

Abstract
The synthesis of 2-alkylated pyridines by the nickel-catalyzed cross-coupling of two different electrophiles, 2-chloropyridines with alkyl bromides, is described. Compared to our previously published conditions for aryl halides, this method uses the different, more rigid bathophenanthroline ligand and is conducted at high concentration in DMF solvent. The method displays promising functional group compatibility and the conditions are orthogonal to the Stille coupling.
Keywords: Nickel, catalysis, pyridine, electrophile, cross-coupling
Alkylated pyridines represent an important class of azines that have appeared in launched drugs such as Nexium, used to treat acid reflux,1 and Lunesta, used in the treatment of insomnia.2 While the Suzuki-Miyaura reaction (Cδ− + Cδ+) is the dominant cross-coupling reaction used in both the discovery and production of pharmaceuticals,3 the coupling of heteroarenes is generally more challenging compared to aryl halides.4 For example, 2-pyridylboronic acid esters are difficult to synthesize and handle.5 In general, the cross-coupling of an alkyl halide with a pyridyl organometallic reagent (diorganozinc or tin reagent) remains challenging.6 A more developed approach is the cross-coupling of 2-halogenated pyridines with alkyl organometallic reagents such as tri-alkyl aluminum reagents,7 alkyl Grignard reagents,8 and alkyl zinc reagents (Scheme 1A).9
Scheme 1.
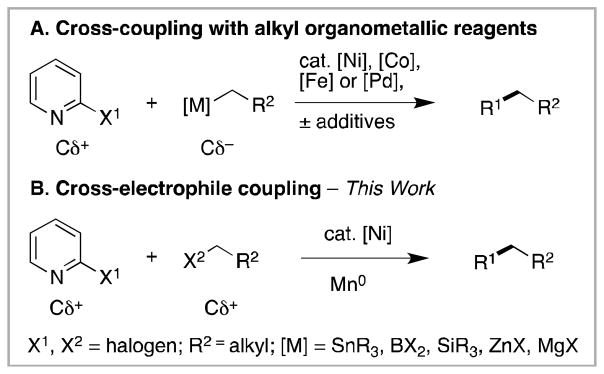
Comparison of cross-coupling with alkyl organometallic reagents (A) with cross-electrophile coupling (B).
In addition to the challenges associated with pyridine substrates, all of the approaches listed above require the synthesis of functionalized organometallic reagents. Besides the additional steps, organometallic reagents can also limit functional-group compatibility. For example, the most general approaches to 2-alkylated pyridines are the Fe8b, c, 10 and Ni8a, 8d, 11 catalyzed coupling of alkyl Grignard reagents with 2-halogenated pyridines (Scheme 1A, [M] = MgX). The synthesis of the Grignard reagents adds an extra step, and the high reactivity of alkyl Grignard reagents places limitations on electrophilic and acidic functional groups. The anionic Mg-C bond also causes problems with β-elimination of leaving groups.12 While methods for the synthesis of functionalized Grignard reagents have advanced considerably,13 it would be easier to avoid the challenge entirely.
One underexplored approach that avoids organometallic reagent synthesis is the direct cross-coupling of 2-halopyridines (Cδ+) with alkyl halides (Cδ+) (Scheme 1B). While we,14 and others15, 16, 17 have made great progress on cross-coupling methods that catalytically join two electrophiles, the best reported yield for pyridine alkylation is only 26% (for 6-methyl-2-chloropyridine).14a While Gong reported the alkylation of 8-bromoquinoline in good yield (96%), the one alkylation of 2-bromopyridine afforded 38% of the desired product, and no 2-chloropyridines were examined.15c We present here our progress towards a more general solution for 2-halopyridine alkylation.
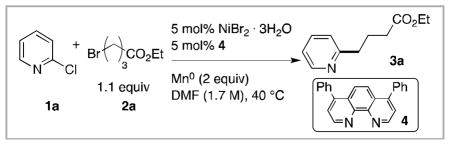
Optimization for the cross-coupling of 2-chloropyridine (1a) with ethyl 4-bromobutyrate (2a) to give 3a yielded the conditions in Table 1 (See SI Table S1 for product distribution data). Comparable yields were obtained at substrate concentrations ranging from 0.25–1.7 M, but concentrations higher than 1.7 M resulted in gells that complicated workup and provided inconsistent results. To ensure complete conversion of 1a, reactions were conducted with a slight excess of alkyl halide. Reactions run with equimolar amounts of each reagent or a slight excess of 1a provided similar results (Table 1 entries 3–4).
