

Abstract
Major alterations of the hypothalamic–pituitary–adrenocortical (HPA) system that can be reversed by successful antidepressant therapy are often seen in depressed patients. Persuasive evidence points to the involvement of a dysfunctional glucocorticoid receptor (GR) system in these changes. Support for this also comes from studies of transgenic mice that express an antisense RNA, complementary to the GR mRNA, and have numerous neuroendocrine characteristics of human depression as well as altered behaviour. Many of these neuroendocrine and behavioural characteristics of the transgenic mice can be reversed by antidepressants. A possible explanation for this is that the antidepressant-induced increase in GRs renders the HPA axis more sensitive to glucocorticoid feedback. This new insight into antidepressant drug action suggests a novel approach to the development of antidepressant drugs.
Medical subject headings: antidepressive agents; corticotropin; depression; hypothalamus; models, animal; pituitary–adrenal system; receptors, glucocorticoid
Abstract
On constate souvent chez les patients déprimés des altérations majeures de l'axe hypothalamo- hypophyso-surrénalien (HPA) qu'il est possible d'inverser par une thérapie fructueuse aux antidépresseurs. Des données probantes convaincantes incriminent dans ces changements un système dysfonctionnel de récepteurs des glucocorticoïdes (RG). Des études portant sur des souris transgéniques qui expriment un ARN antisens, complémentaire de l'ARNm des RG, qui présentent de nombreuses caractéristiques neuroendocriniennes de la dépression humaine, ainsi que des altérations du comportement, appuient cette affirmation. Les antidépresseurs permettent d'inverser un grand nombre de ces caractéristiques neuroendocriniennes et comportementales des souris transgéniques. Le phénomène pourrait s'expliquer par l'augmentation, provoquée par les antidépresseurs, des RG qui sensibilise l'axe HPA à la rétroaction des glucocorticoïdes. Cette nouvelle connaissance de l'action des antidépresseurs indique une nouvelle façon d'aborder la mise au point de ces médicaments.
Introduction
The main driving force for hypothalamic–pituitary–adrenocortical (HPA) activation is hypothalamic corticotropin-releasing hormone (CRH) acting in synergy with vasopressin, which is produced in either the same or distinct neurons of the paraventricular nucleus, to enhance release of pituitary pro-opiomelanocortin (POMC)-derived peptides (corticotropin [ACTH] and endorphins). Adrenal glucocorticoid hormone secretion (corticosterone in rodents, cortisol in humans) is stimulated mainly by ACTH, although adrenocortical sensitivity to ACTH may be modified by sympathetic innervation of the adrenal gland.1 Different regulatory forces are superimposed on this system to coordinate adrenal secretions during periods of inactivity and stress. The first of these is a circadian rhythm of basal activity derived from the suprachiasmatic nucleus.2 Stress-induced responses of the HPA system involve afferent inputs from numerous other brain regions including noradrenergic innervation from the brainstem A1 and A2 cell groups and the pontine locus coeruleus,3 the amygdala,4,5 cerebral cortex and hippocampus.6 In general, the septum and hippocampus have inhibitory actions on HPA activity, whereas the effect of the amygdala is largely permissive. Another regulatory force on HPA system activity is provided by inhibitory feedback actions of adrenal steroids exerted through corticosteroid receptors located in different brain areas.
Glucocorticoid hormones terminate the stress response by negative feedback action at the level of the pituitary, hypothalamus and limbic brain areas, including the hippocampus, amygdala and septum. This action is mediated by 2 types of corticosteroid receptor that have been identified:7 the type I or mineralocorticoid receptor (MR) and the type II or glucocorticoid receptor (GR). The MR is mainly expressed either alone or together with the GR in hippocampal neurons, whereas the GR is more ubiquitously distributed in the brain,8 particularly in neurons. This dual system may be necessary to cope with corticosteroid concentrations ranging from 0.5 nmol/L to 50 nmol/L during the diurnal cycle and up to 100 nmol/L or more in response to stress.9 An adequate physiologic response to a wide hormone concentration range can be achieved by these 2 receptors, because the MR mediates the effects of, and possibly controls, low basal circadian levels of circulating glucocorticoids, and the GR appears to mediate the effects of high-stress levels of glucocorticoids and to be responsible for the negative feedback effects of glucocorticoids on the HPA system.7,9,10,11,12,13
HPA disturbances in affective disorders
Hypersecretion of ACTH and glucocorticoids at baseline and several neuroendocrine function tests convincingly indicate profound alterations of the HPA system in patients with severe depression. Features of affective disorders, secondary to Cushing's syndrome, are also frequently indistinguishable from those of patients with primary psychiatric illness.14 This role of the HPA system in both stress, a major problem of modern-day (and probably past) lifestyles, and its deregulation in mood disorders, which may, at least once during their lifetime, disrupt the lives of up to 15% of the population, underlies the importance of an understanding of the integrated central nervous system mechanisms responsible for HPA system regulation. In addition, it is now becoming apparent that many of the beneficial effects of antidepressants could be exerted through their actions on the HPA system. It is thus imperative to understand how normal HPA system regulation is disturbed in affective disorders and how antidepressants modulate the HPA system.
In certain depressed patients, cortisol secretion may be increased and may show an abnormal 24-hour secretory pattern and, unless exogenous CRH is administered, this pattern may be resistant to suppression by exogenous steroids.15,16,17 Somewhat surprisingly, very similar changes are also seen in the manic phase of bipolar illness.18 Lack of cortisol suppression by dexamethasone in depressed patients could indicate a primary hyperactivity of the adrenal glands rather than a central defect, but recent studies using CRH have eliminated this possibility.19,20,21,22 A key role of elevated CRH secretion in the development of depression is indicated by both clinical23 and preclinical24,25 studies. In patients with depression, elevated concentrations of CRH in the cerebrospinal fluid,26 increased numbers of CRH-containing cells in the paraventricular nucleus,27 decreased CRH binding in the frontal cortex28 and a blunted ACTH response to an intravenously administered test dose of CRH21,29 have been seen. These results are thought to reflect the desensitization of CRH receptors at corticotrophic cells and/or a restricted secretory response of ACTH to CRH caused by elevated basal cortisol levels. The latter mechanism is probably the most important, underscored by a normalized net ACTH output in patients with depression pretreated with metyrapone.30,31 Despite a blunted ACTH response to CRH, the associated cortisol response is unchanged, because ongoing HPA overactivity gradually produces adrenocortical hyperplasia rendering the gland hypersensitive to ACTH. Other non-ACTH mechanisms such as neural sympathetic factors or humoral factors from the immune system may also contribute to the dissociation between ACTH and cortisol in depression.
