

Abstract
Candida glabrata currently ranks as the second most frequent cause of invasive candidiasis. Our previous work has shown that C. glabrata is adapted to intracellular survival in macrophages and replicates within non-acidified late endosomal-stage phagosomes. In contrast, heat killed yeasts are found in acidified matured phagosomes. In the present study, we aimed at elucidating the processes leading to inhibition of phagosome acidification and maturation. We show that phagosomes containing viable C. glabrata cells do not fuse with pre-labeled lysosomes and possess low phagosomal hydrolase activity. Inhibition of acidification occurs independent of macrophage type (human/murine), differentiation (M1-/M2-type) or activation status (vitamin D3 stimulation). We observed no differential activation of macrophage MAPK or NFκB signaling cascades downstream of pattern recognition receptors after internalization of viable compared to heat killed yeasts, but Syk activation decayed faster in macrophages containing viable yeasts. Thus, delivery of viable yeasts to non-matured phagosomes is likely not triggered by initial recognition events via MAPK or NFκB signaling, but Syk activation may be involved. Although V-ATPase is abundant in C. glabrata phagosomes, the influence of this proton pump on intracellular survival is low since blocking V-ATPase activity with bafilomycin A1 has no influence on fungal viability. Active pH modulation is one possible fungal strategy to change phagosome pH. In fact, C. glabrata is able to alkalinize its extracellular environment, when growing on amino acids as the sole carbon source in vitro. By screening a C. glabrata mutant library we identified genes important for environmental alkalinization that were further tested for their impact on phagosome pH. We found that the lack of fungal mannosyltransferases resulted in severely reduced alkalinization in vitro and in the delivery of C. glabrata to acidified phagosomes. Therefore, protein mannosylation may play a key role in alterations of phagosomal properties caused by C. glabrata.
Introduction
Candida spp. are the most frequent causes of invasive fungal infections in the United States [1], [2], with an associated mortality rate of 30% to 50% [3]. The Candida species distribution has shifted in recent years: C. albicans remains the most frequently isolated species, but an increasing fraction of cases is caused by non-albicans species. Of particular concern is the emergence of C. glabrata as the second most frequent cause of invasive candidiasis [4]. Fungal tolerance to azole-class antifungals and the tendency of C. glabrata to acquire drug resistance during antifungal therapy are associated with treatment failure and death [5]–[8].
Surprisingly, and despite its medical importance, C. glabrata is non-lethal and elicits a low inflammatory immune response in systemic models of mouse infection, even following intravenous infection with high inocula [9]–[11]. Nevertheless, viable fungi can readily be isolated from organs of immunocompetent animals several weeks after infection, indicating that even a fully functional immune system cannot efficiently clear C. glabrata [11], [12]. To explain these surprising findings, we proposed that C. glabrata employs an immune evasion strategy, possibly via concealment in intracellular niches. Of note, we found C. glabrata cells associated with mononuclear cell infiltrates in all mouse organs tested (spleen, liver, brain, kidney, lung and heart), whereas no neutrophil infiltration was observed [11]. Using macrophages as a niche is a strategy that has been described for other pathogenic fungi, such as Histoplasma capsulatum and Cryptococcus neoformans [13], [14]. Our and others’ previous research showed that C. glabrata replicates within human and murine macrophages after phagocytosis [15]–[17], an ability which requires distinct attributes, which are only partially understood. For example, a family of glycosylphosphatidylinositol-anchored aspartyl proteases (YPS proteins) is required for survival of C. glabrata in macrophages [15]. Furthermore, the fungus can adapt its metabolism to starvation and can use endogenous resources to overcome nutrient limitation [15], [16]. Moreover, chromatin remodeling and DNA damage repair was shown to be crucial for viability within a macrophage phagosome [18]. Finally, by screening a set of defined C. glabrata mutants for reduced macrophage survival, we recently identified a series of genes required to resist intracellular killing [19]. These data support the view that immune evasion, stress resistance and nutrient acquisition are key aspects for intracellular survival.
Importantly, we found that C. glabrata containing phagosomes are less acidified and blocked at a late-endosomal state [17]. Mature phagolysosomes are normally strongly acidified by proton-pumping activity of vacuolar ATPase (V-ATPase). This acidic pH promotes antimicrobial effector mechanisms such as the activity of hydrolytic enzymes [20]. How phagosome maturation is blocked and acidification is prevented by C. glabrata and whether an immature, non-acidified phagosome is necessary for intracellular survival of the fungus is unknown.
The aim of this work was therefore to define the conditions under which non-acidified C. glabrata containing phagosomes are formed. We sought to find out whether this is a process, which might be dependent on C. glabrata initial recognition and host signaling, or whether fungal cells may actively modify their host compartment. We show that C. glabrata localization in non-phagolysosomal compartments is independent of the macrophage type and activation status. Further, we provide evidence for environmental alkalinization as a possible new strategy of C. glabrata to actively raise phagosome pH.
Materials and Methods
Ethics Statement
Blood was obtained from healthy human donors with written informed consent. The blood donation protocol and use of blood for this study were approved by the Jena institutional ethics committee (Ethik-Kommission des Universitätsklinikum Jena, Permission No 2207-01/08).
Strains and Growth Conditions
Laboratory strain ATCC2001 or its GFP-expressing derivative [17] were used for characterization of macrophage – C. glabrata wild type interaction. C. glabrata mutant strains are derivatives of the laboratory strain ATCC2001, harboring auxotrophies for histidine, leucine and tryptophan (wt hltΔ). Mutant strains were obtained from a novel genome-scale collection of C. glabrata deletion mutants. In each strain of the collection, a single open reading frame (ORF) was replaced with a bar-coded NAT1 resistance cassette (T. Schwarzmüller, B. Cormack, K. Haynes and K. Kuchler, unpublished data). The wt hltΔ strain was used for comparison. Mutant strains anp1Δ and mnn11Δ [21] were generated in a tryptophan-auxotrophic background and were thus compared to a tryptophan-auxotrophic ATCC2001 strain (wt tΔ).
All yeast strains used in this study were routinely grown overnight in YPD (1% yeast extract, 1% peptone, 2% glucose) at 37°C in a shaking incubator. For preparation of heat killed yeast cells, 500 µl overnight culture was incubated at 70°C for 10 min. Growth curves were monitored in a 96 well plate format by recording the absorption at 600 nm at 37°C as a function of time in an ELISA reader (Tecan Infinite 200).
Preparation of Monocyte-Derived Macrophages (MDMs)
Human peripheral blood mononuclear cells (PBMC) were isolated by Histopaque-1077 (Sigma-Aldrich) density centrifugation from buffy coats donated by healthy volunteers. To differentiate PBMC into monocyte-derived macrophages (MDMs), 4×107 PBMC were plated in RPMI 1640 media (with L-glutamine and 25 mM HEPES) (PAA Laboratories, Inc., Austria) in cell culture dishes. After 1–2 hours, non-adherent cells were washed away and 5 ng/ml M-CSF (ImmunoTools, Germany) was included in the cultures to favor the differentiation of M2-type macrophages. In selected experiments, 5 ng/ml GM-CSF was added instead to favor differentiation of M1-type macrophages. After two days at 5% CO2 and 37°C, medium was exchanged to RPMI 1640 containing 10% heat-treated fetal bovine serum (PAA Laboratories, Inc., Austria) and M-CSF or GM-CSF. After four more days, adherent MDMs were detached with 50 mM EDTA and plated in flat-bottom 96 and 24 well plates to give a final concentration of approximately 4×104 MDMs/well and 1×105 MDM/well, respectively, in RPMI 1640 with serum. Macrophage infection experiments were performed in RPMI 1640 without serum. All experiments were performed with PBMCs isolated from at least three different donors.
Macrophage Cell Lines
The murine RAW264.7 macrophage-like cell line [22] used in this study was routinely cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 4 mM L-glutamine and 4.5 g/l glucose (PAA Laboratories, Inc., Austria) and supplemented with 10% heat-treated fetal bovine serum (PAA Laboratories, Inc., Austria) at 37°C and 5% CO2. For infection experiments, RAW264.7 cells were inoculated in 6 or 24 well plates at an initial concentration of approximately 1.5×106 cells/well or 2×105 cells/well, respectively, in DMEM with serum and then incubated overnight at 37°C and 5% CO2 to near confluency (60%–80%).
A stable J774E macrophage-like cell line expressing the subunit E (vatE) of the V1-subcomplex of V-ATPase as a green fluorescent protein fusion construct was constructed as follows. A cDNA encoding murine vatE was purchased from RZPD (Deutsches Ressourcenzentrum für Genomforschung GmbH, Berlin, Germany). This cDNA was PCR-amplified. The reverse primer introduced a change of the stop codon (TGA) into a serine (TCA) codon, extending the vatE coding region by six amino acid residues. The introduction of EcoRI and KpnI restriction sites by the primer pair allowed in-frame cloning of the PCR product cleaved with EcoRI and KpnI in the vector pEGFP-N1 (Addgene, Cambridge, MA). Correct in-frame cloning and point mutagenesis were confirmed by nucleotide sequencing of the product. The vatE-EGFP construct was propagated in E. coli and used to transfect J774E macrophages [23] by electroporation following the protocol by Schneider et al. [24]. Selection was done by Geneticin (0.3 mg/ml) for two weeks. Resulting clones were cultivated and frozen. Frozen stocks were thawed and transfectants cloned by dilution into 96 well plates. Five resulting clones were pooled and used for further analysis. It should be noted that the stable transfectants do not express bright vatE-EGFP in accordance with the relative scarcity of V-ATPase in the cell and regular re-selection steps are necessary. To eventually enhance the weak signal, monoclonal (not cross-reacting) murine monoclonal IgG anti-EGFP antibodies are used (see below). The resulting vatE-EGFP staining was predominantly congruent with LysoTracker staining for acidic compartments, yet not with staining of early endosome antigen-1 (EEA-1).