Table 1.
Optimization results for the cross-coupling of 2-chloropyridine (1a) with ethyl 4-bromobutyrate (2a).a
| ||
|---|---|---|
| Entry | Change from above conditions | Yield (%)b |
| 1 | None | 82 |
| 2 | 10 mol% NiBr2•3H2O/4 | 74c |
| 3 | 1 equiv each 1a and 2a | 78 |
| 4 | 1.1 equiv 1a | 71 |
| 5 | 1,10-phenanthroline (5) in place of 4 | 64 |
| 6 | 4,4′-di-t-butyl-2,2′-bipyridine (6) in place of 4 | 66 |
| 7 | 4,4′-di-methoxy-butyl-2,2′-bipyridine (7) in place of 4 | 69 |
| 8 | 4,4′,4″-tri-tert-butyl-2,2′,:6′,2″-terpyridine (8) in place of 4 | 15 |
| 9 | NiCl2(glyme) in place of NiBr2•3H2O | 79 |
| 10 | Reaction run at 20 °C | 55c |
| 11 | Reaction run at 60 °C | 70c |
| 12 | Reaction run at 80 °C | 62c |
| 13 | 25% DMA in THF in place of DMF | 15d |
| 14 | Zn0 (<10 μm) in place of Mn0 | 19e |
| 15 | Al0/PbBr2 in place of Mn0 | 2e,g |
Reaction conditions: DMF (1 mL), NiBr2•3H2O (0.15 mmol), 1a (3.00 mmol), 2a (3.30 mmol), ligand (0.15 mmol), and Mn0 (6.00 mmol) were added to a 1 dram vial on the bench top and heated under air for 4–22 h.
GC yield corrected vs. dodecane internal standard.
Isolated yield.
Observed partial conversion of starting material at 24 h.
Major coupled product was the alkyl dimer.
No reaction of 2a was observed.
Several ligands were examined, but bathophenanthroline (4) provided the highest yields of cross-coupled product 3a. Substitution of 4 for the less expensive 1,10-phenanthroline (5) does produce product in appreciable yield and should be considered in process applications where ligand cost can become a limiting factor (Table 1, entry 1 vs. 5). Other bi-dentate imine ligands gave lower yield (Table 1, entries 6–7), the tri-dentate imine ligand 4,4′,4″-tri-tert-butyl-2,2′,:6′,2″-terpyridine (8) gave a very low yield of alkylated pyridine and favored dimerization of 2a instead.14c Replacing NiBr2•3H2O with NiCl2(glyme) gave similar results (entries 1 and 9), while other nickel sources (NiI2•xH2O, NiI2, NiBr2, NiCl2) resulted in lower yields (35–58% yield). Lowering the temperature to 20 °C resulted in slow reactions and partial conversion of starting materials, while raising the temperature to 60 or 80 °C decreased selectivity for 3a (entries 1 and 10). The higher temperature reactions displayed an increase in hydrodehalogenated 2-chloropyridine (entries 11 and 12). Solvents other than DMF (DMA, DMPU, NMP, NEP, THF) resulted in either poor selectivity for 3a, partial conversion after 24 h, or both (4–45% yield). Lastly, the use of Zn0 or Al0/PbBr218 in place of Mn0 as the reducing agent resulted in lower yield and selectivity for 3a (entries 13 and 14). Specifically, the use of Zn0 quickly produced hydrodehalogenated 2-chloropyridine, possibly through direct insertion of Zn0 into the C-Cl bond followed by protonation, and this in turn resulted in dimerization of 2a once 1a was consumed. The use of Al0/PbBr2 gave only the dimer product of 2a with almost no conversion of 1a (entry 15).
To examine the scope of this new method, several different alkyl halides were coupled with 1a to give 2-alkylated pyridines (Table 2). Unfunctionalized alkyl bromides coupled with 2-chloropyridine efficiently under the optimized conditions giving 3b in 72% yield (Table 2, entry 2). As we discovered during optimization, alkyl bromides bearing ester functionality couple efficiently (Table 2, entry 1), and those bearing a Boc-protected primary amine also coupled well (Table 2, entry 3). An alkyl bromide with a tri-substituted olefin gave a lower yield (Table 2, entry 4), consistent with the challenge we observed in coupling this bromide with bromobenzene.14a
Table 2.
