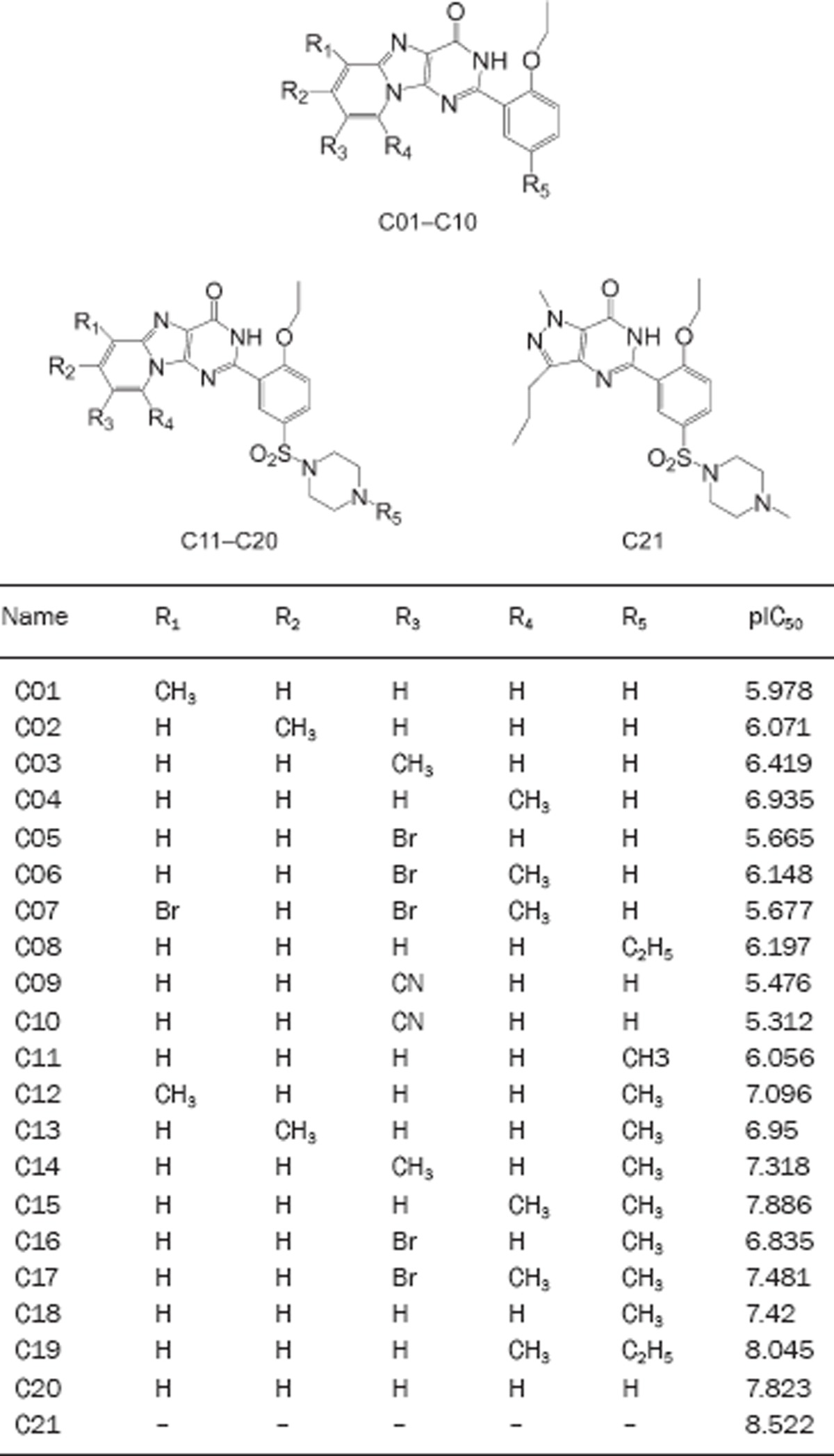
Table S2. The molecular properties of QSAR model.