Genetics of affective disorders
Twin, adoption and family studies point to the inescapable conclusion of a genetic factor in affective disorders. Between 65% and 70% of monozygotic twins are concordant for the illness, whereas only 1%–15% of dizygotic twins are concordant.32,33
Is disturbed glucocorticoid feedback a trait of affective disorders?
Although a genetic predisposition may be necessary for the development of affective disorders, stressful life events often trigger their onset. To avoid detrimental consequences, the cascade of effects that constitute the adaptive response to this stress must be terminated effectively. The mechanisms for this appear to be defective in patients with depression.34,35,36,37
Elimination of primary adrenal gland hyperactivity19,20,21,22 now points to a key role for elevated CRH secretion23,24,25 in the mechanism leading to the failure of dexamethasone to suppress cortisol in depressed patients.
Glucocorticoids terminate the stress response by negative feedback actions mediated by corticosteroid receptor(s), and it is thus apparent that an understanding of their role in both stress and affective disorders is essential. This is underlined by the findings of Sapolsky et al,38 who demonstrated that elevated glucocorticoid levels lead to loss of the GR-containing cells in the hippocampus that mediate the glucocorticoid-induced suppression of CRH neurons in the paraventricular nucleus. Dexamethasone resistance in monkeys was also shown to be associated with a selective decrease of hippocampal GR.39 Loss of hippocampal GR in aged rats40,41 may also be pertinent to the increased corticosterone levels of these animals and can be compared to the higher cortisol levels of older depressed patients compared with younger ones.42
Diminished corticosteroid receptor concentrations caused by a malfunctioning of systems involved in the regulation of corticosteroid receptor gene expression could be a causative factor in the defective feedback action of cortisol seen in patients with severe depression and could thus explain their altered HPA system. Circumstantial findings of decreased GR concentrations in the lymphocytes of patients with depression underline this possibility.43 What is not known is whether a primary disturbance occurs at the level of corticosteroid receptors in limbic brain (MR and GR in the hippocampus, GR in the hypothalamus), thus modifying the fine tuning of ACTH secretion via CRH, vasopressin and POMC regulation, or whether other factors that drive expression of CRH and vasopressin and their release into hypophyseal portal blood result in an excess of ACTH and cortisol that secondarily decreases corticosteroid receptor capacity and function.
Transgenic mouse model of the neuroendocrine changes associated with affective disorders
Introduction of genes into the germ line of mice offers the possibility of generating animal models for human genetic disease. Because the GR is essential for feedback inhibition by glucocorticoids and because in depression adrenal steroids are unable to shut down HPA activity, a malfunction of the GR system may be present in depression, a view supported by the absence of any of the classic physical stigmata associated with Cushing's disease. If, in fact, a defective GR system (this does not indicate a defective receptor, only its regulation) is one characteristic of depressive disorders, an animal model of this disease could be produced, and Pepin et al44 made a transgenic mouse with decreased GR in the brain. We hoped that this transgenic model would shed light on whether the apparent lack of sensitivity to corticosteroids seen in the majority of patients with depression could result in neuroendocrine changes that are causally linked to pathogenesis as well as to the therapeutic effectiveness of antidepressant drugs. To make this transgenic mouse, we inverted an 1815-bp fragment of the 3' noncoding region of the type II GR cDNA downstream from a 2.3-kb EcoR1/Hind III human neurofilament gene promoter element so that it would generate an antisense RNA. When we used this same strategy and inserted the antisense RNA-generating transgene into the mouse genome, we succeeded in producing an animal with defective glucocorticoid feedback.44,45 The transgenic mice have HPA system changes characteristic of those seen in depression, including increased HPA activity (shown by increased plasma ACTH and corticosterone levels), a resistance to suppression of adrenocortical secretions by dexamethasone,45,46,47 feeding disturbances48 and cognitive deficits.49 We have shown that many of the behavioural changes in our transgenic mice are reversed after antidepressant treatment.50,51,52 Similar behavioural changes have also been noted in a more recently developed mouse with brain-specific loss of GR function.53 The HPA system of these mice is activated in the early morning, at a time when, in rodents, it is normally quiescent, and this sort of phase shift is also a characteristic of depressed patients.54 Findings in these transgenic mice thus support the hypothesis that a dysfunctional GR system could be involved in the production of the neuroendocrine components of affective disorders. Genetic studies do not implicate the GR itself to be a cause of affective disorders. It remains possible, however, that a brain-specific regulator of corticosteroid gene expression could be a plausible factor. These mice are, thus, appropriate for use as a model for studying the neuroendocrine characteristics of affective disorders and could help in evaluating the mechanism of action of antidepressant drugs.
Although a primary decrease in functional corticosteroid receptors can produce HPA system activation,45,47,55 the increased glucocorticoid levels could subsequently lead to secondary activation (or inhibition) of glucocorticoid-sensitive gene expression, and this could also lead to neurotransmitter imbalance.