J774-V-ATPase-GFP cells were routinely cultured in DMEM with 4 mM L-glutamine and 4.5 g/l glucose (PAA Laboratories, Inc., Austria), supplemented with 10% heat-treated fetal bovine serum (PAA Laboratories, Inc., Austria) and 0.3 mg/ml G418 at 37°C and 5% CO2. For infection experiments, J774-V-ATPase-GFP cells were inoculated in 24 well plates at an initial concentration of approximately 1×105 cells/well in DMEM with serum and then incubated overnight at 37°C and 5% CO2 to near confluency (60%–80%).
Fluorescence Microscopy
For microscopic analysis MDM, RAW264.7 or J774-V-ATPase-GFP cells were allowed to adhere to coverslips within a 24 well plate. Overnight yeast cultures were washed with PBS, counted using a hemacytometer and adjusted to the desired concentration in cell culture medium without serum. Macrophages were then infected with yeast cells at a multiplicity of infection (MOI) of 5–10 or with latex beads or 1 µg/ml LPS. Synchronization of phagocytosis (not performed in NFκB localization study) was performed by placing the 24 well plate on ice for 30 min after infection until yeast cells settled. Unbound yeast cells were removed by washing with pre-warmed (37°C) medium, and phagocytosis was initiated by incubating at 37°C and 5% CO2. Co-incubation times varied between 10 and 180 min.
Acidification of the phagosomes was assessed by use of the acidotropic dye LysoTracker Red DND-99 (Life Technologies, USA). LysoTracker (diluted 1∶10,000 in cell culture medium) was added 1 h prior to infection and during co-incubation with the yeast cells. In selected experiments, 50–100 nM calcitriol (1α,25-dihydroxy vitamin D3, Cayman, USA) was added one night before the experiment and in parallel with LysoTracker. TROV (texas red ovalbumin, 100 µg/ml [Life Technologies, USA]) was added 2 h prior to infection, followed by a wash step and 30 min incubation in cell culture medium without TROV (chase). DQ-BSA (DQ™ Red BSA, 10 µg/ml [Life Technologies, USA]) was added 1 h prior to infection and during co-incubation with the yeast cells.
Coverslips were fixed with 4% paraformaldehyde (10 min, 37°C) at indicated time points and stained with Alexa Fluor 647-conjugated Concanavalin A (Life Technologies, USA) for 30 min. For antibody staining, macrophages were then permeabilized with 0.5% Triton-X-100 in PBS, and blocked with 2% BSA in PBS (30 min at room temperature). Cells were then incubated with a primary antibody (anti-Rab7, D95F2 XP Rabbit mAb, 1∶100, Cell Signaling, Inc., USA; anti-GFP, rat IgG2a monoclonal, 1∶500, Nacalai Tesque, Inc., Japan; NFκB (p65), 1∶100, Cell Signaling, Inc., USA) diluted in 1% BSA in PBS for 2 h, followed by incubation with a secondary Alexa Fluor-conjugated antibody (Life Technologies, USA) diluted 1∶500 in PBS. Cover slips were mounted and percentage of co-localization was calculated manually by analyzing a minimum of 100 yeast cells per well or in the case of NFκB by scoring a minimum of 100 nuclei (Leica DM5500B, Zeiss Axio Observer, Zeiss LSM).
Western Blot Analysis
RAW264.7 macrophages were seeded in 6 well plates and infected with C. glabrata (viable or heat killed) at a MOI of 5 or treated with 1 µg/ml LPS. At indicated time points macrophages were lysed with 120 µl RIPA-buffer (50 mM Tris-HCl, 150 mM NaCl, 0.1% SDS, pH 7.5) containing protease and phosphatase inhibitors (complete protease inhibitor cocktail tablets, phosSTOP phosphatase inhibitor cocktail tablets [Roche, Switzerland]) for 30 min on ice. After centrifugation the supernatant was used for measurement of protein concentration (BCA protein assay kit [Thermo Fisher Scientific, Inc., USA]). 30 µg proteins were separated by SDS-PAGE and electrophoretically transferred to a nitrocellulose membrane. The membranes were blocked with 5% I-Block protein-based blocking reagent (Life Technologies, USA), solved in TBS-T (50 mM Tris-HCl [ph 7.6], 0.15 M NaCl, 0.05% Tween-20) and then incubated with primary antibodies for phosphorylated proteins in TBS-T overnight at 4°C. Antibodies specific for P-p38, P-p44/42 (P-ERK1/2), P-SAPK/JNK, P-IKKαβ, P-IκB, P-p65 were all purchased from Cell Signaling, Inc., USA. After washing three times with TBS-T, the membrane was incubated with a horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibody (Santa Cruz Biotechnology, Inc., USA) followed by three washing steps. Immunoreactivity was detected by enhanced chemiluminescence (ECL Plus Western Blotting Substrate [Thermo Fisher Scientific, Inc., USA]). Membranes were then stripped with 0.5 M Tris-HCl (pH 6.7), 2% SDS and 100 mM β-mercaptoethanol for 30 min at 56°C. To ensure application of equal protein amount, membranes were again incubated with primary antibodies for unphosphorylated proteins in TBS-T overnight at 4°C (antibodies specific for p38, p44/42 (ERK1/2), SAPK/JNK, IKKα, IκB, p65 and were all purchased from Cell Signaling, Inc., USA) and detected as described above. All experiments have been performed at least in triplicate. Western Blot analysis for detection of Syk activation were performed as described elsewhere [25]. β-actin detection was included as loading control.
Survival Assays
MDMs were seeded in 96 well plates. If necessary, cells were pre-treated with 50 µM chloroquine, 50 nM bafilomycin A1 (Sigma) or 20 µM iron nitriloacetate (FeNTA) one hour before infection. MDMs were infected at a MOI of 1 with wild type or mutant C. glabrata strains and incubated at 37°C and 5% CO2 with or without chloroquine, bafilomycin A1 or FeNTA. Survival of macrophage-internalized yeasts was assessed after 3 h or 24 h by removing non-cell-associated yeasts by washing with RPMI, subsequent lysis of MDMs with 20 µl 0.5% Triton-X-100 per well and plating lysates on YPD plates to determine colony forming units (cfus). For survival experiments comparing M1- and M2-type macrophages, macrophage-associated plus non-associated yeasts were plated and cfus were compared to wells containing yeasts only.
Alkalinization Experiments
Liquid alkalinization-promoting medium contained 1×YNB without amino acids and ammonium sulfate (BD Bioscience, USA), 1% casamino acids (BD Bioscience, USA) and 20 mg/l phenol red. Solid alkalinization-promoting medium contained 1×YNB without amino acids and ammonium sulfate, 1% casamino acids, 2% agar and 0.01% bromocresol green. Both media were adjusted to pH 4. For experiments with triple-auxotrophic strains, histidine (0.1 g/l), leucine (0.02 g/l) and tryptophan (0.05 g/l) were added to the media.
Alkalinization in liquid media was assayed by inoculating 1×106 C. glabrata cells/ml in a 24 well plate and incubating at 37°C while shaking at 180 rpm. pH indicator color was photographed after 20–24 hours. Growth controls were performed by measuring OD600 in alkalinization-promoting medium without pH indicator or in YPD medium using an ELISA reader (Tecan Infinite 200).
Alkalinization on solid media was assayed in a 96 well format with incubation at 37°C. pH indicator color was photographed after 9 hours. Growth controls were performed by monitoring colony size on solid alkalinization-promoting medium without pH indicator or on solid YPD medium.
For screening of auxotrophic C. glabrata deletion mutants (T. Schwarzmüller, B. Cormack, K. Haynes and K. Kuchler, unpublished data) for alkalinization defects, cells were inoculated from glycerol stocks and subcultured twice over night at 37°C in liquid YPD. Then 6 µl of the C. glabrata cultures were spotted on solid medium containing bromocresol green as described above. C. glabrata mutants that did not show a pH indicator change from green to blue, but were forming colonies on control plates, were considered as alkalinization-defective. Every assay contained a triple-auxotrophic wild type and medium alone as controls. Alkalinization defects were verified with defined inoculum cell numbers in liquid alkalinization medium containing phenol red as described above.
Statistical Analysis
All experiments were performed at least in triplicate (n≥3). All data are reported as the mean ± SD. The data were analyzed using two-tailed, unpaired Student’s t-test for inter-group comparisons. For data sets based on microscopic quantification (percentage of counts), an arcsine transformation was performed prior to the t-test. A minimum of 100 yeast cells per sample or in the case of NFκB a minimum of 100 nuclei were counted. Statistical significant results were marked with a single asterisk meaning P value<0.05, double asterisks meaning P value<0.01 or triple asterisks meaning P value<0.005.
Results
C. glabrata Containing Phagosomes do not Reach the Phagolysosomal State
Our previous analyses on maturation of phagosomes containing viable C. glabrata in macrophages revealed a compartment positive for the late endosome marker LAMP1 but less acidified than phagolysosomes containing heat killed yeasts [17]. Here we aimed at a more detailed characterization of the C. glabrata containing vacuole to better understand the composition of phagosomes, in which C. glabrata is able to survive. We therefore analyzed further markers of phagosome maturation in infected monocyte-derived macrophages (MDMs) as well as a murine macrophage-like cell line. As with LAMP1, the majority of phagosomes containing viable and heat killed yeasts acquired the small GTPase Rab7 as a marker protein of late endosomes (Fig. 1A).