Scope of the electrophile cross-coupling of 2-chloropyridines with alkyl halides.a

| ||||
|---|---|---|---|---|
| Entry | Py-Cl (1) | Alkyl-X (2) | Product (3) | Yield (%)b |
| 1 | 1a | 2a |
3a |
72 |
| 2 | 1a | 2b |
 3b |
72c |
| 3 | 1a | 2c |
3c |
60d |
| 4 | 1a | 2d |
3d |
33c |
| 5 | 1a | 2e |
 3e |
48e |
| 6 | 1a | 2f |
 3f |
45f |
| 7 | 1b | 2a |
3g |
48 |
| 8 | 1c | 2a |
 3h |
50 |
| 9 | 1d | 2a |
3i |
46g |
Reaction conditions: Reaction conditions: DMF (1 mL), NiBr2•3H2O (0.15 mmol), 4 (0.15 mmol), chloropyridine (3.00 mmol), alkyl bromide (3.30 mmol), and Mn0 (6.00 mmol) were added to a 1 dram vial on the bench top and heated under air for 4–22 h.
Yield of isolated and purified product.
Reaction run with 10 mol% NiBr2•3H2O/4, yield 65% at 5 mol%.
Reaction run on 0.75 mmol scale with 1.1 equiv 1a and 10 mol% NiBr2•H2O/4.
Reaction run with 2 equiv 2e, yield was 33% with 1.1 equiv.
Reaction run with 10 mol% NiBr2•3H2O/4, yield is 37% with 5 mol%.
Reaction run with 10 mol% NiBr2•3H2O/4, 25 mol% NaI, and 10 mol% AIBN as additives, yield under standard conditions was 27%.
In addition to these primary halides, cyclohexyl bromide (2e) coupled in reasonable yield showing the promise of this method to couple secondary alkyl bromides, which are challenging substrates because of their propensity for β-hydrogen elimination (Table 2, entry 5).
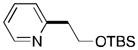
An alkyl bromide bearing a β-silyloxy leaving group (2f) also coupled in reasonable yield if a higher catalyst loading was used (10 mol %, Table 2, entry 6). The corresponding organometallic reagent (TBSOCH2CH2-[M]) is prone to β-elimination and presents a particular challenge.12 In our case, the parent alkane of 2f was the predominant by-product rather than the β-elimination product. The product, 3f, is a precursor to enediynes of tetrahydropyridine that exhibit antitumor activity.19
The synthetically useful tri-butyltin group on the pyridyl chloride (1b) was tolerated with no observable de-stannylation (Table 2, entry 7). The tri-butyl tin functional group can be used in subsequent steps for poly functionalization of the pyridine core with the well-established Stille reaction.20
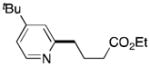
Finally, electronics on the pyridine core were briefly explored. The coupling of 2-chloro-4-tert-butylpyridine (1c) with 2a provided a 50% yield of the desired alkylated pyridine (Table 2, entry 8). Electron-poor pyridine 1d was plagued by long induction periods and long reaction times that allowed for competing side reactions that resulted in a low yield (27%) without additives. Halogen exchange that converted the alkyl bromide to the much less reactive alkyl chloride was the major competing reaction. The yield of 3i could be improved from 27% to 46% by the addition of catalytic amounts (25 mol%) of sodium iodide that converted the alkyl chloride into alkyl iodide in situ,14c and the addition of catalytic amounts (10 mol%) of azobisisobutyronitrile, AIBN, reduced the reaction time from 48 to 19 h. The observation that a radical initiator, AIBN, significantly decreased the reaction time is suggestive of a mechanism that contains radical steps. AIBN may decrease reaction times by generating alkyl radicals.