Normalization of HPA activity by antidepressants
Depressed patients with symptoms of HPA malfunction respond to antidepressants with temporally associated changes in both mood and hormones. This challenged us to question whether antidepressants could elevate mood in patients with depression through their long-term effects on HPA system regulation.56,57
Two major classes of antidepressant drugs exist: monoamine oxidase inhibitors and monoamine reuptake inhibitors. This latter class is the most extensively used group of drugs to treat unipolar affective disorders. It is a structurally heterogeneous group of drugs that act by blocking the reuptake transporters of either, or both, norepinephrine and serotonin (5-HT).58 Normalization of the hyperactive HPA system occurs during successful antidepressant pharmacotherapy of depressive illness.34,59,60 One possible mechanism for this action of antidepressants could be an increase in the cellular corticosteroid receptor concentration rendering the HPA system more susceptible to feedback inhibition by cortisol. Work from my laboratory first showed that different types of antidepressant could in fact alter GR mRNA levels.61,62 Subsequent work has indicated that the capacity of brain tissues to bind corticosteroids63,64 and corticosteroid receptor immunoreactivity in the brain65 are also increased by antidepressants. Although it was known that antidepressants produce changes in monoaminergic neurotransmitter systems, these changes could not be correlated with therapeutic efficiencies or measures of hypothalamic–pituitary–adrenal function,66 suggesting that as-yet-unknown mechanisms of action may be operative. Although some actions of antidepressants on the GR may be exerted through postsynaptic or presynaptic influences on monoamine concentrations, an additional mechanism of action appears completely unrelated to this. Thus, we found increased GR mRNA levels irrespective of the preferential inhibitory action of antidepressants on the reuptake of different classes of monoamine neurotransmitters. Furthermore, we showed increased GR gene transcription in antidepressant-treated mouse fibroblast cells67 that do not possess monoamine reuptake mechanisms. We hypothesized that one action of antidepressants may be exerted at the genomic level (but not necessarily directly) to stimulate the transcription of the GR gene and that this action may not be limited to neurons. We investigated this in mouse fibroblast Ltk– cells and in the mouse neuroblastoma Neuro 2A cell line transfected with a plasmid DNA vector consisting of the glucocorticoid-responsive mouse mammary tumour virus (MMTV) promoter–enhancer element fused to a reporter gene (CAT). This allowed measurement of antidepressant-induced changes in glucocorticoid response because, at constant glucocorticoid concentrations, a linear relation between MMTV–CAT activity and GR concentrations existed.68 With this chimeric construct, we observed increased glucocorticoid-stimulated CAT activity when the cells were treated with the antidepressant desipramine. A different chimeric gene construct consisting of a 2.7-kb fragment of the GR gene promoter region fused to the CAT gene (pHGR2.7CAT) allowed more direct measurement of antidepressant action. Increased CAT activity was also seen when cells transfected with pHGR2.7CAT were treated with desipramine.69 Finally, GR mRNA concentrations and glucocorticoid-binding activity of neuroblastoma and fibroblast cells increased after treatment of cells with antidepressant.67
The time course of hippocampal and hypothalamic MR and GR concentration changes in rats treated with a tricyclic antidepressant,63 or with moclobemide,64 a reversible inhibitor of monoamine oxidase A, showed that both the MR and the GR are elevated between 2 and 5 weeks after the start of treatment. This suggests that antidepressant-induced changes in brain corticosteroid receptor capacity may underlie the observed simultaneous decrease in basal circulating ACTH and corticosterone levels and decreased adrenal size.63,64 Furthermore, when challenged by a stressor, antidepressant-treated rats showed a decreased ACTH and corticosterone response, possibly induced through enhanced effectiveness of negative corticosteroid feedback because of re-established GR and MR capacity.63,64 Some of these effects may be mediated through CRH because in rats that were treated daily with imipramine,69 or in amitriptyline-treated transgenic mice, the CRH mRNA levels in the paraventricular nucleus were decreased by 26%–37%.70,71
The fact that antidepressant therapy can normalize cortisol levels and restore the suppressibility of cortisol by dexamethasone34,59 suggests that the negative feedback action of cortisol at the limbic–hypothalamic level (possibly acting on CRH and arginine vasopressin secretion) is less effective in depressed patients and is restored to full efficiency by antidepressant therapy. One possible explanation for this is that neurons involved in the central control of CRH production have reduced GR concentrations and are therefore less able to curtail stress-evoked cortisol levels, leading to a deficient negative feedback effect on the secretion of CRH and arginine vasopressin. The time course of antidepressant actions on corticosteroid receptors coincides with their long-term actions on HPA system activity and follows closely that of clinical improvement of depression.34,59,60,61,62,70 The tissue specificity of antidepressant action on corticosteroid receptors is only partly known but, as indicated by actions in nonneuronal cell lines, may be more widespread than the brain areas involved in neuroendocrine regulation. If this action is also found in lymphocytes, a predictive test for antidepressant action could be envisaged.
Rationale and new hypothesis for antidepressant action
As described earlier, antidepressants clearly increase mRNA levels of the MR and the GR and hormone-binding activities.63,64,65,72 In transfected cells, GR gene promoter activity is stimulated by antidepressants,67 and several different antidepressants can modulate this GR in vivo.71 On the basis of these findings, we hypothesize that a primary action of antidepressants could be the stimulation of corticosteroid receptor gene expression with a resultant decrease in HPA system activity, including reduced expression of CRH, which has been implicated in the pathogenesis of depression. Antidepressants could thus increase the capacity of neurons involved in the regulation of the HPA system to respond to feedback inhibition by glucocorticoids, and they may have a common mechanism of action at the level of the corticosteroid receptor genes. Secondary effects, resulting from a reduction in glucocorticoid concentrations, would be exerted on the expression of genes susceptible to glucocorticoid control such as enzymes involved in neurotransmitter biosynthesis.73 The necessity of HPA system normalization for mood improvement remains speculative, although the fact that patients with hypercortisolemia (e.g., Cushing's syndrome) or on glucocorticoid therapy often have depressive symptoms, in addition to the poor treatment response in depressed patients with persistent HPA alteration, supports this hypothesis. If additional evidence could be accumulated that neuroendocrine alterations are causally involved in affective disorders, as some preliminary data suggest,17,74 this would provide a lead for the development of more effective drugs. Nevertheless, this work opens up a completely new insight into antidepressant drug action and suggests a line of approach to the development of new drugs by focusing on this action.