Figure 1. C. glabrata resides in non-matured macrophage phagosomes.

Representative DIC and fluorescence microscopy images of viable or heat killed C. glabrata 90 min post infection phagocytosed by human MDMs (left panels) showing marker proteins in red, while GFP-expressing C. glabrata are indicated in green. To detect non-phagocytosed yeasts, samples were stained with Concanavalin A (ConA, shown in yellow). Phagocytosed yeasts are labeled with a white arrow while non-phagocytosed yeasts are marked with red arrows. Co-localization with fluorescence markers was quantified for phagosomes containing viable or heat killed (Hk) C. glabrata at indicated time points (right panel). (A) Phagosomes containing viable and heat killed C. glabrata co-localize with the late endosome marker Rab7. (B) In contrast to heat killed C. glabrata containing phagosomes, compartments containing viable yeasts show low phagosomal proteolytic activity as measured by co-localization with the fluorogenic protease substrate DQ-BSA. (C) Heat killed but not viable C. glabrata containing phagosomes acquire the lysosomal tracer texas red ovalbumin (TROV). Statistical analysis was performed comparing heat killed with viable C. glabrata at indicated time points (n≥3; *p<0.05, ***p<0.005 by unpaired Student’s t test).
DQ-BSA is a tracer for proteolytic activities. Once cleaved in acidic intracellular lysosomes, it generates a highly fluorescent product that can be monitored by microscopy. As our previous data showed viable C. glabrata to be localized in non-acidified phagosomes, we expected a low DQ-BSA staining for these compartments. Indeed, viable C. glabrata cells are located in less degradative phagosomes as compared to heat killed cells (Fig. 1B). A similar result was obtained when the fluid phase tracer texas red ovalbumin (TROV) had been chased into lysosomes. The number of TROV-positive yeast-containing phagosomes was dramatically increased for macrophages infected with heat killed as compared to viable cells (Fig. 1C). We conclude that viable C. glabrata containing phagosomes reach the late endosomal stage but do not fuse with lysosomes, resulting in an environment with low degradative activity. Similar data demonstrating the disparity in maturation of viable and heat killed yeast containing phagosomes were obtained with murine RAW264.7 macrophages (Fig. S1).
Phagosome Maturation Arrest Occurs Independent of Macrophage Type, Differentiation or Activation Status and is Specific for C. glabrata Containing Compartments
In the human body, macrophages change their physiology in response to environmental stimuli such as innate and adaptive immune responses. This generates different populations of macrophages with distinct functions. M1-type or classically activated macrophages are often associated with higher microbicidal activity, while M2-type or alternatively activated macrophages are more connected to regulatory functions [26]. To determine whether the initial macrophage differentiation status has an influence on C. glabrata-macrophage interaction, we tested whether GM-CSF stimulation, resulting in M1-polarized macrophages would enhance fungicidal activity as compared to M-CSF stimulation, resulting in M2-polarized macrophages. We detected no difference in phagocytosis rate, phagosome acidification or fungal survival in M1- or M2-polarized macrophages (Fig. 2A).
Figure 2. Phagosome maturation arrest occurs in different macrophage differentiation or activation states and is yeast phagosome-specific.
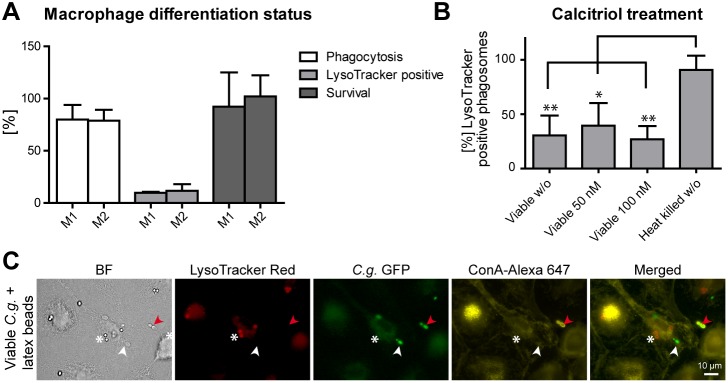
(A) Human M1-polarized and M2-polarized MDMs do not differ in central aspects of C. glabrata-macrophage interaction: phagocytosis, phagosome acidification and killing. Phagocytosis (MOI of 5) was quantified microscopically by determining the percentage of internalized (Concanavalin A stain-negative) yeasts out of total yeasts after 90 min. Phagosome acidification was quantified microscopically by determining the percentage of LysoTracker-positive phagosomes after 90 min. Survival of C. glabrata was determined by cfu-plating of macrophage lysates after 3 h of co-incubation and comparing to yeasts incubated without macrophages. (B) Treatment with vitamin D3 (calcitriol) has no influence on the number of LysoTracker-positive viable C. glabrata containing phagosomes of human MDMs. (C) MDMs co-infected with C. glabrata and latex beads show a acidification defect specific to C. glabrata containing phagosomes (LysoTracker-negative staining; white arrow) but acidify latex bead containing phagosomes (LysoTracker-positive staining; white asterisk). Representative image 90 min post infection. GFP-expressing C. glabrata is indicated in green and non-phagocytosed yeasts stained with Concanavalin A (ConA) are in yellow (marked with red arrows). Statistical analysis was performed comparing M1-type with M2-type macrophages (A) or drug treated with untreated viable C. glabrata (B) (n≥3; *p<0.05, **p<0.01 by unpaired Student’s t test).
Besides cytokines, other endogenous factors can regulate macrophage functions. Vitamin D3 (calcitriol) is known to activate antimicrobial activity of macrophages against the intracellular pathogen Mycobacterium tuberculosis [27], [28]. To find out whether C. glabrata containing macrophages can be activated in a similar way, we tested intracellular survival of C. glabrata and acidification of C. glabrata phagosomes in calcitriol-treated macrophages. No differences between treated and untreated macrophages were observed (Fig. 2B).
Next, we sought to evaluate whether phagocytosis of C. glabrata by a macrophage globally modifies phagosome maturation of neighboring, non-fungal containing phagosomes in the same macrophage. We therefore analyzed phagosome acidification in macrophages that had taken up C. glabrata in combination with latex beads. Lack of phagosome acidification was only observed for C. glabrata containing phagosomes, while neighboring latex-bead containing phagosomes in the same macrophage were acidified (Fig. 2C).
In summary, C. glabrata persistence in macrophages in non-mature, non-acidified phagosomes is not affected by different macrophage differentiation programs and activation types, and is specific to fungus containing phagosomes.
Viable or Heat Killed C. glabrata do not Evoke Different Intracellular Signaling Pathways
Recognition of ligands by receptors at the macrophage membrane activates a series of intracellular signaling pathways that lead to both, reorganization of the actin cytoskeleton, which is essential for the phagocytic uptake, and expression of immunomodulatory proteins, such as cytokines [29]. Thus, variations of intracellular signaling within macrophages may hint towards a different recognition of viable vs. heat killed C. glabrata cells, which may have an impact on the maturation of phagosomes. We therefore analyzed the activation of three different MAP-kinases induced upon recognition of microorganisms by macrophages [30]–[32]. Moreover, components of the nuclear factor-κB (NFκB) pathway, which have been shown to be central in the expression of immunomodulatory factors, were examined upon infection of RAW264.7 macrophages with viable or heat killed C. glabrata cells. A treatment with LPS served as a positive control.
The three major subgroups of MAP-kinases, comprising of the extracellular signal-regulated kinases (ERKs), the stress-activated protein kinases/c-jun amino-terminal kinases (SAPK/JNKs) and the p38 MAP-kinases, are all activated by phosphorylation of a common threonine-X-tyrosine regulatory motif. The activation state can be monitored by Western Blot analyses with the appropriate anti-phospho MAP-kinase antibody. As expected, LPS enhanced the phosphorylation of all three types of MAP-kinases as early as 10 to 20 min after treatment, whereas neither viable nor heat killed C. glabrata cells induced a strong MAP-kinase phosphorylation even at a high MOI of 5 (Fig. 3A). Only p38 was slightly activated by both cell types. Activation of the NFκB signaling involves activation of an IκB kinase (IKK) complex catalyzing a phosphorylation-induced, proteasome-mediated degradation of inhibitory protein IκB. This leads to the release and activation of the transcription factor NFκB [33]. LPS treatment induced phosphorylation of the IKKα/β catalytic subunits of the IKK complex after 10 min (Fig. 3A). As a consequence, the NFκB binding protein IκB was phosphorylated and thus degraded within 45 min of treatment, indicated by a signal reduction of the phosphorylated and unphosphorylated form. This suggests that the NFκB transcription factor is released and activated. Besides release of sequestration of NFκB in the cytoplasm, stimulus-induced phosphorylation of the p65 subunit plays a key role in activation and nuclear translocation of NFκB [33]. We therefore analyzed phosphorylation of serine 536 of the NFκB subunit p65 upon infection of macrophages with LPS or C. glabrata, either viable or heat killed. Compared to the uninfected control, only LPS induced a detectable increase of the phosphorylated form of p65 (Fig. 3A).
Figure 3. C. glabrata does not induce MAP-kinase or NFκB signaling cascades upon phagocytosis but activates Syk.