| Name | Activ | ALogP | Energy | MW | MS | MV | NR Bonds | N Rings | NA Rings | NH Acceptors | PS Area | NH Donors | MLR Temp Model |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C01 | 5.978 | 3.02 | 75.34 | 320.345 | −4.02 | 204.42 | 3 | 4 | 3 | 4 | 1 | 67.98 | 6.217 |
| C02 | 6.071 | 3.02 | 57.01 | 320.345 | −4.048 | 203.39 | 3 | 4 | 3 | 4 | 1 | 67.98 | 6.209 |
| C03 | 6.419 | 3.02 | 75.93 | 320.345 | −4.037 | 204.08 | 3 | 4 | 3 | 4 | 1 | 67.98 | 6.287 |
| C04 | 6.935 | 2.816 | 78.77 | 320.345 | −3.988 | 205.79 | 3 | 4 | 3 | 4 | 1 | 67.98 | 6.297 |
| C05 | 5.665 | 3.282 | 75.74 | 385.215 | −4.753 | 220.54 | 3 | 4 | 3 | 4 | 1 | 67.98 | 5.403 |
| C06 | 6.148 | 3.565 | 80.13 | 399.241 | −5.207 | 231.18 | 3 | 4 | 3 | 4 | 1 | 67.98 | 6.214 |
| C07 | 5.677 | 4.313 | 80.52 | 478.137 | −6.413 | 259.65 | 3 | 4 | 3 | 4 | 1 | 67.98 | 6.072 |
| C08 | 6.197 | 3.483 | 78.55 | 334.372 | −4.356 | 217.11 | 4 | 4 | 3 | 4 | 1 | 67.98 | 6.389 |
| C09 | 5.476 | 2.413 | 83.24 | 331.328 | −4.054 | 201.68 | 3 | 4 | 3 | 5 | 1 | 91.78 | 5.173 |
| C10 | 5.312 | 2.695 | 81.01 | 345.355 | −4.425 | 220.2 | 3 | 4 | 3 | 5 | 1 | 91.78 | 5.615 |
| C11 | 6.056 | 1.608 | 90.4 | 468.529 | −3.094 | 299.09 | 5 | 5 | 3 | 7 | 1 | 116.98 | 6.868 |
| C12 | 7.096 | 2.094 | 90.28 | 482.555 | −3.529 | 315.21 | 5 | 5 | 3 | 7 | 1 | 116.98 | 7.385 |
| C13 | 6.95 | 2.094 | 88.17 | 482.555 | −3.56 | 314.53 | 5 | 5 | 3 | 7 | 1 | 116.98 | 7.495 |
| C14 | 7.318 | 2.094 | 91.05 | 482.555 | −3.545 | 303.89 | 5 | 5 | 3 | 7 | 1 | 116.98 | 7.457 |
| C15 | 7.886 | 1.891 | 93.36 | 482.555 | −3.491 | 309.72 | 5 | 5 | 3 | 7 | 1 | 116.98 | 7.446 |
| C16 | 6.835 | 2.356 | 87.67 | 547.425 | −4.271 | 322.41 | 5 | 5 | 3 | 7 | 1 | 116.98 | 6.589 |
| C17 | 7.481 | 2.639 | 95.04 | 561.451 | −4.698 | 331.68 | 5 | 5 | 3 | 7 | 1 | 116.98 | 7.313 |
| C18 | 7.42 | 2.557 | 55.89 | 496.582 | −3.826 | 321.39 | 6 | 5 | 3 | 7 | 1 | 116.98 | 7.164 |
| C19 | 8.045 | 2.239 | 96.91 | 496.582 | −3.808 | 319.67 | 6 | 5 | 3 | 7 | 1 | 116.98 | 7.65 |
| C20 | 7.823 | 2.617 | 94.28 | 510.608 | −4.064 | 339.91 | 6 | 5 | 3 | 7 | 1 | 116.98 | 7.543 |
| C21 | 8.522 | 2.247 | 50.58 | 474.576 | −3.978 | 313.5 | 7 | 4 | 2 | 7 | 1 | 117.5 | 8.522 |
Activ: pIC50; MW: molecular weight; MS: molecular solubility; MV: molecular volume; NR Bonds: number of rotatable bonds; N Rings: number of rings; NA Rings: number of aromatic rings; NH Acceptors: number of hydrogen bond acceptors; PS Area: polar surface area; NH Donors: number of hydrogen bond donors.
Results
Docking screening and binding free energy calculation
Compared the docking scores and binding energies of the control group with the components of Epimedium sagittatum, we found that ES03b had a high score the same as tadalafil (Table 2). The binding energy of ES03a was much better than ES03b. However, docking score was considered as the degree of difficulty about ligands moving into the binding site, but the binding energy was the stability of ligand-receptor complex. Therefore, ES03b was selected to design derivatives.