Antidepressants can also influence the HPA system by actions other than those mediated via corticosteroid receptors. Antidepressant action on monoamines, their receptors and transporters, and the second messenger systems involved, is a vast field and one that is being extensively studied by many different groups. The effects of various stressors on ACTH secretion and corticosterone concentrations are well documented, and many investigators have explored the role of monoaminergic neurotransmitters in these processes. Antidepressants do not, however, appear to have effects on the mRNA of serotonergic receptors, biosynthetic enzymes or transporters,75,76 suggesting that their action on the HPA system is exerted by other mechanisms. The role of brain 5-HT in the regulation of adrenocortical activity has been extensively investigated, but no consensus of opinion exists other than that it represents an essential regulatory component of the pituitary–adrenocortical response to stress. We have measured 5-HT concentrations and turnover in response to immobilization stress and found no modification in numerous hypothalamic and amygdaloid nuclei.77 Lesion of the central nucleus of the amygdala, however, increased 5-HT turnover in most hypothalamic nuclei and this was reversed by stress, suggesting an inverse relation between serotonergic activity and ACTH secretion.77 Increased HPA activity does indeed appear to coincide with changes in the 5-HT system, including increased 5-HT turnover,78 increased postsynaptic 5-HT2 receptors79 and decreased 5-HT1A receptors.80,81,82,83 These same changes are noted in depression,84 and tryptophan depletion leads to mood lowering.85 Perhaps the most important aspect of the 5-HT system is in relation to the negative feedback action of glucocorticoids.86 The retroinhibitory action of corticosterone is exerted via receptor sites localized in neurons of the hypothalamus, hippocampus, septum, amygdala and reticular formation. Innervation of these areas is provided by noradrenergic efferents from the locus coeruleus and the serotonergic raphe neurons. Although adrenalectomy or corticosteroid administration do not alter norepinephrine metabolism, corticosterone elevates 5-HT synthesis and concentrations and alters the dynamics of 5-HT release and uptake by synaptosomes.87 Lesion of the raphe nuclei by 5,7-dihydroxytryptamine decreased the GR and MR capacity of the hippocampus88,89 by a mechanism distinct from the downregulation that can be caused by high glucocorticoid concentrations,90 but lesion of the locus coeruleus by 6-hydroxydopamine had no effect on GR.91 The possibility remains that norepinephrine may exert actions on the HPA axis, but this is not well demonstrated by lesion studies. It is interesting to note that the antidepressant mirtazapine that acts as antagonist to presynaptic α2 receptors significantly reduces the hypercortisolemia associated with depression.92 Although the availability of new selective noradrenergic reuptake inhibitors with antidepressant profiles such as reboxetine could allow clarification of the role of norepinephrine, the results must be treated with caution because of the simultaneous effects on other monoaminergic systems.93 Lesion of cholinergic inputs to the hippocampus has the opposite effect on corticosteroid receptors,94 but this is probably not a component of antidepressant action considering their low reactivity with the cholinergic system. The action of antidepressants on the HPA axis, other than that exerted directly on corticosteroid receptor genes, is thus most likely to be exerted via the serotonergic and noradrenergic systems, and antidepressants could exert action on the HPA system through increased serotonergic/noradrenergic postsynaptic activation, leading to stimulation of corticosteroid receptor gene expression and increased sensitivity of the HPA system to retroinhibiton by corticosteroids. There would thus be a dual system for activation of corticosteroid receptor genes: directly, via unknown mechanisms, and via 5-HT (Fig. 1).
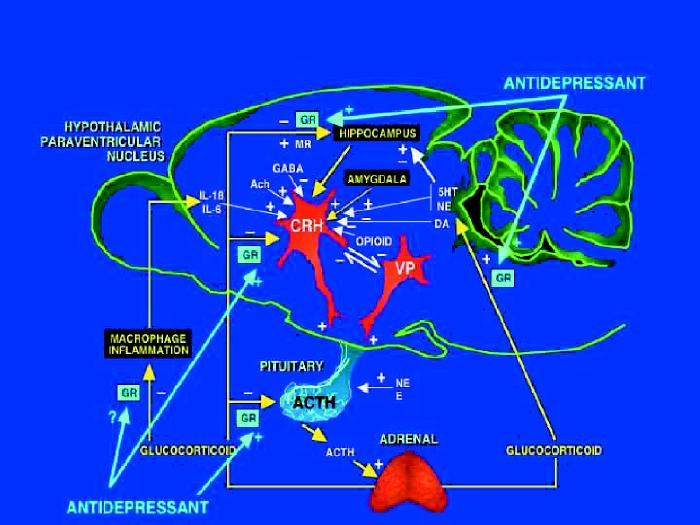
Fig. 1: Schematic representation of the hypothalamic–pituitary–adrenocortical (HPA) axis. Stimulatory (+) and inhibitory (–) actions of neural inputs to brain regions involved in HPA system regulation and the sites of corticosteroid regulation are shown. The sites at which antidepressants have stimulatory actions on the glucocorticoid receptor (GR), mineralocorticoid receptor (MR), or both, are indicated. IL = interleukin, GABA = γ-aminobutyric acid, Ach = acetylcholine, CRH = corticotropin-releasing hormone, VP = vasopressin, ACTH = corticotropin, NE = norepinephrine, E = epinephrine, 5HT = serotonin, DA = dopamine.
Footnotes
Competing interests: None declared.