RAW264.7 macrophages were stimulated with LPS (1 µg/ml) or infected with viable or heat killed (Hk) C. glabrata (MOI of 5) for indicated time points. (A) Cell lysates were subjected to Western Blot analyses by using antibodies detecting either the phosphorylated or unphosphorylated form (as a loading control) of p38, p44/42 (Erk1/2), SAPK/JNK, IKKαβ, IκBα and p65. Only LPS treatment induced changes in phosphorylation patterns of analyzed proteins. Data shown are representatives of three independent experiments. (B) Cell lysates were resolved on SDS-PAGE and membranes blotted for phosphorylated Syk (P-Syk) as described in [25] (C) Localization of the NFκB subunit p65 was analyzed by immunofluorescence microscopy. Representative pictures of macrophages treated with LPS or viable C. glabrata for 10 min are shown on the left site, a quantification of indicated time points on the right site. Percentage of NFκB nuclear localization was quantified for all macrophages (LPS) or for yeast-bound macrophages (viable, heat killed). While LPS induced the translocation of p65 to the nucleus, C. glabrata independent of its viability, did not. Statistical analysis was performed for C. glabrata-infected versus LPS-treated macrophages at the indicated time points (n≥3; *p<0.05, ***p<0.005 by unpaired Student’s t test).
In addition to the above-mentioned pathways, signaling downstream of the β-glucan receptor dectin-1, via activation of the spleen tyrosine kinase (Syk), has recently been described to influence phagosome maturation of C. albicans containing vacuoles [25]. Western Blot analysis detected Syk phosphorylation immediately after both, heat killed and viable C. glabrata infection (Fig. 3B). Of note, activation was retained longer for heat killed cells (up to 120 min) as compared to viable cells (40–60 min). LPS treatment did not induce Syk phosphorylation (data not shown).
In addition to Western Blot analyses, immunofluorescence staining of the p65 subunit of NFκB confirmed its translocation to the nucleus of macrophages upon treatment with LPS as early as 10 min after addition (Fig. 3C). Viable or heat killed C. glabrata, however, did not induce a shuttling of NFκB from the cytoplasm to the nucleus at any time point investigated. Taken together, these data show that viable and heat killed yeasts do not induce a strong or differential activation of three major MAP-kinase pathways and the NFκB pathway. In contrast, Syk activation is evident and prolonged after infection with heat killed as compared to viable cells.
Effect of Phagosome pH on C. glabrata Survival
Maturing phagosomes become increasingly acidic due to delivery of H+ into the phagosomal lumen via the vacuolar ATPase (V-ATPase). To elucidate whether reduced acidification of C. glabrata containing phagosomes may be a consequence of reduced V-ATPase accumulation on phagosome membranes, we used J774E macrophages expressing a GFP-tagged V-ATPase. Using anti-GFP antibody staining, we detected tagged V-ATPase on membranes of about 50% of viable C. glabrata containing phagosomes after 180 min of co-incubation, but also on acidified, heat killed yeast containing phagosomes (Fig. 4A). Thus, a reduced accumulation of V-ATPase is likely not the reason for reduced phagosome acidification.
Figure 4. Effect of phagosome pH on C. glabrata survival.

(A) Viable and heat killed C. glabrata containing phagosomes acquire similar levels of V-ATPase. Representative fluorescence microscopy images of viable or heat killed C. glabrata 180 min post-infection phagocytosed by murine J774E cells expressing a V-ATPase-GFP fusion protein (left panel). V-ATPase is shown in green while non-phagocytosed yeasts (stained with concanavalin A [ConA]) are indicated in yellow (marked with red arrows). Phagocytosed yeasts are labeled with white arrows. Co-localization with V-ATPase was quantified for phagosomes containing viable or heat killed C. glabrata at indicated time points (right panel). (B) Rising phagosome pH with chloroquine but not bafilomycin A1 reduces C. glabrata survival in MDMs. Survival of C. glabrata was determined by cfu-plating of macrophage lysates after 24 h. Co-incubation samples contained no drug (untreated), chloroquine (50 µM), chloroquine plus iron nitriloacetate (20 µM, FeNTA) or bafilomycin A1 (50 nM). (C) Chloroquine or bafilomycin A1 are not toxic to C. glabrata in vitro. Growth in presence of the drugs is comparable to untreated cultures. Statistical analysis was performed comparing heat killed with viable C. glabrata at indicated time points (A) or comparing untreated and drug-treated samples (B) (n≥3; *p<0.05, ***p<0.005 by unpaired Student’s t test).
We next sought to determine whether artificial elevation of phagosome pH or inhibition of V-ATPase activity would affect C. glabrata survival in macrophages. For this, we added the weak base chloroquine or the V-ATPase inhibitor bafilomycin A1 to macrophages infected with C. glabrata. The addition of both drugs raised the pH of heat killed yeast containing phagosomes, as observed by loss of a LysoTracker signal, but did not induce macrophage damage (data not shown) or inhibit in vitro growth of C. glabrata (Fig. 4C). Neutralizing the pH of macrophage phagosomes with chloroquine significantly reduced the survival of C. glabrata (Fig. 4B). However, this survival defect was rescued by the addition of FeNTA, an iron containing compound soluble at neutral pH [34] (Fig. 4B), arguing for an iron-dependent inhibitory effect of chloroquine on fungal survival.
In contrast, when adding bafilomycin A1, we observed no effect on survival of the whole population of C. glabrata after phagocytosis by macrophages (Fig. 4B), indicating that acidification by V-ATPase is not involved in C. glabrata killing. However, video microscopy of untreated RAW264.7 macrophages in presence of LysoTracker showed that a small subset of viable yeast cells was delivered to acidic phagosomes, which then resulted in degradation of the respective cells (Video S1).
Together, these findings support the view that the majority of viable C. glabrata cells are able to efficiently counteract V-ATPase proton pumping activity and that additional chemical inhibition of the proton pump has no impact on fungal survival.
Environmental Alkalinization by C. glabrata
We reasoned that the lack of acidification of C. glabrata containing phagosomes may be due to fungal metabolic processes that actively raise the phagosome pH. We found that similar to C. albicans, C. glabrata is able to alkalinize an originally acidic minimal medium when grown with 1% casamino acids as the sole carbon and nitrogen source [35]. The pH of the medium increased from pH 4 to a pH above 6.8, as indicated by a color change of the pH indicator phenol red after 24 hours (Fig. 5A). A subsequent direct pH measurement showed a medium pH value of around 7.5. A similar result was obtained when using solid media with bromocresol green as a pH indicator (Fig. 5B). These pH changes were not observed when heat killed C. glabrata cells were incubated in the same media.
Figure 5. Environmental alkalinization by C. glabrata.

(A) Viable, but not heat killed C. glabrata were able to alkalinize an acidic medium when grown with amino acids as the sole carbon and nitrogen source. 1×106 C. glabrata cells/ml were inoculated in a 24 well plate with liquid YNB medium with 1% casamino acids and 20 mg/l phenol red. Within 24 h the pH rose from pH 4 to a pH above 6.8, as indicated by a color change of the pH indicator. The mutant put3Δ was incapable of alkalinization, the mutant bcy1Δ showed an intermediate phenotype of reduced alkalinization. (B) Similar results were obtained when using a solid medium (YNB with 1% casamino acids, 2% agar and 0.01% bromocresol green).
To identify C. glabrata genes involved in the in vitro alkalinization process, we screened a C. glabrata mutant library containing 647 mutant strains (T. Schwarzmüller, B. Cormack, K. Haynes and K. Kuchler, unpublished data) for alkalinization on solid alkalinization-promoting medium. Mutants that did not grow on parallel YPD plates were excluded. With this first screening round, we identified 32 deletion mutants to be deficient in environmental alkalinization. For verification, defined cell numbers of the 32 identified mutants were grown in liquid alkalinization-promoting medium and two genetically independent clones were tested. This way, the alkalinization defect was confirmed for 19 out of 32 mutants. For some of these mutants, a growth defect in YPD or alkalinization-promoting medium without pH indicator was observed (Table 1).
Table 1. Alkalinization-defective C. glabrata mutants.
| Gene designation/ORF | Alkalini-zation defectA | Growth defectin YPDB | Growth defect in YNB+1% casaminoacids pH = 4C | LysoTrackersignalD | Function(based on homology toS. cerevisiae) |
| PUT3/CAGL0L09691g | ++ | − | + | − | transcriptional activator, regulates proline utilization genes |
| ADA2/CAGL0K06193g | ++ | + | − | + | transcription coactivator |
| SPT8/CAGL0F01837g | ++ | + | + | + | subunit of the SAGA transcriptional regulatory complex |
| HAP2/CAGL0H07843g | ++ | − | − | + | transcriptional activator, subunit of Hap2/3/4/5 CCAAT binding complex |
| HAP3/CAGL0J04400g | ++ | − | − | + | transcriptional activator, subunit of Hap2/3/4/5 CCAAT binding complex |
| HAP4/CAGL0K08624g | ++ | − | + | ++ | transcriptional activator, subunit of Hap2/3/4/5 CCAAT binding complex |
| HAP5/CAGL0K09900g | ++ | − | − | ++ | transcriptional activator, subunit of Hap2/3/4/5 CCAAT binding complex |
| EMI1/CAGL0K05797g | ++ | − | ++ | + | transcriptional regulator |
| ADR1/CAGL0E04884g | ++ | − | − | + | carbon source-responsive transcription factor |
| SIP4/CAGL0L03377g | ++ | − | + | ++ | transcriptional activator, binds carbon source-responsive elements |
| SNF1/CAGL0M08910g | ++ | + | ++ | − | serine/threonine protein kinase, transcription of glucose-repressed genes |
| BCY1/CAGL0I05236g | + | ++ | + | − | cAMP-dependent protein kinase |
| EMI5/CAGL0I08085g | ++ | − | ++ | + | succinate dehydrogenase activity |
| CIT1/CAGL0H03993g | ++ | − | ++ | − | citrate synthase (glyoxylate cycle) |
| MDH2/CAGL0E01705g | + | − | + | − | malate dehydrogenase (glyoxylate cycle, gluconeogenesis) |
| PCK1/CAGL0H06633g | ++ | − | ++ | − | phosphoenolpyruvate carboxykinase (gluconeogenesis) |
| MNN10/CAGL0K11231g | ++ | + | + | ++ | mannosyltransferase activity |
| KRE1/CAGL0M04169g | + | + | − | ++ | cell wall glycoprotein |
| CAGL0A01892g | + | + | + | ++ | uncharacterized |
Listed are mutants that showed reduced in vitro alkalinization of phenol red containing YNB medium with 1% casamino acids as sole carbon and nitrogen source in a screen of 647 mutants. Alkalinization defects were verified in independent assays and with two independent clones.