Table 2. The results of docking.
| Name | LigS1 | LigS2 | −PLP1 | −PLP2 | Jain | −PMF | DS | LIE | Ludi_1 | Ludi_2 | Ludi_3 | Binding energy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cGMP | 4.51 | 5.69 | 79.95 | 75.79 | 2.59 | 65.88 | 55.670 | −4.84 | 410 | 333 | 459 | −233.96 |
| Tadalafil | 2.31 | 5.35 | 88.24 | 78.53 | 4.54 | 101.90 | 53.271 | −4.373 | 479 | 420 | 964 | −129.99 |
| Sildenafil citrate | 5.51 | 6.94 | 108.72 | 110.40 | 7.3 | 122.45 | 64.055 | −4.427 | 687 | 556 | 491 | −148.33 |
| E S02 | 7.18 | 7.42 | 135.02 | 132.62 | 5.73 | 184.65 | 57.864 | 0.034 | 894 | 695 | 955 | −324.07 |
| E S03a | 7.17 | 8.06 | 165.41 | 156.11 | 6.15 | 176.44 | 64.260 | −5.467 | 1047 | 743 | 952 | −351.74 |
| E S03b | 8.18 | 8.02 | 141.15 | 131.44 | 6.35 | 168.93 | 76.915 | −18.302 | 854 | 641 | 727 | −256.70 |
The unit of binding energy was kcal/mol. DS was docking score and LIE was Lig internal energy.
To observe the binding site of PDE-5, we found that the binding site was a typical pocket conformation and had a narrow entry but a spacious inside. At the bottom of the inside, Phe786, Asn662, and Leu804 built a wall (Figure 2); it implied that ligands should twist around the wall but could not dock into the depths of the pocket. The structure of ES01 was similar to ES02, but ES01 had only one rhamnosyl groups (Table 1), which were difficult to twist to detour the wall and to touch the depths of the pocket. Accordingly, ES01 was non available in docking scoring. Unfortunately, the flexibility of receptor was set as rigid body in our program. Thus, the possibility of ES01 can't perform in our study.
Figure 2.

The conformation of binding site in PDE 5 structure. The green region constructed the binding site, which is a typical pocket conformation. The three amino acids (Asn662, Phe786, and Leu804) built a wall at the bottom of the pocket.
ES03s had two long arms at R5 site in their structure (Table 1). They could detour the wall and dock into the depths of the pocket but would be difficult to deviate from binding site. It was correspondent to the results of binding energies. Compared with sildenafil citrate and tadalafil, ES03s had higher stability than sildenafil citrate and tadalafil (Figure 3).
Figure 3.

The docking poses of cGMP (A), sildenafil citrate (B), tadalafil (C), ES02 (D), ES03a (E), and ES03b (F) in PDE5.
Comparing ES03a and ES03b, the hydroxyl group on type1 was at R5 site. This conformation was set by the two arms. In fact, the hydroxyl group at R5 site was more difficult to form a hydrogen bond than in ES03b.
Ligand de novo evolution and scoring function analysis
Ligscore 1 and Ligscore 2 were calculated by the descriptors of polar surface in receptor-ligand interactions. As Table 3, there was no significant difference in Ligscore 1 and Ligscore 2. Compared with Table 2 and 3, Ligscore 1 and Ligscore 2 in the evolution results were indeed higher than the control group. It was thought that the Evo series could have more polar interaction area than cGMP. Piecewise Linear Potential (PLP) scores were calculated by the descriptors about hydrogen bonds forming. Higher PLP scores indicated stronger receptor-ligand binding. The expressions of hydrogen bonds could be observed in Figure 3 and Table 2. In PLP scores, the values of the Evo series were higher than the control group as well. Potential of mean force (PMF) scores were calculated by summing pairwise interaction terms over all interatomic pairs of the receptor-ligand complex. Increased PMF scores were important in the competition of cGMP with PDE-5 inhibitors (Table 2). According to PMF scores and docking scores, Evo 45 and 48 were elevated into pharmacophore analysis. Jain, Lig internal energy (LIE), Ludi scores, and binding energy were consulted in hydrophobic interaction, entropy, degree of freedom, and energy change. They were employed for insuring the binding stability in receptor-ligand complex of Evo series higher than the control group.