Correspondence to: Dr. Nicholas Barden, Neuroscience Research Department, Centre Hospitalier de l'Université Laval Research Centre, 2705 boul. Laurier, RC-9800, Sainte-Foy QC G1V 4G2; fax 418 654-2753; barden@crchul.ulaval.ca
Submitted July 4, 2003; Revised Nov. 26, 2003; Accepted Dec. 1, 2003
References
- 1.Jasper MS, Engeland WC. Splanchnic neural activity modulates ultradian and circadian rhythms in adrenocortical secretion in awake rats. Neuroendocrinology 1994;59:97-109. [DOI] [PubMed]
- 2.Cascio CS, Shinsako J, Dallman MF. The suprachiasmatic nuclei stimulate evening ACTH secretion in the rat. Brain Res 1987;423:173-9. [DOI] [PubMed]
- 3.Szafarczyk A, Alonso G, Ixart G, Malaval F, Assenmacher I. Diurnal-stimulated and stress-induced ACTH release in rats is mediated by ventral noradrenergic bundle. Am J Physiol 1985;249:E219-26. [DOI] [PubMed]
- 4.Beaulieu S, Pelletier G, Vaudry H, Barden N. Influence of the central nucleus of the amygdala on the content of corticotropin-releasing factor in the median eminence. Neuroendocrinology 1989;49:255-61. [DOI] [PubMed]
- 5.Beaulieu S, De Paolo T, Côté J, Barden N. Participation of the central amygdaloid nucleus in the response of adrenocorticotropin secretion to immobilization stress: opposing roles of the noradrenergic and dopaminergic systems. Neuroendocrinology 1987;45:37-46. [DOI] [PubMed]
- 6.Jacobson L, Sapolsky R. The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocr Rev 1991;12:118-34. [DOI] [PubMed]
- 7.Reul JMHM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology 1985;117:2505-12. [DOI] [PubMed]
- 8.Cintra A, Zoli M, Rosen L, Agnati LF, Okret S, Wikstrom AC, et al. Mapping and computer assisted morphometry and microdensitometry of glucocorticoid receptor immunoreactive neurons and glial cells in the rat central nervous system. Neuroscience 1994;62:843-97. [DOI] [PubMed]
- 9.de Kloet ER. Brain corticosteroid receptor balance and homeostatic control. Front Neuroendocrinol 1991;12:95-164. [DOI] [PubMed]
- 10.Funder JW. Corticosteroid receptors and the central nervous system. J Steroid Biochem Mol Biol 1994;49:381-4. [DOI] [PubMed]
- 11.Ratka A, Sutanto W, Bloemers M, de Kloet ER. On the role of brain mineralocorticoid (type I) and glucocorticoid (type II) receptors in neuroendocrine regulation. Neuroendocrinology 1989;50:117-23. [DOI] [PubMed]
- 12.Bradbury MJ, Akana SF, Cascio CS, Levin N, Jacobson L, Dallman MF. Regulation of basal ACTH secretion by corticosterone is mediated by both type I (MR) and type II (GR) receptors in rat brain. J Steroid Biochem Mol Biol 1991;40:133-42. [DOI] [PubMed]
- 13.Dallman MF, Levin N, Cascio CS, Akana SF, Jacobson L, Kuhn RW. Pharmacological evidence that the inhibition of diurnal adrenocorticotropin secretion by corticosteroids is mediated via type I corticosterone-preferring receptors. Endocrinology 1989;124:2844-50. [DOI] [PubMed]
- 14.Regestein QR, Rose LI, Williams GH. Psychopathology in Cushing's syndrome. Arch Intern Med 1972;130:114-7. [PubMed]
- 15.Carroll BJ, Curtis GC, Mendels J. Neuroendocrine regulation in depression I. Limbic system-adrenocortical dysfunction. Arch Gen Psychiatry 1976;33:1039-50. [DOI] [PubMed]
- 16.Von Bardeleben U, Holsboer F. Cortisol response to a combined dexamethasone–human corticotropin-releasing- hormone challenge in patients with depression. J Neuroendocrinol 1989;1:485-8. [DOI] [PubMed]
- 17.Murphy BE. Steroids and depression. J Steroid Biochem Mol Biol 1991;38:537-59. [DOI] [PubMed]
- 18.Linkowski P, Kerkhofs M, Van Onderbergen A, Hubain P, Copinschi G, l'Hermite-Baleriaux M, et al. The 24-hour profiles of cortisol, prolactin, and growth hormone secretion in mania. Arch Gen Psychiatry 1994;51:616-24. [DOI] [PubMed]
- 19.Holsboer F, von Bardeleben U, Gerken A, Stalla GK, Müller OA. Blunted corticotropin and normal cortisol response to human corticotropin-releasing factor (h-CRF) in depression. N Engl J Med 1984;311:1127. [DOI] [PubMed]
- 20.Holsboer F, Gerken A, Stalla GK, Müller OA. ACTH, cortisol and corticosterone output after ovine corticotropin-releasing factor challenge during depression and after recovery. Biol Psychiatry 1985;20:276-86. [DOI] [PubMed]
- 21.Holsboer F, Gerken A, von Bardeleben U, Grimm W, Beyer H, Müller OA, et al. Human corticotropin-releasing hormone in depression. Biol Psychiatry 1986;21:601-11. [DOI] [PubMed]
- 22.Gold PW, Chrousos G, Kellner C, Post R, Roy A, Augerinos P, et al. Psychiatric implications of basic and clinical studies with corticotropin-releasing factor. Am J Pyschiatry 1984;141:619-27. [DOI] [PubMed]