With the exception of 3f the major challenge to overcome in the cross-coupling reactions of 2-chloropyridines with alkyl bromides is competing dimerization of the alkyl bromide (See SI, Table S2, which contains product distribution data and the structures of 9–13). The synthesis of 3i that employed AIBN as an additive to decrease the reaction time exhibited poor mass balance, only 50% with respect to the alkyl bromide, and only 41% with respect to the chloropyridine (See SI, Table S2). Unproductive side reactions with AIBN may account for the missing mass, but no such products were identified by GC analysis of crude reaction mixtures. The propensity of these coupling reactions to, in some cases, be selective for alkyl bromide dimerization over the cross-coupling reaction suggests the chemistry of nickel terpyridine complexes.14c, 15g Nickel complexes (14) or (15), formed under these conditions, might have similar reactivity to terpyridine nickel complexes, such as (16),21 that are efficient alkyl dimerization catalysts (Figure 1).14c
Figure 1.
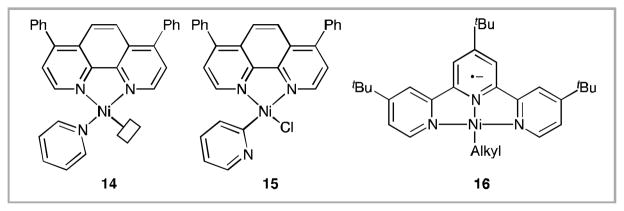
Postulated nickel complexes and terpyridine complex 16.
Our success with AIBN made us consider the possibility that Minisci chemistry,22 and not cross-electrophile coupling, was responsible for the observed products (Scheme 2). However, no product (3a or 3i) are formed in reactions performed with added AIBN but no nickel. In fact, the addition of AIBN lowered the yield of 3a (Table 2, entry 1 vs. Scheme 2) because of increased alkyl dimerization, consistent with the over-production of alkyl radicals being detrimental.
Scheme 2.
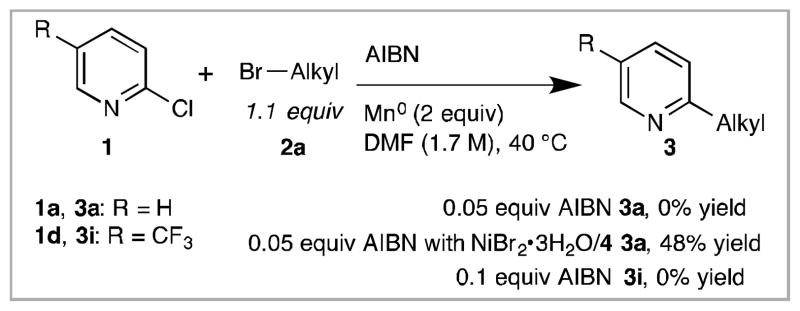
Reactions with AIBN require nickel to form cross-product.
Pyridines halogenated at the 3- or 4-postion do not currently couple in acceptable yields under the reaction conditions developed here (eq. 1). Similarly, a few other heterocycles that were examined also did not couple in high yield (2-chlorothiophene, 2-chlorobenzo[d]oxazole, 2-chloro-1H-benzo[d]imidazole, and 2-bromopyrazine). New reaction conditions and catalysts for these couplings are an active area of research in our lab.
The cross-electrophile coupling approach to 2-alkylated pyridines enables the synthesis of functionalized molecules, not easily accessible from coupling reactions of Grignard reagents, in a single step from easily available organic halides and 2-chloropyridines. Although future studies will seek to improve yields and expand substrate scope, these conditions should already prove helpful in synthesis.
General Experimental Procedure
In a well-ventilated fume hood, a 15 mL round bottom flask equipped with a Teflon coated magnetic stir bar was charged with NiBr2•3H2O (40.9 mg, 0.150 mmol, 0.05 equiv), bathophenanthroline (49.9 mg, 0.150 mmol, 0.05 equiv), DMF (2.0 mL), and alkyl bromide (3.3 mmol, 1.1 equiv). The vessel was stoppered with a rubber septum and heated to 40 °C in a fume hood until a green homogenous solution resulted (approx. 20 min). Once homogeneity was achieved the vessel was removed from the heat. The 2-halogenated pyridine (3.00 mmol, 1.00 equiv) and Mn0 (−325 mesh, 330 mg, 6.00 mmol, 2.00 equiv) were added, after which, the vessel was resealed with the septum, purged with argon gas, and heated again to 40 °C for the duration of the reaction. Reaction progress was monitored by GC analysis of aliquots of crude reaction mixture. In general the reactions turn dark brown or black in color when complete. Upon completion the reaction was cooled to room temperature, diluted with ether (10 mL) and filtered through a short pad of Celite 545 (approx. 1″ × 1″ × 1″) that had been wetted with ether (approx. 10 mL) to remove metal salts. The celite pad was washed with additional ether (2 × 10 mL). The filtrate was transferred to a separatory funnel and washed with 1M aqueous NH4Cl (10 mL). The layers were separated and the aqueous layer was washed with additional ether (3 × 10 mL). The combined organic extracts were washed with brine (10 mL), dried over MgSO4, filtered, and evaporated under reduced pressure. The crude products were purified by silica gel flash column chromatography.