Table 3. The better results from 49 compounds of de novo evolution results.
| Name | LigS1 | LigS2 | −PLP1 | −PLP2 | Jain | −PMF | DS | LIE | Ludi_1 | Ludi_2 | Ludi_3 | Binding energy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Evo_4 | 7.96 | 7.65 | 155.06 | 146.11 | 6.54 | 173.03 | 84.983 | 12.137 | 934 | 726 | 814 | −313.93 |
| Evo_6 | 8.25 | 7.58 | 157.98 | 154.04 | 8.11 | 193.52 | 89.923 | 1.070 | 896 | 732 | 818 | −332.88 |
| Evo_9 | 7.89 | 7.70 | 159.77 | 150.27 | 6.58 | 179.7 | 84.031 | 14.601 | 887 | 706 | 788 | −315.64 |
| Evo_10 | 8.02 | 7.64 | 157.63 | 149.94 | 6.98 | 174.42 | 87.701 | 12.052 | 869 | 709 | 786 | −320.44 |
| Evo_12 | 7.86 | 7.51 | 151.48 | 143.00 | 6.71 | 170.99 | 81.758 | 13.481 | 900 | 706 | 792 | −307.47 |
| Evo_15 | 8.12 | 7.94 | 156.22 | 145.27 | 6.5 | 184.05 | 88.842 | 12.431 | 841 | 689 | 760 | −315.77 |
| Evo_16 | 8.05 | 7.53 | 143.19 | 143.0 | 6.23 | 179.02 | 106.79 | −13.72 | 884 | 702 | 792 | −315.20 |
| Evo_17 | 8.05 | 7.86 | 159.73 | 150.90 | 6.83 | 180.08 | 88.826 | 11.188 | 870 | 708 | 777 | −317.99 |
| Evo_22 | 7.94 | 7.64 | 158.68 | 150.45 | 7.36 | 176.00 | 86.105 | 12.077 | 920 | 728 | 814 | −313.34 |
| Evo_24 | 8.09 | 7.58 | 159.88 | 149.57 | 7.95 | 185.43 | 106.351 | −11.257 | 966 | 762 | 848 | −365.11 |
| Evo_25 | 8.36 | 7.45 | 160.15 | 154.69 | 9.06 | 177.54 | 109.045 | −13.805 | 1005 | 807 | 891 | −377.74 |
| Evo_28 | 7.97 | 7.51 | 152.88 | 145.81 | 8.18 | 179.35 | 103.493 | −12.376 | 980 | 753 | 841 | −342.05 |
| Evo_35 | 8.37 | 7.82 | 161.39 | 152.67 | 8.44 | 185.71 | 108.98 | −14.67 | 961 | 772 | 892 | −369.77 |
| Evo_36 | 7.75 | 7.51 | 144.96 | 138.06 | 6.14 | 171.5 | 111.53 | −12.224 | 858 | 679 | 756 | −311.08 |
| Evo_38 | 7.82 | 7.57 | 144.42 | 135.3 | 5.98 | 193.83 | 83.302 | 17.028 | 822 | 675 | 769 | −306.05 |
| Evo_39 | 8.07 | 7.58 | 158.57 | 149.73 | 8.04 | 181.25 | 108.11 | −13.78 | 940 | 759 | 893 | −347.30 |
| Evo_41 | 8.2 | 7.74 | 144.61 | 134.78 | 5.9 | 173.7 | 109.581 | −11.854 | 775 | 648 | 732 | −310.22 |
| Evo_42 | 7.89 | 7.50 | 139.58 | 131.11 | 5.47 | 168.46 | 109.825 | −13.267 | 765 | 621 | 709 | −306.24 |
| Evo_44 | 7.73 | 7.47 | 143.62 | 133.43 | 5.32 | 178.97 | 108.429 | −10.803 | 705 | 592 | 683 | −311.86 |
| Evo_45 | 7.94 | 7.51 | 141.53 | 132.98 | 5.19 | 173.20 | 113.536 | −13.352 | 700 | 604 | 679 | −315.57 |
| Evo_47 | 7.99 | 7.87 | 144.43 | 133.86 | 5.39 | 179.68 | 113.39 | −12.973 | 730 | 617 | 684 | −314.22 |
| Evo_48 | 7.99 | 7.68 | 144.31 | 135.63 | 5.68 | 180.12 | 114.461 | −14.216 | 727 | 607 | 676 | −310.99 |
| Evo_49 | 7.90 | 7.5 | 143.14 | 134.83 | 6.16 | 174.45 | 111.962 | −13.327 | 762 | 629 | 719 | −307.23 |
The unit of binding energy was kcal/mol. LigS was Ligscore, DS was docking score and LIE was Lig internal energy.
Pharmacophore analysis and functional prediction
By pharmacophore analysis, the results were suggested that type2 formed a hydrogen bond in the depths of the pocket by the hydroxyl group on R4 site. Because the hydroxyl group could be hydrogen bond donor or acceptor, the interaction map about hydrogen bond acceptor would be the same as the map about hydrogen bond donor on the hydroxyl group (Figure 4).
Figure 4.