+reduced alkalinization (same phenotype as bcy1Δ in Fig. 5A), ++ strongly reduced alkalinization (same phenotype as put3Δ in Fig. 5A) as compared to the wild type.
Growth was monitored in parallel in YPD and in YNB medium with 1% casamino acids without phenol red by measuring absorption at 600 nm. ++ strong growth defect, + weak growth defect, - unaltered growth as compared to the wild type.
Mutants were co-incubated with MDMs for 90 min and phagosome acidification was monitored by LysoTracker staining. At least three independent microscopic fields were scored per mutant. ++ strong increase in LysoTracker signal, + medium increase in LysoTracker signal, - no change in LysoTracker signal as compared to the wild type.
Influence of Environmental Alkalinization on Phagosome Acidification and Maturation
If environmental alkalinization is causing an active elevation of the phagosome pH, we would expect to find alkalinization-defective mutants in more acidified phagosomes as compared to the wild type. We therefore performed LysoTracker staining on macrophages incubated with the identified alkalinization-defective mutants. Indeed, out of 19 mutants, 13 strains showed a higher number of LysoTracker-positive phagosomes as compared to the wild type (Table 1).
The strongest LysoTracker signal was observed for the C. glabrata mnn10Δ mutant (Fig. 6A). CAGL0K11231g (MNN10) codes for a putative α-1,6-mannosyltransferase, that is involved in glycosylation of cell wall components [36]. In contrast to wild type cells, a significantly higher percentage of mnn10Δ cells showed an accumulation of LysoTracker around phagocytosed yeast cells (Fig. 6B). These data suggest that mnn10Δ cell containing phagosomes are indeed acidified. The deletion of MNN10 also impaired fungal survival in MDMs. Survival of the mnn10Δ mutant was slightly, but significantly, reduced as compared to the wild type (mutant survival 85.3% [±21.4] of wt).
Figure 6. The influence of mannosyltransferases of C. glabrata on environmental alkalinization and acidification of phagosomes.

(A) Representative fluorescence microscopy images of wild type (wt hltΔ) C. glabrata and mnn10Δ mutant 90 min post infection, phagocytosed by human MDMs (left panels). LysoTracker staining is shown in red, while non-phagocytosed yeasts, stained with Concanavalin A (ConA), are shown in yellow. Phagocytosed yeasts are labeled with a white arrow while non-phagocytosed yeasts are marked with red arrows. (B) Co-localization with LysoTracker was quantified for phagosomes containing wild type (wt hltΔ or wt tΔ) or mutant (mnn10Δ, mnn11Δ, anp1Δ) C. glabrata at 90 min post infection. Statistical analysis was performed comparing mutant with wild type C. glabrata (n≥3; **p<0.01, ***p<0.005 by unpaired Student’s t test). (C) mnn10Δ and mnn11Δ mutants showed severe defects in environmental alkalinization in vitro, while alkalinization by the anp1Δ mutant was comparable to isogenic wild type levels. 1×106 C. glabrata cells/ml were inoculated in a 24 well plate with liquid YNB medium with 1% casamino acids and 20 mg/l phenol red and incubated for 24 h.
Mnn10 is predicted to act in a complex with the mannosyltransferases Anp1 and Mnn11 [36]. We therefore tested C. glabrata mutants lacking ANP1 and MNN11 for in vitro alkalinization, phagosome acidification and survival in MDMs [21]. The mnn11Δ mutant phenocopied the mnn10Δ mutant in showing a drastic alkalinization defect (Fig. 6C) and an increased number of acidified phagosomes (Fig. 6B). In contrast, the anp1Δ mutant showed wild type-like alkalinization but resembled mnn10Δ and mnn11Δ phenotypes regarding phagosome acidification. Survival of C. glabrata in macrophages was not affected by deletion of the MNN11 gene, while ANP1 deletion lead to reduced survival as compared to the wild type. Survival rates, in percentage of wt, were 81.9% [±11.9] for anp1Δ and 111.6% [±23.3] for mnn11Δ.
Discussion
Successful elimination of pathogens relies on the rapid actions of phagocytes of the innate immune system, such as macrophages, dendritic cells and neutrophils. Upon phagocytosis, the break-up of internalized microbes is carried out in phago (lyso) somes - specialized compartments in which oxidative and non-oxidative mechanisms kill and degrade microbes [37]–[39]. Therefore, immune evasion and survival strategies are critical for successful pathogens when infecting a host.
C. glabrata is a fungal pathogen which survives inside macrophages [15], [17], [40]. We recently showed that C. glabrata infection of macrophages leads to altered phagosome maturation, characterized by the arrest in a late endosomal, less acidified stage [17]. However, the mechanisms associated with the inhibited maturation and the lack of acidification were unknown. In our current study we gained further insights into the composition of C. glabrata containing phagosomes by analyzing markers of phagosome maturation.
Immunofluorescence microscopy demonstrated the presence of the late endosome marker Rab7, while DQ-BSA, a fluorogenic substrate for proteases, and the lysosomal tracer TROV were absent in the majority of phagosomes containing viable C. glabrata in MDMs and murine macrophages. These data confirmed and extended our previous results, allowing the conclusion that viable C. glabrata are found in phagosomes with late endosomal characteristics but with reduced acidification, reduced lysosomal fusion and low degradative activity.
Several studies have shown an impact of macrophage activation or differentiation on phagosome maturation and/or killing of intracellular pathogens. To mention a few examples, a study by Marodi et al. highlights the importance of INFγ to enhance clearance capacity of macrophages [41]. Further, recent studies on the fungal pathogen Cryptococcus neoformans or the bacterium Chlamydia muridarum stated an influence of macrophage differentiation: while M1 macrophages suppressed fungal and bacterial growth, M2 macrophages were less effective [42], [43]. In addition, the regulatory compound calcitriol (also called 1,25-dihydroxy vitamin D3), has been shown to directly promote phagocyte functions [27], [28], [44]. Pre-treatment of THP-1 macrophages with calcitriol abolished the inhibitory effect of mycobacterial cell wall glycolipid lipoarabinomannan on phagolysosome fusion [27]. Moreover, incubation of monocytes with cholecalciferol (the unhydroxylated form of vitamin D3) metabolites induced anti-tuberculosis activity [28].
In our previous experiments, however, we saw no influence of INFγ on replication of C. glabrata within MDMs, macrophage ROS production and cytokine release [17]. Differentiation of MDMs to M1 or M2 polarized macrophages did not measurably affect phagocytosis, phagosome maturation or killing of fungal cells (this study). Also, pre-treatment of MDMs with calcitriol did not enhance phagosome acidification of C. glabrata containing phagosomes. In addition, we found the altered fungus containing phagosome properties not only in human but also in mouse macrophages. Consequently, under the conditions investigated so far, modification of phagosome maturation seems to be a conserved feature of different types and differentiation states of C. glabrata-infected macrophages.
Finally, upon simultaneous infection of MDMs with viable C. glabrata and latex beads, phagosome acidification of vesicles containing latex beads progressed normally, while C. glabrata containing phagosomes within the same macrophage were not acidified. An influence of a pathogen containing vesicle on neighboring phagosomes would be expected if any secreted factor of a C. glabrata cell would affect a macrophage beyond its own compartment. For example, lipoarabinomannan of M. tuberculosis interferes with phagosome maturation via insertion into macrophage cell membranes [45]. Thus, our results do not support the presence of such a secreted fungal factor.