- 23.Holsboer F, Spengler D, Heuser I. The role of corticotropin- releasing hormone in the pathogenesis of Cushing's disease, anorexia nervosa, alcoholism, affective disorders and dementia. In: Swaab DF, Hofman MA, Mirmiran M, Ravid R, van Leeuwen FW, editors. Progress in brain research. Vol. 93. Amsterdam: Elsevier; 1992. p. 385-417. [DOI] [PubMed]
- 24.Dunn AJ, Berridge CW. Physiological and behavioral responses to corticotropin-releasing factor administration: Is CRF a mediator of anxiety or stress responses? Brain Res Rev 1990;15:71-100. [DOI] [PubMed]
- 25.Owens MJ, Nemeroff CB. Physiology and pharmacology of corticotropin-releasing factor. Pharmacol Rev 1991;43:425-73. [PubMed]
- 26.Nemeroff CB, Widerlov E, Bisette G, Walléus H, Karlson I, Eklund K, et al. Elevated concentrations of CSF corticotropin- releasing factor-like immunoreactivity in depressed patients. Science 1984;226:1342-3. [DOI] [PubMed]
- 27.Raadsheer FC, Hoogendijk WJG, Stam FC, Tilders FJH, Swaab DF. Increased numbers of corticotropin-releasing hormone expressing neurons in the hypothalamic paraventricular nucleus of depressed patients. Neuroendocrinology 1994;60:436-44. [DOI] [PubMed]
- 28.Nemeroff CB, Owens MJ, Bissette G, Andorn AC, Stanley M. Reduced corticotropin releasing factor binding sites in the frontal cortex of suicide victims. Arch Gen Psychiatry 1988; 45:577-9. [DOI] [PubMed]
- 29.Gold PW, Loriaux DL, Roy A. Responses to corticotropin- releasing hormone in the hypercortisolism of depression and Cushing's disease. Pathophysiologic and diagnostic implications. N Engl J Med 1986;314:1329-35. [DOI] [PubMed]
- 30.Von Bardeleben U, Stalla GK, Müller OA, Holsboer F. Blunting of ACTH response to human CRH in depressed patients is avoided by metyrapone pre-treatment. Biol Psychiatry 1988; 24:782-6. [DOI] [PubMed]
- 31.Lisansky J, Peake GT, Strassman RJ, Qualls C, Meikle AW, Risch C, et al. Augmented pituitary corticotropin response to a threshold dosage of human corticotropin-releasing hormone in depressives pre-treated with metyrapone. Arch Gen Psychiatry 1989;46:641-9. [DOI] [PubMed]
- 32.Gershon ES, Targum SD, Kessler LR, Mazure CM, Bunney WE Jr. Genetic studies and biologic strategies in the affective disorders. Prog Med Genet 1977;2:101-64. [PubMed]
- 33.Bertelsen A, Harvald B, Hauge M. A Danish twin study of manic-depressive disorders. Br J Psychiatry 1977;130:330-51. [DOI] [PubMed]
- 34.Holsboer-Trachsler E, Stohler R, Hatzinger M. Repeated administration of the combined dexamethasone-human corticotropin releasing hormone stimulation test during treatment of depression. Psychiatry Res 1991;38:163-71. [DOI] [PubMed]
- 35.Young EA, Haskett RF, Murphy-Weinberg V, Watson SJ, Akil H. Loss of glucocorticoid fast feedback in depression. Arch Gen Psychiatry 1991;48:693-9. [DOI] [PubMed]
- 36.Gotthardt U, Schweiger U, Fahrenberg J, Lauer C, Holsboer F, Heuser I. Cortisol, ACTH and cardiovascular resposes to a cognitive challenge paradigm in aging and depression. Am J Physiol 1995;268:R865-73. [DOI] [PubMed]
- 37.Heuser I, Gotthardt U, Schweiger, U, Schmider J, Lammers CH, Dettling M, et al. Age-associated changes of pituitary-adrenocortical hormone regulation in humans: importance of gender. Neurobiol Aging 1994;15:227-31. [DOI] [PubMed]
- 38.Sapolsky RM, Krey LC, McEwen BS. Glucocorticoid-sensitive hippocampal neurons are involved in terminating the adrenocortical stress response. Proc Natl Acad Sci U S A 1984;81:6174-7. [DOI] [PMC free article] [PubMed]
- 39.Brooke SM, Haasjohnson AM, Kaplan JR, Manuck SB, Sapolsky RM. Dexamethasone resistance among nonhuman primates associated with a selective decrease of glucocorticoid receptors in the hippocampus and a history of social instability. Neuroendocrinology 1994;60:134-40. [DOI] [PubMed]
- 40.Peiffer A, Barden N, Meaney MJ. Age-related changes in glucocorticoid receptor binding and mRNA levels in the rat brain and pituitary. Neurobiol Aging 1991;12:475-9. [DOI] [PubMed]
- 41.Sapolsky RM, Krey LC, McEwen BS. The adrenocortical stress-response in the aged male rat: impairment of recovery from stress. Exp Gerontol 1983;18:55-64. [DOI] [PubMed]
- 42.Halbreich U, Asnis GM, Zumoff B, Nathan RS, Shindledecker R. Effect of age and sex on cortisol secretion in depressives and normals. Psychiatry Res 1984;13:221-9. [DOI] [PubMed]
- 43.Whalley LJ, Borthwick N, Copolov D, Dick H, Christie JE, Fink G. Glucocorticoid receptors and depression. BMJ 1986;292:859-61. [DOI] [PMC free article] [PubMed]