The purple ball revealed the favor of hydrogen bond donor on PDE5. The interaction maps of hydrogen bond donor about the hydroxyl group were revealed in ES03a (A) and ES03b (B). The results of phamacophore were revealed in (C). The green balls labeled the region of hydrogen bond accepter. The purple balls labeled the regions of hydrogen bond donor. The blue balls labeled the hydrophobic regions.
The results of multiple linear regression analysis
The molecular properties from Xia's study37 were associated to QSAR model by followed equation:
[MLRTempModel]=−3.259−5.359*[Molecular_Weight]−0.9236 *[ALogP]+6.434*[Energy]−3.959*[Molecular_Solubility]+5.149* [Num_Rings]+8.689*[Num_H_Acceptors]−4.693*[Num_AromaticRings]−0.415*[Molecular_PolarSurfaceArea]
The values of MLRTempModel had high correlation (R2=0.848) with drug activities in this QSAR model (Figure 5). This correlation was employed to predict pIC50 of the ES series (Cialis=7.988, ES02=6.31, ES03a=9.78, ES03b=10.03). The trend of this result consisted with the docking consequence (Table 2).
Figure 5.

The docking poses of ES03b, No45, and No48 in PDE5. The green one was ES03b, the light blue one was No45, and the purple one is No48. The main different side chain between them was labeled by red circle.
Discussion
The center of binding site was filled with hydrophobic regions (Figure 4C). It meant that hydrophobic compounds could have higher stability in PDE-5. ES03b was more agreeable to the phamacophore map than ES03a, because of the hydroxyl group on R5 site nearby the regions of hydrogen bond donor at the bottom pocket (Figure 4A, 4B). At the gate of the pocket, there was the region of hydrogen bond accepter, and the pose of ES03b was more suitable than type1 as well (Figure 4A, 4B). According to those results, in MLR, ES03b was selected into the drug design procedure.
By Ligand De Novo Evolution, 49 kinds of derivatives of ES03b were designed. We also calculated the docking scores and binding energies for 49 compounds, whose docking scores and binding energies were better than ES03b and would be selected (Table 2). No 45 and 48 were the best result in this study; their poses were very similar to ES03b, but docking scores and binding energies were better than ES03b. The different side chain was pointed out that the change of this site might improve binding affinity (Figure 6).
Figure 6.

The docking poses of ES003-2 (A) and No 48 (B) in PDE5. The green dotted lines were hydrogen bonds.
Compared the poses of ES03b, Evo 45 and Evo 48, the derivatives of ES03b binding to PDE-5 were by four key residuals on PDE-5: Asn661, Tyr664, Asp803, and Lys812 (Figure 7). We suggested that a new inhibitor of PDE-5 be designed from this viewpoint, because the four hydrogen bonds could tightly grasp to ES03b derivatives, which were like a claw to catch the pocket of PDE-5.
Figure 7.

The results of QSAR model of multiple linear regression. The values of Activ were pIC50.
In the results of MLR analysis, Evo48 was overestimated by calculation (Table S3). We suggested that the value of prediction (pIC50=29.582) should not be absurd, but it was far from the truth indeed. However, the prediction was considered with higher activity than the control group significantly. To rank the consequence of the prediction, the trend consisted with the docking results (Table 3 and Table S3). In other words, MLR analysis ranged the potent inhibitors clearly, but it could not calculate the prediction value precisely. It required more information to adjust the QSAR model to increase the accuracy of prediction.
Table S3. The prediction results of QSAR model.
| Name | MW | ALogP | Energy | MS | NR Bonds | NH Acceptors | NA Rings | PS Area | MLRTemp-Model | Predict activ |
|---|---|---|---|---|---|---|---|---|---|---|
| Cialis | 389.404 | 2.183 | 70.44 | −3.288 | 6 | 4 | 3 | 74.87 | 7.794 | 7.989 |
| ES02 | 692.661 | 0.741 | 181.42 | −3.539 | 5 | 14 | 2 | 254.51 | 6.375 | 6.315 |
| ES03a | 724.663 | 5.369 | 84.38 | −7.657 | 6 | 12 | 4 | 218.73 | 9.319 | 9.787 |
| ES03b | 724.663 | 5.369 | 102.43 | −7.68 | 6 | 12 | 4 | 218.73 | 9.526 | 10.031 |
| Evo48 | 798.1 | 6.239 | 204.29 | −8.97 | 6 | 15 | 4 | 244.03 | 26.106 | 29.582 |
Conclusion
In Traditional Chinese Medicines (TCM), Epimedium sagittatum was well known for treating erectile dysfunction. Four components from Epimedium sagittatum (ES01, ES02, ES03a, and ES03b) which docked to PDE-5 and ES03b had a high docking score as tadalafil had. The possibly active component could be analyzed by CADD. MLR was applied to build a QSAR model, which was employed to predict activities of the ES series. The trend of this prediction consisted with the docking consequences. According to the consequences, we suggest that ES03b should be a potent inhibitor of PDE-5. 49 kinds of derivatives of ES03b were designed and docking scores were calculated, which were compared to those of tadalafil and sildenafil citrate. 23 derivatives were selected to be leading drugs for treating erectile dysfunction.