Supplementary Material
Equation 1.
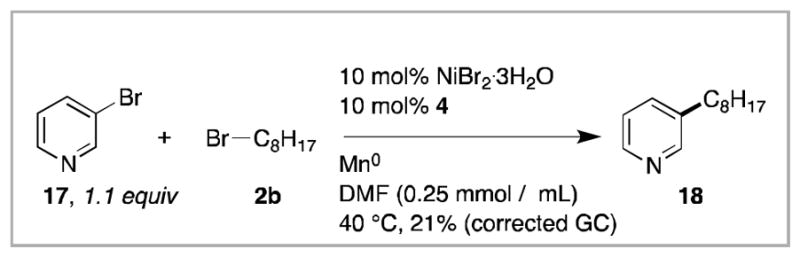
Electrophile cross-coupling of 3-bromopyridine (17) with 1-bromooctane (2b).
Acknowledgments
This work was supported by the University of Rochester, the NIH (R01 GM097243), and the NSF (Graduate Research Fellowship to DAE and Research Experience for Undergraduates Fellowship to JAB). Analytical data were obtained from the CENTC Elemental Analysis Facility at the University of Rochester, funded by NSF CHE-0650456. We thank David George (Univ. of Rochester) for running the control reactions in Scheme 2. We thank the reviewers for helpful suggestions.
Footnotes
Primary Data for this article are available online at http://www.thieme-connect.com/ejournals/toc/synlett and can be cited using the following DOI: (number will be inserted prior to online publication).
Supporting Information for this article is available online at http://www.thieme-connect.com/ejournals/toc/synlett.
References
- 1.Raju SVN, Purandhar K, Reddy PP, Reddy GM, Reddy LA, Reddy KS, Sreenath K, Mukkanti K, Reddy GS. Org Process Res Dev. 2005;10:33–35. [Google Scholar]
- 2.Turner K. Org Process Res Dev. 2009;13:381–390. [Google Scholar]
- 3.(a) Roughley SD, Jordan AM. J Med Chem. 2011;54:3451–3479. doi: 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]; (b) Cooper TWJ, Campbell IB, Macdonald SJF. Angew Chem Int Ed. 2010;49:8082–8091. doi: 10.1002/anie.201002238. [DOI] [PubMed] [Google Scholar]; (c) Dugger RW, Ragan JA, Ripin DHB. Org Process Res Dev. 2005;9:253–258. [Google Scholar]; (d) Carey JS, Laffan D, Thomson C, Williams MT. Org Biomol Chem. 2006;4:2337–2347. doi: 10.1039/b602413k. [DOI] [PubMed] [Google Scholar]; (e) Laird T. Org Process Res Dev. 2006;10:851–852. [Google Scholar]
- 4.Slagt VF, de Vries AHM, de Vries JG, Kellogg RM. Org Process Res Dev. 2009;14:30–47. [Google Scholar]
- 5.Dick GR, Knapp DM, Gillis EP, Burke MD. Org Lett. 2010;12:2314–2317. doi: 10.1021/ol100671v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Nakamura M, Ito S, Matsuo K, Nakamura E. Synlett. 2005:1794–1798. [Google Scholar]; (b) Bourdier T, Huiban M, Huet A, Sobrio F, Fouquet E, Perrio C, Barré L. Synthesis. 2008:978–984. [Google Scholar]; (c) Suzuki M, Sumi K, Koyama H, Siqin, Hosoya T, Takashima-Hirano M, Doi H. Chem – Eur J. 2009;15:12489–12495. doi: 10.1002/chem.200901145. [DOI] [PubMed] [Google Scholar]; (d) Vechorkin O, Proust V, Hu X. J Am Chem Soc. 2009;131:9756–9766. doi: 10.1021/ja9027378. [DOI] [PubMed] [Google Scholar]
- 7.(a) Huang Y, Bennett F, Girijavallabhan V, Alvarez C, Chan T-M, Osterman R, Senior M, Kwong C, Bansal N, George Njoroge F, MacCoss M. Tetrahedron Lett. 2010;51:2800–2802. [Google Scholar]; (b) Girijavallabhan V, Arasappan A, Bennett F, Huang Y, George Njoroge F, MacCoss M. Tetrahedron Lett. 2010;51:2797–2799. [Google Scholar]; (c) Joubert N, Pohl R, Klepetářová B, Hocek M. J Org Chem. 2007;72:6797–6805. doi: 10.1021/jo0709504. [DOI] [PubMed] [Google Scholar]