- 44.Pepin MC, Pothier F, Barden N. Impaired type II glucocorticoid receptor function in transgenic mice expressing antisense RNA. Nature 1992;355:725-8. [DOI] [PubMed]
- 45.Stec I, Barden N, Reul JMHM, Holsboer F. Dexamethasone nonsuppression in transgenic mice expressing antisense RNA to the glucocorticoid receptor. J Psychiatr Res 1994;28:1-5. [DOI] [PubMed]
- 46.Barden N, Stec ISM, Montkowski A, Holsboer F, Reul JMHM. Endocrine profile and neuroendocrine challenge tests in transgenic mice expressing antisense RNA against the glucocorticoid receptor. Neuroendocrinology 1997;66:212-20. [DOI] [PubMed]
- 47.Barden N. Corticosteroid receptor modulation in transgenic animals. Ann N Y Acad Sci 1994;746:89-100. [DOI] [PubMed]
- 48.Richard D, Chapdelaine S, Deshaies Y, Pepin MC, Barden N. Energy balance and lipid metabolism in transgenic mice bearing an antisense glucocorticoid receptor gene construct. Am J Physiol 1993;265:146-50. [DOI] [PubMed]
- 49.Montkowski A, Barden N, Wotjak C, Stec I, Ganster J, Meaney M, et al. Long-term antidepressant treatment reduces behavioural deficits in transgenic mice with impaired glucocorticoid receptor function. J Neuroendocrinol 1995;7:841-5. [DOI] [PubMed]
- 50.Rousse I, Beaulieu S, Rowe W, Meaney MJ, Barden N, Rochford J. Spatial memory in transgenic mice with impaired glucocorticoid receptor function. NeuroReport 1997;8:841-5. [DOI] [PubMed]
- 51.Rochford J, Beaulieu S, Rousse I, Glowa JR, Barden N. Behavioral reactivity to aversive stimuli in a transgenic mouse model of impaired glucocorticoid (type II) receptor function: effects of diazepam and FG-7142. Psychopharmacology 1997;132:145-52. [DOI] [PubMed]
- 52.Rousse I, Beaulieu S, Barden N, Rochford J. Spatial memory in transgenic mice with impaired glucocorticoid receptor function. Neuroreport 1997;8:841-5. [DOI] [PubMed]
- 53.Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 1999; 23:99-103. [DOI] [PubMed]
- 54.Young EA, Haskett RF, Grunhaus L, Pande A, Weinberg VM, Watson SJ, et al. Increased evening activation of the hypothalamic-pituitary-adrenal axis in depressed patients. Arch Gen Psychiatry 1994;51:701-7. [DOI] [PubMed]
- 55.Vamvakopoulos NC, Chrousos GP. Structural organization of the 5' flanking region of the human corticotropin releasing hormone gene. DNA Seq 1993;4:197-206. [DOI] [PubMed]
- 56.Barden N, Reul JMHM, Holsboer F. Do antidepressants stabilize mood through actions on the hypothalamic-pituitary-adrenocortical system? Trends Neurosci 1995;18:6-11. [DOI] [PubMed]
- 57.Holsboer F, Barden N. Antidepressants and hypothalamic- pituitary-adrenocortical regulation. Endocr Rev 1996;17:187-205. [DOI] [PubMed]
- 58.Ban TA. Psychopharmacology of depression. New York: Karger; 1981.
- 59.Greden JF, Gardner R, King D, Grunhaus L, Carroll BJ, Kronfol Z. Dexamethasone suppression test in antidepressant treatment of melancholia: the process of normalization and test-retest reproducibility. Arch Gen Psychiatry 1983;40:493-500. [DOI] [PubMed]
- 60.Hoslboer F, Liebl R, Hifschuster E. Repeated dexamethasone suppression test during depressive illness. Nomalization of test result compared with clinical improvement. J Affect Disord 1982;4:93-101. [DOI] [PubMed]
- 61.Beaulieu S, Barden N. Antidepressant, serotoninergic and glucocorticoid modulation of glucocorticoid receptor messenger RNA in hypothalamic neurons. Neuroendocrinol Lett 1987;9:186.
- 62.Pepin MC, Beaulieu S, Barden N. Antidepressants regulate glucocorticoid receptor messenger RNA concentrations in primary neuronal cultures. Mol Brain Res 1989;6:77-83. [DOI] [PubMed]
- 63.Reul JMHM, Stec I, Söder M, Holsboer F. Chronic treatment of rats with the antidepressant amitriptyline attenuates the activity of the hypothalamic-pituitary-adrenocortical system. Endocrinology 1993;133:312-20. [DOI] [PubMed]
- 64.Reul JMHM, Labeur MS, Grigoriadis DE, De Souza EB, Holsboer F. Hypothalamic-pituitary-adrenocortical axis changes in the rat after long-term treatment with the reversible monoamine oxidase-A inhibitor moclobemide. Neuroendocrinology 1994; 60:509-19. [DOI] [PubMed]
- 65.Kitayama I, Janson AM, Cintra A, Fuxe K, Agnati LF, Ogren SO, et al. Effects of chronic imipramine treatment on glucocorticoid receptor immunoreactivity in various regions of the rat brain. J Neural Transm 1988;73:191-203. [DOI] [PubMed]
- 66.Hanin I, Frazer A, Croughan J, Davis JM, Katz MM, Koslow SH, et al. Depression: implications of clinical studies for basic research. Fed Proc 1985;44:85-90.