Phagocytosis is initiated by individual receptors or receptor complexes, which not only bind different ligands, but also trigger different signals. Many of these signals are controlled by kinases, including Syk and MAP-kinases that regulate phosphorylation cascades leading to effector responses including inflammatory mediators, cytokine production and antigen presentation [30], [46]–[49]. Moreover, effects of signaling mediators on maturation of phagosomes have recently been described [25]. Hence, analysis of kinase phosphorylation events in macrophages initiated by phagocytosis of C. glabrata could be instrumental in understanding recognition and activation of macrophages as well as alterations in phagosome maturation. Interestingly, neither viable nor heat killed yeasts induced a strong activation of three major MAP-kinases ERK1/2, SAPK/JNK or p38. In addition, even at high infectious doses, activation and translocation of NFκB, a critical transcription factor for maximal expression of many immunoregulatory molecules such as cytokines [32], [33], [50], was not observed. In line with this, previous analysis of cytokine production by MDMs revealed overall low levels of pro-inflammatory cytokines produced and no strong differences upon infection with viable or heat killed C. glabrata cells [17]. Thus, despite replication inside the phagosome, C. glabrata does not induce major signaling pathways and macrophage activation remains low. In contrast, S. cerevisiae, a close relative of C. glabrata, induces lectin- (such as dectin-1) and toll-like receptor (TLR) dependent recognition and subsequent pro-inflammatory cytokine production [51]. As determined by immunoblotting, signal transduction pathways activated in response to S. cerevisiae involve phosphorylation of ERK1/2, but not of p38 or SAPK/JNK [30]. Soluble β-glucan of S. cerevisiae leads to a significant decrease in cytosolic IκBα levels and an increase in nuclear p65 protein levels. These data and the difference to S. cerevisiae leads us to propose that reduced macrophage activation is a immune evasion mechanism of C. glabrata which contributes to persistence and low inflammatory immune responses in the systemic mouse model [11], [12].
Interestingly, we detected Syk kinase activation which was prolonged after infection with heat killed as compared to viable C. glabrata. When activation of Syk kinase downstream of the β-glucan receptor dectin-1 is blocked, compartments harboring C. albicans cells are blocked in their progression of phagosome maturation [25]. A faster release from Syk activation, by a so far unknown mechanism, may therefore be a further factor preventing full maturation of viable C. glabrata containing phagosomes. Syk activation further suggests dectin-1 or other Syk-coupled receptors such as dectin-2 as pattern recognition receptors mediating recognition of C. glabrata by macrophages. In agreement with this, a recent study has shown a role of dectin-2 for host defense against systemic C. glabrata infection of mice [52].
One main aim of our study was to analyze the correlation between phagosome pH, phagosome maturation and C. glabrata survival. Phagosomes, when undergoing maturation from early endosomal to phagolysosomal stages, accumulate the phagosomal proton pump V-ATPase, coinciding with a gradual drop in pH. This controls membrane trafficking in the endocytic pathway and may thus have an influence on phagosome maturation [53]. Consequently, the elevated pH of C. glabrata phagosomes may either be the cause for or the consequence of a phagosome maturation arrest.
Inhibition of phagosome acidification is a common microbial strategy to avoid destructive activities of macrophage phagosomes [54]. One possible way is the exclusion of V-ATPases from phagosome membranes to manipulate phagosome pH and maturation, as demonstrated for M. tuberculosis and Rhodococcus equi [55], [56]. This is likely not the case for C. glabrata, as we detected similar co-localization patterns for phagosomal V-ATPase for viable and heat killed yeast containing phagosomes. It is yet not clear whether the observed block of phagosome acidification by C. glabrata is a prerequisite for intracellular fungal replication or whether growth would also be possible in an acidified phagosome. In fact, in vitro growth of the fungus is possible at acidic pH down to pH 2 [57]. Moreover, none of the C. glabrata mutants identified in a large scale screening for reduced intracellular survival in MDMs lost the ability to inhibit acidification [19], which argues for pH-independent killing mechanisms. However, our observation that a small proportion of yeast cells was delivered to acidified phagosomes and degraded, suggests that an acidic phagosome at least indicates full antifungal properties.
In line with this, we showed that the proton pumping activity of V-ATPase is not required for killing of the majority of C. glabrata cells, as bafilomycin A1-induced inhibition of V-ATPase activity had no significant influence on overall fungal survival rates. Artificially rising phagosome pH with the weak base chloroquine, however, reduced fungal survival in macrophages. Since the reduced fungal survival rate in the presence of chloroquine was reversed by iron nitriloacetate, an iron compound soluble at neutral to basic pH, we conclude that chloroquine effects on C. glabrata survival are rather iron-utilization-related. A possible explanation may be that C. glabrata needs a slightly acidified compartment to utilize phagosomal iron sources that are crucial for intracellular survival. In presence of bafilomycin A1 that only targets V-ATPase proton pumping activity, the fungus may still be able to slightly acidify its environment to a pH value allowing iron utilization. In contrast, the weak base chloroquine may buffer such fungal activity and prevent slight acidification. A similar strategy has been suggested for intracellular survival of H. capsulatum [34], [58].
Besides exclusion of V-ATPase from phagosomes, there are more possible strategies to avoid phagosome acidification. First, C. glabrata may directly inhibit V-ATPase activity as shown for Legionella pneumophila [59] and other pathogens. Second, containment of viable C. glabrata may lead to permeabilization of phagosomal membranes, resulting in proton leakage, as observed for other fungi [60]. Third, other ion pumps that counteract V-ATPase activities, such as Na+-K+-ATPases, may be upregulated in viable yeast containing phagosomes. Finally, metabolic processes of the engulfed pathogen leading to an alkalinization of the environment, such as production of ammonia may contribute to the elevation of phagosome pH.
To test for the latter hypothesis, we set up an in vitro assay to determine the ability of C. glabrata to raise the pH of its environment. We found that environmental alkalinization by C. glabrata occurred within hours with similar kinetics and under similar conditions to those published by Vylkova et al. studying alkalinization by C. albicans [35]. Alkalinization took place in media lacking glucose and containing exogenous amino acids as the sole carbon source. Transcriptional profiling of C. glabrata phagocytosed by macrophages suggests that this yeast is exposed to similar nutritional conditions, namely glucose deprivation, inside macrophage phagosomes [15], [18]. Alkalinization by C. albicans relied on amino acid uptake and catabolism [35]. Mutants of C. glabrata lacking predicted homologous genes of the main identified C. albicans alkalinization factors with functions in amino acid metabolism alkalinized without any impairment (data not shown), suggesting that either other genes or other mechanisms are required for alkalinization by C. glabrata. In fact, C. glabrata shows differences in up-take and metabolism of certain amino acids as compared to C. albicans or S. cerevisiae and, for example, can grow with histidine as a sole nitrogen source by using an aromatic aminotransferase, instead of a histidinase (Brunke et al., unpublished data).
A screen of a deletion mutant library (T. Schwarzmüller, B. Cormack, K. Haynes and K. Kuchler, unpublished data) for defects in alkalinization of culture medium in vitro identified 19 mutants. Of these, 13 mutants co-localized more frequently with LysoTracker in MDMs as compared to the wild type, indicating a possible correlation between the potential for environmental alkalinization and the elevation of phagosome pH. For most of these mutants a more or less pronounced growth defect in complete and/or minimal medium was observed, suggesting a physiological activity to be necessary to grow and alkalinize under the conditions used.
Interestingly, single mutants lacking all four components of the HAP complex, a heteromeric transcriptional regulator with a complex position in the global transcriptional regulation of the cell, showed up in the screening (Table 1). The HAP complex was originally identified as regulator of the ‘diauxic shift’ of S. cerevisiae, a reprogramming of respiratory metabolism when yeasts adapt to glucose-limiting conditions [61]. Additionally, mutants lacking genes encoding the protein kinase Snf1 and its target, the transcriptional activator Sip4, were identified. Both proteins play a role in expression of glucose-repressed genes in response to glucose deprivation [62]. Moreover, the lack of glucose is reflected by the appearance of mutants, which lack genes involved in the glyoxylate cycle and gluconeogenesis (cit1Δ, mdh2Δ; pck1Δ). Thus, metabolic processes that enable C. glabrata to adapt to nutrient limitation are vital to grow in the alkalinization medium, which consequentially raises the extracellular pH. Also, the functional divergence of alkalinization-defective mutants identified suggests that more than one distinct pathway may be involved raising extracellular pH in C. glabrata.
Thirteen out of 19 alkalinization-defective mutants were more frequently found in LysoTracker-positive phagosomes, suggesting that environmental alkalinization enables C. glabrata to actively modify phagosome pH after macrophage phagocytosis. Similarly, C. albicans has recently been shown to neutralize the macrophage phagosome [63]. The C. glabrata mutant with the strongest LysoTracker phenotype identified in our study was mnn10Δ, lacking a putative Golgi-localized α-1,6-mannosyltransferase. As in S. cerevisiae, Mnn10 is believed to act in an α-1,6-mannosyltransferase complex with Anp1 and Mnn11 on the extension of N-linked mannose backbones in C. glabrata [21], [36]. In our study, alkalinization and phagosome acidification phenotypes of the mnn10Δ and mnn11Δ mutants were similar, hinting towards a functional connection and possibly a redundancy of Mnn10 and Mnn11 in C. glabrata. Thus, Mnn10 and Mnn11-related α-1,6-mannosyltransferase functions in environmental alkalinization may enable C. glabrata to elevate the phagosome pH in macrophages. In this context, Mnn10 and Mnn11 glycosylation activities may be important for secretion and/or functionality of either general fungal proteins that ensure fitness and physiological activity of C. glabrata, of alkalinization-specific proteins or of other proteins that counteract a drop in phagosome pH. In S. cerevisiae, MNN10 and MNN11 deletion has been shown to cause a hyper-secretory phenotype [64]. Another possibility, however, would be an alkalinization-independent effect by Mnn10- and Mnn11-mediated surface modifications that influence initial recognition of C. glabrata by macrophages. Such an effect on phagosome pH may be also an explanation for the observed anp1Δ phenotype. ANP1 seems to be dispensable for environmental alkalinization in vitro, while still having an influence on phagosome acidification. In addition, our data suggest an alkalinization-independent function of Anp1 in macrophage survival.