- 8.(a) Johnson S, Drowns M, Tatlock J, Linton A, Gonzalez J, Hoffman R, Jewell T, Patel L, Blazel J, Tang M, Li H. Synlett. 2010:796–800. [Google Scholar]; (b) Fürstner A, Leitner A, Méndez M, Krause H. J Am Chem Soc. 2002;124:13856–13863. doi: 10.1021/ja027190t. [DOI] [PubMed] [Google Scholar]; (c) Fürstner A, Leitner A. Angew Chem Int Ed. 2002;41:609–612. [Google Scholar]; (d) Hintermann L, Dang TT, Labonne A, Kribber T, Xiao L, Naumov P. Chem– Eur J. 2009;15:7167–7179. doi: 10.1002/chem.200900563. [DOI] [PubMed] [Google Scholar]
- 9.(a) Hoekstra WJ, Patel HS, Liang X, Blanc JBE, Heyer DO, Willson TM, Iannone MA, Kadwell SH, Miller LA, Pearce KH, Simmons CA, Shearin J. J Med Chem. 2004;48:2243–2247. doi: 10.1021/jm040154f. [DOI] [PubMed] [Google Scholar]; (b) Pompeo M, Froese RDJ, Hadei N, Organ MG. Angew Chem Int Ed. 2012;51:11354–11357. doi: 10.1002/anie.201205747. [DOI] [PubMed] [Google Scholar]; (c) Hendricks RT, Spencer SR, Blake JF, Fell JB, Fischer JP, Stengel PJ, Leveque VJP, LePogam S, Rajyaguru S, Najera I, Josey JA, Swallow S. Bioorg Med Chem Lett. 2009;19:410–414. doi: 10.1016/j.bmcl.2008.11.060. [DOI] [PubMed] [Google Scholar]
- 10.Sherry BD, Fürstner A. Acc Chem Res. 2008;41:1500–1511. doi: 10.1021/ar800039x. [DOI] [PubMed] [Google Scholar]
- 11.Tamao K, Sumitani K, Kumada M. Journal of the American Chemical Society. 1972;94:4374–4376. [Google Scholar]
- 12.Fleury-Brégeot N, Presset M, Beaumard F, Colombel V, Oehlrich D, Rombouts F, Molander GA. J Org Chem. 2012;77:10399–10408. doi: 10.1021/jo3021665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knochel P. Handbook of functionalized organometallics: applications in synthesis. Wiley-VCH; Weinheim: 2005. p. 653. [Google Scholar]
- 14.(a) Everson DA, Jones BA, Weix DJ. J Am Chem Soc. 2012;134:6146–6159. doi: 10.1021/ja301769r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Everson DA, Shrestha R, Weix DJ. J Am Chem Soc. 2010;132:920–921. doi: 10.1021/ja9093956. [DOI] [PubMed] [Google Scholar]; (c) Prinsell MR, Everson DA, Weix DJ. Chem Commun. 2010;46:5743–5745. doi: 10.1039/c0cc01716g. [DOI] [PubMed] [Google Scholar]; (d) Shrestha R, Dorn SCM, Weix DJ. J Am Chem Soc. 2012;135:751–762. doi: 10.1021/ja309176h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Anka-Lufford LL, Prinsell MR, Weix DJ. J Org Chem. 2012;77:9989–10000. doi: 10.1021/jo302086g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Shrestha R, Weix DJ. Org Lett. 2011;13:2766–2769. doi: 10.1021/ol200881v. [DOI] [PubMed] [Google Scholar]; (g) Wotal AC, Weix DJ. Org Lett. 2012;14:1476–1479. doi: 10.1021/ol300217x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Everson DA, George DT, Weix DJ, Buergler JF, Wood JL. Org Synth. 