Epimedium sagittatum is not expensive and could be a potential herb to extract leading compound for treating erectile dysfunction. We expect this study of TCM could decrease the cost on drug production and synthesis. This research indicated that the hydrophobic core in the binding site and hydrogen bond production on Asn663 played crucial roles in designing PDE-5 inhibitors. The docking score of Evo48 was 114.46, which signified that the structure of Evo48 could be developed for new PDE-5 inhibitor.
Author contribution
Calvin Yu-chian CHEN, Fuu-jen TSAI, and Chang-Hai TSAI designed research; Chien-yu CHEN, Yea-huey CHANG, and Hung-jin HUANG performed research; Da-tian BAU and Chien-yu CHEN analyzed data; Calvin Yu-chian CHEN and Chien-yu CHEN wrote the paper.
Acknowledgments
The research was supported by grants from the National Science Council of China (NSC 94-2213-E-039-002) and China Medical University (CMU97-CMC-014, CMU96-178). We are grateful to the National Center for High-performance Computing for computer time and facilities.
References
- Corbin JD, Francis SH. Cyclic GMP phosphodiesterase-5: target of sildenafil. J Biol Chem. 1999;274:13729–32. doi: 10.1074/jbc.274.20.13729. [DOI] [PubMed] [Google Scholar]
- Yoo HH, Kim NS, Im GJ, Kim DH. Pharmacokinetics and tissue distribution of a novel PDE5 inhibitor, SK-3530, in rats. Acta Pharmacol Sin. 2007;28:1247–53. doi: 10.1111/j.1745-7254.2007.00611.x. [DOI] [PubMed] [Google Scholar]
- Whitehill D. Viagra, levitra, and cialis: What's the difference. S D J Med. 2005;58:129–30. [PubMed] [Google Scholar]
- Weeks JL, Zoraghi R, Beasley A, Sekhar KR, Francis SH, Corbin JD. High biochemical selectivity of tadalafil, sildenafil and vardenafil for human phosphodiesterase 5a1 (pde5) over pde11a4 suggests the absence of pde11a4 cross-reaction in patients. Int J Impot Res. 2005;17:5–9. doi: 10.1038/sj.ijir.3901283. [DOI] [PubMed] [Google Scholar]
- Rao YJ, Xi L. Pivotal effects of phosphodiesterase inhibitors on myocyte contractility and viability in normal and ischemic hearts. Acta Pharmacol Sin. 2009;1:1–24. doi: 10.1038/aps.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughney K, Taylor J, Florio VA. 3',5'-cyclic nucleotide phosphodiesterase 11a: Localization in human tissues. Int J Impot Res. 2005;17:320–5. doi: 10.1038/sj.ijir.3901317. [DOI] [PubMed] [Google Scholar]
- Weeks JL, Blount MA, Beasley A, Zoraghi R, Thomas MK, Sekhar KR, et al. Radiolabeled ligand binding to the catalytic or allosteric sites of pde5 and pde11. Methods Mol Biol. 2005;307:239–62. doi: 10.1385/1-59259-839-0:239. [DOI] [PubMed] [Google Scholar]
- Liao HJ, Chen XM, Li WG. Effect of epimedium sagittatum on quality of life and cellular immunity in patients of hemodialysis maintenance. Zhongguo Zhong Xi Yi Jie He Za Zhi. 1995;15:202–4. [PubMed] [Google Scholar]
- Li RS, Li JL, Xu JJ. Stimulatory action of epimedium sagittatum (sieb. Et zucc.) maxim. On platelet aggregation in rats. Zhong Yao Tong Bao. 1987;12:40–2. [PubMed] [Google Scholar]
- Feng K, Xie W, Chen BL, Wang JZ, Guo JG. Effect of five species of epimedium on growth of cartilage and proliferation of cartilage cell in vitro. Zhongguo Zhong Yao Za Zhi. 2006;31:2065–7. [PubMed] [Google Scholar]
- Liu F, Ding G, Li J.Effects of epimedium sagittatum maxim. Polysaccharides on DNA synthesis of bone marrow cells of “Yang deficiency” Animal model caused by hydroxyurea Zhongguo Zhong Yao Za Zhi 199116620–2.Chinese [PubMed] [Google Scholar]