- 67.Pepin MC, Govindan MV, Barden N. Increased glucocorticoid receptor gene promoter activity following antidepressant treatment. Mol Pharmacol 1992;41:1016-22. [PubMed]
- 68.Pepin MC, Barden N. Decreased glucocorticoid receptor activity following glucocorticoid receptor anti-sense RNA gene fragment transfection. Mol Cell Biol 1991;11:1647-53. [DOI] [PMC free article] [PubMed]
- 69.Brady LS, Whitfield HJ Jr, Fox RJ, Gold PW, Herkenham M. Long-term antidepressant administration alters corticotropin-releasing hormone, tyrosine hydroxylase, and mineralocorticoid receptor gene expression in rat brain. Therapeutic implications. J Clin Invest 1991;87:831-7. [DOI] [PMC free article] [PubMed]
- 70.Barden N. Modulation of glucocorticoid receptor gene expression by antidepressant drugs. Pharmacopsychiatry 1996;29:12-22. [DOI] [PubMed]
- 71.Peiffer A, Veilleux S, Barden N. Antidepressant and other centrally acting drugs regulate glucocorticoid receptor mRNA levels in rat brain. Psychoneuroendocrinology 1991;16:505-15. [DOI] [PubMed]
- 72.Seckl JR, Fink G. Antidepressants increase glucocorticoid and mineralocorticoid receptor mRNA expression in rat hippocampus in vivo. Neuroendocrinology 1992;55:621-6. [DOI] [PubMed]
- 73.Kvetnansky R, Fukuhara K, Pacak K, Cizza G, Goldstein DS, Kopin IJ. Endogenous glucocorticoids restrain catecholamine synthesis and release at rest and during immobilization stress in rats. Endocrinology 1993;133:1411-9. [DOI] [PubMed]
- 74.Murphy BEP, Filipini D, Ghadirian AM. Possible use of glucocorticoid receptor antagonists in the treatment of major depression: preliminary results using RU 486. J Psychiatry Neurosci 1993;18:209-13. [PMC free article] [PubMed]
- 75.Spurlock G, Buckland P, O'Donovan M, McGuffin P. Lack of effect of antidepressant drugs on the levels of mRNAs encoding serotonergic receptors, synthetic enzymes and 5HT transporter. Neuropharmacology 1994;33:433-40. [DOI] [PubMed]
- 76.Benmansour S, Cecchi M, Morilak DA, Gerhardt GA, Javors MA, Gould GG, et al. Effects of chronic antidepressant treatments on serotonin transporter function, density and mRNA level. J Neurosci 1999;19:10494-501. [DOI] [PMC free article] [PubMed]
- 77.Beaulieu S, Di Paolo T, Côté J, Barden N. Participation of the central amygdaloid nuleus in the response of adrenocorticotropin secretion to immobilization stress: opposing roles of the noradrenergic and dopaminergic systems. Neuroendocrinology 1987;45:37-46. [DOI] [PubMed]
- 78.Singh VB, Corley KC, Phan T, Boadle-Biber MC. Increases in the activity of tryptophan hydroxylase from rat cortex and midbrain in response to acute or repeated sound stress are blocked by adrenalectomy and restored by dexamethasone treatment. Brain Res 1990;516:66-76. [DOI] [PubMed]
- 79.Kuroda Y, Mikuni M, Ogawa T, Takahashi K. Effect of ACTH, adrenalectomy and the combination treatment on the density of 5-HT2 receptor binding sites in neocortex of rat forebrain and 5-HT2 receptor-mediated wet-dog shake behaviors. Psychopharmacology 1992;108:27-32. [DOI] [PubMed]
- 80.Mendelson SD, McEwen BS. Autoradiographic analyses of the effects of restraint-induced stress on 5-HT1A, 5-HT1C and 5-HT2 receptors in the dorsal hippocampus of male and female rats. Neuroendocrinology 1991;54:454-61. [DOI] [PubMed]
- 81.Biegon A, Rainbow TC, McEwen B. Corticosterone modulation of neurotransmitter receptors in rat hippocampus: a quantitative autoradiographic study. Brain Res 1985;332:309-14. [DOI] [PubMed]
- 82.Young AH, MacDonald LM, St John H, Dick H, Goodwin GM. The effects of corticosterone on 5-HT receptor function in rodents. Neuropharmacology 1992;31:433-8. [DOI] [PubMed]
- 83.Chalmers DT, Lopez JF, Vazquez DM, Akil H, Watson SJ. Regulation of hippocampal 5-HT1A receptor gene expression by dexamethasone. Neuropsychopharmacology 1994;10:215-22. [DOI] [PubMed]
- 84.Maes M, Meltzer HYM. The serotonin hypothesis of major depression. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: the fourth generation of progress. New York: Raven Press; 1994. p. 933-44.
- 85.Benkelfat C, Ellenbogen MA, Dean P, Palmour RM, Young SN. Mood-lowering effect of tryptophan depletion — enhanced susceptibility in young men at genetic risk for major affective disorders. Arch Gen Psychiatry 1994;51:687-97. [DOI] [PubMed]
- 86.Maes M, Meltzer HY, D'Hondt P, Cosyns P, Blockx P. Effects of serotonin precursors on the negative feedback effects of glucocorticoids on hypothalamic-pituitary-adrenal axis function in depression. Psychoneuroendocrinology 1995;20:149-67. [DOI] [PubMed]
- 87.de Kloet ER, Kovacs GL, Szabo G, Telegdy G, Bohus B, Versteeg DHG. Decreased serotonin turnover shortly after adrenalectomy: selective normalization after corticosterone substitution. Brain Res 1982;239:659-63. [DOI] [PubMed]
- 88.Seckl JR, Dickson KL, Fink G. Central 5,7-dihydroxytryptamine lesions decrease hippocampal glucocorticoid and mineralocorticoid receptor messenger ribonucleic acid expression. J Neuroendocrinol 1990;2:911-6. [DOI] [PubMed]
- 89.Ogawa T, Mikuni M, Kuroda Y, Muneoka K, Mori KJ, Takahashi K. Effects of the altered serotonergic signalling by neonatal treatment with 5,7-dihydroxytryptamine, ritanserin or clomipramine on the adrenocortical stress response and the glucocorticoid receptor binding in the hippocampus in adult rats. J Neural Transm Gen Sect 1994;96:113-23. [DOI] [PubMed]
- 90.Silva CM, Powell-Oliver FE, Jewell CM, Sar M, Allgood VE, Cidlowski JA. Regulation of the human glucocorticoid receptor by long-term and chronic treatment with glucocorticoid. Steroids 1994;59:436-42. [DOI] [PubMed]
- 91.Yau JLW, Seckl JR. Central 6-hydroxydopamine lesions decrease mineralocorticoid, but not glucocorticoid receptor gene expression in the rat hippocampus. Neurosci Lett 1992;142:159-62. [DOI] [PubMed]
- 92.Laakmann G, Hennig J, Baghai T, Schule C. Influence of mirtazapine on salivary cortisol in depressed patients. Neuropsychobiology 2003;47:31-6. [DOI] [PubMed]
- 93.Szabo ST, Blier P. Effects of the selective norepinephrine reuptake inhibitor reboxetine on norepinephrine and serotonin transmission in the rat hippocampus. Neuropsychopharmacology 2001;25:845-57. [DOI] [PubMed]
- 94.Yau JL, Kelly PA, Seckl JR. Increased glucocorticoid receptor gene expression in the rat hippocampus following combined serotonergic and medial septal cholinergic lesions. Mol Brain Res 1994;27:174-8. [DOI] [PubMed]