Finally, the fact that MNN10 deletion reduced the ability of C. glabrata to survive in macrophages suggests that Mnn10 functions in alkalinization and phagosome modification affect the intracellular fate of C. glabrata in macrophages. The wild type-like survival of a mnn11Δ mutant may argue for a redundancy of functions among the different α-1,6-mannosyltransferases in C. glabrata. In addition, further fungal attributes have been shown to affect intracellular survival of C. glabrata in macrophages [15], [16], [19].
In conclusion, we have identified protein mannosylation as an important fungal attribute for C. glabrata to modify phagosome acidification. Moreover, to our knowledge this is the first study to show rapid environmental alkalinization by C. glabrata. This activity may at least be one strategy of C. glabrata to raise phagosome pH in order to create a less hostile compartment for intracellular replication.
Supporting Information
C. glabrata resides in non-matured macrophage phagosomes of murine macrophages. Murine RAW264.7 macrophages were infected with viable or heat killed (Hk) C. glabrata, followed by fluorescence staining for the lysosomal tracer texas red ovalbumin (TROV), the fluorogenic protease substrate DQ-BSA or the acidotropic dye LysoTracker. Co-localization with fluorescence markers was quantified for yeast containing phagosomes at indicated time points. Heat killed but not viable C. glabrata containing phagosomes acquire the lysosomal tracer TROV, show high phagosomal proteolytic activity as measured by co-localization with DQ-BSA, and co-localize with LysoTracker. Statistical analysis was performed comparing heat killed with viable C. glabrata at indicated time points (n = 3; *p<0.05, **p<0.01, ***p<0.005 by unpaired Student’s t test).
(TIF)
C. glabrata replicates in LysoTracker negative compartments. RAW264.7 macrophages were stained with LysoTracker Red (diluted 1∶10.000 in cell culture medium) 1 h prior to infection and during co-incubation with viable C. glabrata cells. After infection (MOI of 4) time-lapse microscopy was started and pictures were taken every 5 min at a magnification of 630×. While the majority of yeasts resides in LysoTracker negative compartments and replicates, killing of yeast cells and subsequent LysoTracker acquisition can be observed within a few macrophages (red arrows 1–3). In macrophage 2, yeast replication in close proximity to a killed C. glabrata cell is shown.
(AVI)
Acknowledgments
We wish to thank members of DFG SPP1580 network for fruitful discussions and helpful suggestions. We wish to thank Nadja Jablonowski, Stephanie Wisgott, Daniela Schulz and Birgit Weber from the Departments of Microbial Pathogenicity Mechanisms and Microbial Immunology at the HKI for excellent technical support.
Funding Statement
This work was mainly supported by grants from the Deutsche Forschungsgemeinschaft to B.H. (Hu 528/15-1 and 16-1), A.H. (Ha 1929/11-1) and N.R. (Re 1228/5-1) within the priority program SPP1580 (www.spp1580.uni-bonn.de). Further, this work was in part supported by National Institutes of Health grants NIAID 1K08AI110655 (M.K.M.), 1R01AI079198 (J.M.V.), and 1R01AI092084 (J.M.V.). K.K. was funded by FWF-I 746-B11-DACH. Work in the Haynes lab was supported by grants from The Wellcome Trust (097377/Z/11/Z) and BBSRC (BB/F005210/1). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Arendrup MC (2010) Epidemiology of invasive candidiasis. Curr Opin Crit Care 16: 445–452. [DOI] [PubMed] [Google Scholar]
- 2. Zaoutis TE, Argon J, Chu J, Berlin JA, Walsh TJ, et al. (2005) The epidemiology and attributable outcomes of candidemia in adults and children hospitalized in the United States: a propensity analysis. Clin Infect Dis 41: 1232–1239. [DOI] [PubMed] [Google Scholar]
- 3. Horn DL, Neofytos D, Anaissie EJ, Fishman JA, Steinbach WJ, et al. (2009) Epidemiology and outcomes of candidemia in 2019 patients: data from the prospective antifungal therapy alliance registry. Clin Infect Dis 48: 1695–1703. [DOI] [PubMed] [Google Scholar]
- 4. Pfaller MA, Diekema DJ (2007) Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev 20: 133–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grim SA, Berger K, Teng C, Gupta S, Layden JE, et al. (2012) Timing of susceptibility-based antifungal drug administration in patients with Candida bloodstream infection: correlation with outcomes. J Antimicrob Chemother 67: 707–714. [DOI] [PubMed] [Google Scholar]
- 6. Gumbo T, Chemaly RF, Isada CM, Hall GS, Gordon SM (2002) Late complications of Candida (Torulopsis) glabrata fungemia: description of a phenomenon. Scand J Infect Dis 34: 817–818. [DOI] [PubMed] [Google Scholar]
- 7. Lewis RE, Viale P, Kontoyiannis DP (2012) The potential impact of antifungal drug resistance mechanisms on the host immune response to Candida . Virulence 3: 368–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Steinbach WJ, Roilides E, Berman D, Hoffman JA, Groll AH, et al. (2012) Results from a prospective, international, epidemiologic study of invasive candidiasis in children and neonates. Pediatr Infect Dis J 31: 1252–1257. [DOI] [PubMed] [Google Scholar]
- 9. Arendrup M, Horn T, Frimodt-Moller N (2002) In vivo pathogenicity of eight medically relevant Candida species in an animal model. Infection 30: 286–291. [DOI] [PubMed] [Google Scholar]
- 10. Haynes K (2001) Virulence in Candida species. Trends Microbiol 9: 591–596. [DOI] [PubMed] [Google Scholar]
- 11. Jacobsen ID, Brunke S, Seider K, Schwarzmuller T, Firon A, et al. (2010) Candida glabrata persistence in mice does not depend on host immunosuppression and is unaffected by fungal amino acid auxotrophy. Infect Immun 78: 1066–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brieland J, Essig D, Jackson C, Frank D, Loebenberg D, et al. (2001) Comparison of pathogenesis and host immune responses to Candida glabrata and Candida albicans in systemically infected immunocompetent mice. Infect Immun 69: 5046–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eissenberg LG, Goldman WE, Schlesinger PH (1993) Histoplasma capsulatum modulates the acidification of phagolysosomes. J Exp Med 177: 1605–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feldmesser M, Tucker S, Casadevall A (2001) Intracellular parasitism of macrophages by Cryptococcus neoformans . Trends Microbiol 9: 273–278. [DOI] [PubMed] [Google Scholar]
- 15. Kaur R, Ma B, Cormack BP (2007) A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata . Proc Natl Acad Sci U S A 104: 7628–7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roetzer A, Gratz N, Kovarik P, Schuller C (2010) Autophagy supports Candida glabrata survival during phagocytosis. Cell Microbiol 12: 199–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Seider K, Brunke S, Schild L, Jablonowski N, Wilson D, et al. (2011) The facultative intracellular pathogen Candida glabrata subverts macrophage cytokine production and phagolysosome maturation. J Immunol 187: 3072–3086. [DOI] [PubMed] [Google Scholar]
- 18. Rai MN, Balusu S, Gorityala N, Dandu L, Kaur R (2012) Functional genomic analysis of Candida glabrata-macrophage interaction: role of chromatin remodeling in virulence. PLoS Pathog 8: e1002863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Seider K, Gerwien F, Kasper L, Brunke S, Jablonowski N, et al. (2014) Immune evasion, stress resistance and efficient nutrient acquisition are crucial for intracellular survival of Candida glabrata within macrophages. Eukaryot Cell 13: 170–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Flannagan RS, Cosio G, Grinstein S (2009) Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol 7: 355–366. [DOI] [PubMed] [Google Scholar]
- 21. West L, Lowman DW, Mora-Montes HM, Grubb S, Murdoch C, et al. (2013) Differential virulence of Candida glabrata glycosylation mutants. J Biol Chem 288: 22006–22018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Raschke WC, Baird S, Ralph P, Nakoinz I (1978) Functional macrophage cell lines transformed by Abelson leukemia virus. Cell 15: 261–267. [DOI] [PubMed] [Google Scholar]
- 23. Diment S, Leech MS, Stahl PD (1987) Generation of macrophage variants with 5-azacytidine: selection for mannose receptor expression. J Leukoc Biol 42: 485–490. [DOI] [PubMed] [Google Scholar]
- 24. Schneider B, Schueller C, Utermoehlen O, Haas A (2007) Lipid microdomain-dependent macropinocytosis determines compartmentation of Afipia felis . Traffic 8: 226–240. [DOI] [PubMed] [Google Scholar]
- 25. Mansour MK, Tam JM, Khan NS, Seward M, Davids PJ, et al. (2013) Dectin-1 activation controls maturation of beta-1,3-glucan-containing phagosomes. J Biol Chem 288: 16043–16054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mosser DM, Edwards JP (2008) Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8: 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hmama Z, Sendide K, Talal A, Garcia R, Dobos K, et al. (2004) Quantitative analysis of phagolysosome fusion in intact cells: inhibition by mycobacterial lipoarabinomannan and rescue by an 1alpha,25-dihydroxyvitamin D3-phosphoinositide 3-kinase pathway. J Cell Sci 117: 2131–2140. [DOI] [PubMed] [Google Scholar]
- 28. Rook GA, Steele J, Fraher L, Barker S, Karmali R, et al. (1986) Vitamin D3, gamma interferon, and control of proliferation of Mycobacterium tuberculosis by human monocytes. Immunology 57: 159–163. [PMC free article] [PubMed] [Google Scholar]