- Iinuma M, Tanaka T, Sakakibara N, Mizuno M, Matsuda H, Shiomoto H, et al. Phagocytic activity of leaves of epimedium species on mouse reticuloendotherial system. Yakugaku Zasshi. 1990;110:179–85. doi: 10.1248/yakushi1947.110.3_179. [DOI] [PubMed] [Google Scholar]
- Chen X, Zhou M, Wang J. Effect of epimedium sagittatum on soluble IL-2 receptor and IL-6 levels in patients undergoing hemodialysis. Zhonghua Nei Ke Za Zhi. 1995;34:102–4. [PubMed] [Google Scholar]
- Chen CYC. A novel perspective on designing the inhibitor of HER2 receptor. J Chin Inst Chem Engrs. 2008;39:291–9. [Google Scholar]
- Chen CYC. Insights into the Suanzaoren mechanism — from constructing the 3D structure of GABA-A receptor to its binding interaction analysis. J Chin Inst Chem Engrs. 2008;39:663–71. [Google Scholar]
- Chen CYC. Discovery of novel inhibitors for c-Met by virtual screening and pharmacophore analysis. J Chin Inst Chem Engrs. 2008;39:617–24. [Google Scholar]
- Chen YC, Chen KT. Novel selective inhibitors of hydroxyxanthone derivatives for human cyclooxygenase-2. Acta Pharmacol Sin. 2007;28:2027–32. doi: 10.1111/j.1745-7254.2007.00663.x. [DOI] [PubMed] [Google Scholar]
- Chen CYC. De novo design of novel selective COX-2 inhibitors: from virtual screening to pharmacophore analysis. J Chin Inst Chem Engrs. 2009;40:55–69. [Google Scholar]
- Chen CYC. Chemoinformatics and pharmacoinformatics approach for exploring the GABA-A agonist from Chinese herb suanzaoren. J Chin Inst Chem Engrs. 2009;40:36–47. [Google Scholar]
- Chen CYC. Pharmacoinformatics approach for mPGES-1 in anti-inflammation by 3D-QSAR pharmacophore mapping. J Chin Inst Chem Engrs. 2009;40:155–61. [Google Scholar]
- Chen CYC, Chen YF, Chen YC, Tsai HY. What is the effective component in Suanzaoren decoction for curing insomnia? Discovery by virtual screening and molecular dynamic simulation. J Biomol struct Dyn. 2008;26:57–64. doi: 10.1080/07391102.2008.10507223. [DOI] [PubMed] [Google Scholar]
- Liu Q, Yu KW, Chang YC, Lukas RJ, Wu J. Agonist-induced hump current production in heterologously-expressed human alpha4beta2-nicotinic acetylcholine receptors. Acta Pharmacol Sin. 2008;29:305–19. doi: 10.1111/j.1745-7254.2008.00760.x. [DOI] [PubMed] [Google Scholar]
- Dong PP, Zhang YY, Ge GB, Ai CZ, Liu Y, Yang L, et al. Modeling resistance index of taxoids to MCF-7 cell lines using ANN together with electrotopological state descriptors. Acta Pharmacol Sin. 2008;29:385–96. doi: 10.1111/j.1745-7254.2008.00746.x. [DOI] [PubMed] [Google Scholar]
- Shi L, Yu HP, Zhou YY, Du JQ, Shen Q, Li JY, Li J. Discovery of a novel competitive inhibitor of PTP1B by high-throughput screening. Acta Pharmacol Sin. 2008;29:278–84. doi: 10.1111/j.1745-7254.2008.00737.x. [DOI] [PubMed] [Google Scholar]
- He HQ, Ma XH, Liu B, Chen WZ, Wang CX, Cheng SH. A novel high-throughput format assay for HIV-1 integrase strand transfer reaction using magnetic beads. Acta Pharmacol Sin. 2008;29:397–404. doi: 10.1111/j.1745-7254.2008.00748.x. [DOI] [PubMed] [Google Scholar]
- Feng X, Wang LN, Zhou YY, Yu HP, Shen Q, Zang Y, et al. Discovery and characterization of a novel inhibitor of CDC25B, LGH00045. Acta Pharmacol Sin. 2008;29:1268–74. doi: 10.1111/j.1745-7254.2008.00841.x. [DOI] [PubMed] [Google Scholar]
- Yan JH, Su HR, Boutin JA, Renard MP, Wang MW. High-throughput screening assay for new ligands at human melatonin receptors. Acta Pharmacol Sin. 2008;29:1515–21. doi: 10.1111/j.1745-7254.2008.00903.x. [DOI] [PubMed] [Google Scholar]
- Fu J, Tang ZM, Gao X, Zhao F, Zhong H, Wen MR, et al. Optimal design and validation of antiviral siRNA for targeting hepatitis B virus. Acta Pharmacol Sin. 2008;29:1529–38. doi: 10.1111/j.1745-7254.2008.00891.x. [DOI] [PubMed] [Google Scholar]
- Taha MO, Bustanji Y, Al-Bakri AG, Yousef AM, Zalloum WA, Al-Masri IM, et al. Discovery of new potent human protein tyrosine phosphatase inhibitors via pharmacophore and QSAR analysis followed by in silico screening. J Mol Graph Model. 2007;25:870–84. doi: 10.1016/j.jmgm.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Srivani P, Srinivas E, Raghu R, Sastry GN. Molecular modeling studies of pyridopurinone derivatives—potential phosphodiesterase 5 inhibitors. J Mol Graph Model. 2007;26:378–90. doi: 10.1016/j.jmgm.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Bhattacharya M, Pillalamari U, Sarkhel S, Ishino T, Urbina C, Jameson B, et al. Recruitment pharmacophore for interleukin 5 receptor alpha antagonism. Biopolymers. 2007;88:83–93. doi: 10.1002/bip.20612. [DOI] [PubMed] [Google Scholar]
- Sung BJ, Hwang KY, Jeon YH, Lee JI, Heo YS, Kim JH, et al. Structure of the catalytic domain of human phosphodiesterase 5 with bound drug molecules. Nature. 2003;425:98–102. doi: 10.1038/nature01914. [DOI] [PubMed] [Google Scholar]
- Lu A, Zhang J, Yin X, Luo X, Jiang H. Farnesyltransferase pharmacophore model derived from diverse classes of inhibitors. Bioorg Med Chem Lett. 2007;17:243–9. doi: 10.1016/j.bmcl.2006.09.055. [DOI] [PubMed] [Google Scholar]
- Doddareddy MR, Choo H, Cho YS, Rhim H, Koh HY, Lee JH, et al. 3d pharmacophore based virtual screening of t-type calcium channel blockers. Bioorg Med Chem. 2007;15:1091–105. doi: 10.1016/j.bmc.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Liu F, You QD, Chen YD. Pharmacophore identification of KSP inhibitors. Bioorg Med Chem Lett. 2007;17:722–6. doi: 10.1016/j.bmcl.2006.10.083. [DOI] [PubMed] [Google Scholar]
- Keith JM, Gomez LA, Letavic MA, Ly KS, Jablonowski JA, Seierstad M, et al. Dual serotonin transporter/histamine h3 ligands: optimization of the h3 pharmacophore. Bioorg Med Chem Lett. 2007;17:702–6. doi: 10.1016/j.bmcl.2006.10.089. [DOI] [PubMed] [Google Scholar]
- Xia G, Li J, Peng A, Lai S, Zhang S, Shen J, et al. Synthesis and phosphodiesterase 5 inhibitory activity of novel pyrido [1,2-e] purin-4(3H)-one derivatives. Bioorg Med Chem Lett. 2005;15:2790–94. doi: 10.1016/j.bmcl.2005.03.102. [DOI] [PubMed] [Google Scholar]
- Keith JM, Gomez LA, Letavic MA, Ly KS, Jablonowski JA, Seierstad M, et al. Dual serotonin transporter/histamine h3 ligands: optimization of the h3 pharmacophore. Bioorg Med Chem Lett. 2007;17:702–6. doi: 10.1016/j.bmcl.2006.10.089. [DOI] [PubMed] [Google Scholar]
- Tataridis D, Fytas G, Kolocouris A, Fytas C, Kolocouris N, Foscolos GB, et al. Influence of an additional 2-amino substituent of the 1-aminoethyl pharmacophore group on the potency of rimantadine against influenza virus a. Bioorg Med Chem Lett. 2007;17:692–6. doi: 10.1016/j.bmcl.2006.10.092. [DOI] [PubMed] [Google Scholar]
- Lenkowski PW, Batts TW, Smith MD, Ko SH, Jones PJ, Taylor CH, et al. A pharmacophore derived phenytoin analogue with increased affinity for slow inactivated sodium channels exhibits a desired anticonvulsant profile. Neuropharmacology. 2007;52:1044–54. doi: 10.1016/j.neuropharm.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]