- 29. Underhill DM, Ozinsky A (2002) Phagocytosis of microbes: complexity in action. Annu Rev Immunol 20: 825–852. [DOI] [PubMed] [Google Scholar]
- 30. Ibata-Ombetta S, Jouault T, Trinel PA, Poulain D (2001) Role of extracellular signal-regulated protein kinase cascade in macrophage killing of Candida albicans . J Leukoc Biol 70: 149–154. [PubMed] [Google Scholar]
- 31. Kong L, Ge BX (2008) MyD88-independent activation of a novel actin-Cdc42/Rac pathway is required for Toll-like receptor-stimulated phagocytosis. Cell Res 18: 745–755. [DOI] [PubMed] [Google Scholar]
- 32. Roeder A, Kirschning CJ, Schaller M, Weindl G, Wagner H, et al. (2004) Induction of nuclear factor- kappa B and c-Jun/activator protein-1 via toll-like receptor 2 in macrophages by antimycotic-treated Candida albicans . J Infect Dis 190: 1318–1326. [DOI] [PubMed] [Google Scholar]
- 33. Kingeter LM, Lin X (2012) C-type lectin receptor-induced NF-kappaB activation in innate immune and inflammatory responses. Cell Mol Immunol 9: 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Newman SL, Gootee L, Brunner G, Deepe GS Jr (1994) Chloroquine induces human macrophage killing of Histoplasma capsulatum by limiting the availability of intracellular iron and is therapeutic in a murine model of histoplasmosis. J Clin Invest 93: 1422–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vylkova S, Carman AJ, Danhof HA, Collette JR, Zhou H, et al. (2011) The fungal pathogen Candida albicans autoinduces hyphal morphogenesis by raising extracellular pH. MBio 2: e00055–00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jungmann J, Rayner JC, Munro S (1999) The Saccharomyces cerevisiae protein Mnn10p/Bed1p is a subunit of a Golgi mannosyltransferase complex. J Biol Chem 274: 6579–6585. [DOI] [PubMed] [Google Scholar]
- 37. Haas A (2007) The phagosome: compartment with a license to kill. Traffic 8: 311–330. [DOI] [PubMed] [Google Scholar]
- 38. Underhill DM (2005) Phagosome maturation: steady as she goes. Immunity 23: 343–344. [DOI] [PubMed] [Google Scholar]
- 39. Vieira OV, Botelho RJ, Grinstein S (2002) Phagosome maturation: aging gracefully. Biochem J 366: 689–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Otto V, Howard DH (1976) Further studies on the intracellular behavior of Torulopsis glabrata . Infect Immun 14: 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marodi L, Schreiber S, Anderson DC, MacDermott RP, Korchak HM, et al. (1993) Enhancement of macrophage candidacidal activity by interferon-gamma. Increased phagocytosis, killing, and calcium signal mediated by a decreased number of mannose receptors. J Clin Invest 91: 2596–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Davis MJ, Tsang TM, Qiu Y, Dayrit JK, Freij JB, et al. (2013) Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. MBio 4: e00264–00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gracey E, Lin A, Akram A, Chiu B, Inman RD (2013) Intracellular survival and persistence of Chlamydia muridarum is determined by macrophage polarization. PLoS One 8: e69421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mangelsdorf DJ, Koeffler HP, Donaldson CA, Pike JW, Haussler MR (1984) 1,25-Dihydroxyvitamin D3-induced differentiation in a human promyelocytic leukemia cell line (HL-60): receptor-mediated maturation to macrophage-like cells. J Cell Biol 98: 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Welin A, Winberg ME, Abdalla H, Sarndahl E, Rasmusson B, et al. (2008) Incorporation of Mycobacterium tuberculosis lipoarabinomannan into macrophage membrane rafts is a prerequisite for the phagosomal maturation block. Infect Immun 76: 2882–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Filler SG (2006) Candida-host cell receptor-ligand interactions. Curr Opin Microbiol 9: 333–339. [DOI] [PubMed] [Google Scholar]
- 47. McGreal EP, Miller JL, Gordon S (2005) Ligand recognition by antigen-presenting cell C-type lectin receptors. Curr Opin Immunol 17: 18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Netea MG, Van Der Graaf CA, Vonk AG, Verschueren I, Van Der Meer JW, et al. (2002) The role of toll-like receptor (TLR) 2 and TLR4 in the host defense against disseminated candidiasis. J Infect Dis 185: 1483–1489. [DOI] [PubMed] [Google Scholar]
- 49. Rogers NC, Slack EC, Edwards AD, Nolte MA, Schulz O, et al. (2005) Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity 22: 507–517. [DOI] [PubMed] [Google Scholar]
- 50. Naumann M (2000) Nuclear factor-kappa B activation and innate immune response in microbial pathogen infection. Biochem Pharmacol 60: 1109–1114. [DOI] [PubMed] [Google Scholar]
- 51. Jouault T, El Abed-El Behi M, Martinez-Esparza M, Breuilh L, Trinel PA, et al. (2006) Specific recognition of Candida albicans by macrophages requires galectin-3 to discriminate Saccharomyces cerevisiae and needs association with TLR2 for signaling. J Immunol 177: 4679–4687. [DOI] [PubMed] [Google Scholar]
- 52. Ifrim DC, Bain JM, Reid DM, Oosting M, Verschueren I, et al. (2014) Role of Dectin-2 for Host Defense against Systemic Infection with Candida glabrata . Infect Immun 82: 1064–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Huynh KK, Grinstein S (2007) Regulation of vacuolar pH and its modulation by some microbial species. Microbiol Mol Biol Rev 71: 452–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seider K, Heyken A, Luttich A, Miramon P, Hube B (2010) Interaction of pathogenic yeasts with phagocytes: survival, persistence and escape. Curr Opin Microbiol. [DOI] [PubMed]
- 55. Fernandez-Mora E, Polidori M, Luhrmann A, Schaible UE, Haas A (2005) Maturation of Rhodococcus equi-containing vacuoles is arrested after completion of the early endosome stage. Traffic 6: 635–653. [DOI] [PubMed] [Google Scholar]
- 56. Wong D, Bach H, Sun J, Hmama Z, Av-Gay Y (2011) Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc Natl Acad Sci U S A 108: 19371–19376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bairwa G, Kaur R (2011) A novel role for a glycosylphosphatidylinositol-anchored aspartyl protease, CgYps1, in the regulation of pH homeostasis in Candida glabrata . Mol Microbiol 79: 900–913. [DOI] [PubMed] [Google Scholar]
- 58. Strasser JE, Newman SL, Ciraolo GM, Morris RE, Howell ML, et al. (1999) Regulation of the macrophage vacuolar ATPase and phagosome-lysosome fusion by Histoplasma capsulatum . J Immunol 162: 6148–6154. [PubMed] [Google Scholar]
- 59. Xu L, Shen X, Bryan A, Banga S, Swanson MS, et al. (2010) Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog 6: e1000822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tucker SC, Casadevall A (2002) Replication of Cryptococcus neoformans in macrophages is accompanied by phagosomal permeabilization and accumulation of vesicles containing polysaccharide in the cytoplasm. Proc Natl Acad Sci U S A 99: 3165–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Buschlen S, Amillet JM, Guiard B, Fournier A, Marcireau C, et al. (2003) The S. cerevisiae HAP complex, a key regulator of mitochondrial function, coordinates nuclear and mitochondrial gene expression. Comp Funct Genomics 4: 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lesage P, Yang X, Carlson M (1996) Yeast SNF1 protein kinase interacts with SIP4, a C6 zinc cluster transcriptional activator: a new role for SNF1 in the glucose response. Mol Cell Biol 16: 1921–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vylkova S, Lorenz MC (2014) Modulation of Phagosomal pH by Candida albicans Promotes Hyphal Morphogenesis and Requires Stp2p, a Regulator of Amino Acid Transport. PLoS Pathog 10: e1003995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bartkeviciute D, Sasnauskas K (2004) Disruption of the MNN10 gene enhances protein secretion in Kluyveromyces lactis and Saccharomyces cerevisiae . FEMS Yeast Res 4: 833–840. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
C. glabrata resides in non-matured macrophage phagosomes of murine macrophages. Murine RAW264.7 macrophages were infected with viable or heat killed (Hk) C. glabrata, followed by fluorescence staining for the lysosomal tracer texas red ovalbumin (TROV), the fluorogenic protease substrate DQ-BSA or the acidotropic dye LysoTracker. Co-localization with fluorescence markers was quantified for yeast containing phagosomes at indicated time points. Heat killed but not viable C. glabrata containing phagosomes acquire the lysosomal tracer TROV, show high phagosomal proteolytic activity as measured by co-localization with DQ-BSA, and co-localize with LysoTracker. Statistical analysis was performed comparing heat killed with viable C. glabrata at indicated time points (n = 3; *p<0.05, **p<0.01, ***p<0.005 by unpaired Student’s t test).
(TIF)
C. glabrata replicates in LysoTracker negative compartments. RAW264.7 macrophages were stained with LysoTracker Red (diluted 1∶10.000 in cell culture medium) 1 h prior to infection and during co-incubation with viable C. glabrata cells. After infection (MOI of 4) time-lapse microscopy was started and pictures were taken every 5 min at a magnification of 630×. While the majority of yeasts resides in LysoTracker negative compartments and replicates, killing of yeast cells and subsequent LysoTracker acquisition can be observed within a few macrophages (red arrows 1–3). In macrophage 2, yeast replication in close proximity to a killed C. glabrata cell is shown.
(AVI)