2013;90:200–214. doi: 10.15227/orgsyn.090.0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Yin H, Zhao C, You H, Lin K, Gong H. Chem Commun. 2012;48:7034–7036. doi: 10.1039/c2cc33232a. [DOI] [PubMed] [Google Scholar]; (b) Dai Y, Wu F, Zang Z, You H, Gong H. Chem Eur J. 2012;18:808–812. doi: 10.1002/chem.201102984. [DOI] [PubMed] [Google Scholar]; (c) Wang S, Qian Q, Gong H. Org Lett. 2012;14:3352–3355. doi: 10.1021/ol3013342. [DOI] [PubMed] [Google Scholar]; (d) Wu F, Lu W, Qian Q, Ren Q, Gong H. Org Lett. 2012;14:3044–3047. doi: 10.1021/ol3011198. [DOI] [PubMed] [Google Scholar]; (e) Yu X, Yang T, Wang S, Xu H, Gong H. Org Lett. 2011;13:2138–2141. doi: 10.1021/ol200617f. [DOI] [PubMed] [Google Scholar]; (f) Amatore M, Gosmini C. Chem Eur J. 2010;16:5848–5852. doi: 10.1002/chem.201000178. [DOI] [PubMed] [Google Scholar]; (g) Goldup SM, Leigh DA, McBurney RT, McGonigal PR, Plant A. Chem Sci. 2010;1:383–386. [Google Scholar]; (h) Amatore M, Gosmini C. Synlett. 2009:1073–1076. [Google Scholar]; (i) Kim H, Lee C. Org Lett. 2011;13:2050–2053. doi: 10.1021/ol200455n. [DOI] [PubMed] [Google Scholar]
- 16.Yan CS, Peng Y, Xu XB, Wang YW. Chem–Eur J. 2012;18:6039–6048. doi: 10.1002/chem.201200190. [DOI] [PubMed] [Google Scholar]
- 17.Amatore M, Gosmini C. Angew Chem Int Ed. 2008;47:2089–2092. doi: 10.1002/anie.200704402. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka H, Kuroboshi M. Curr Org Chem. 2004;8:1027–1056. [Google Scholar]
- 19.Braña MF, Morán M, Pérez de Vega MJ, Pita-Romero I. J Org Chem. 1996;61:1369–1374. [Google Scholar]
- 20.(a) Trost BM, Cook GR. Tetrahedron Lett. 1996;37:7485–7488. [Google Scholar]; (b) Sirisoma NS, Johnson CR. Tetrahedron Lett. 1998;39:2059–2062. [Google Scholar]; (c) Barros MT, Maycock CD, Ventura MR. Tetrahedron Lett. 1999;40:557–560. [Google Scholar]; (d) Barros MT, Maycock CD, Ventura MR. J Chem Soc, Perkin Trans 1. 2001;0:166–173. [Google Scholar]; (e) Lee SJ, Lin W. J Am Chem Soc. 2002;124:4554–4555. doi: 10.1021/ja0256257. [DOI] [PubMed] [Google Scholar]
- 21.(a) Anderson TJ, Jones G, Mcfarland C, Vicic D. Chem Commun. 2005:4211. doi: 10.1039/b504996b. [DOI] [PubMed] [Google Scholar]; (b) Ciszewski JT, Mikhaylov DY, Holin KV, Kadirov MK, Budnikova YH, Sinyashin O, Vicic DA. Inorg Chem. 2011;50:8630–8635. doi: 10.1021/ic201184x. [DOI] [PubMed] [Google Scholar]; (c) Anderson T, Jones G, Vicic D. J Am Chem Soc. 2004;126:8100–8101. doi: 10.1021/ja0483903. [DOI] [PubMed] [Google Scholar]
- 22.(a) Fontana F, Minisci F, Nogueira Barbosa MC, Vismara E. Tetrahedron. 1990;46:2525–2538. [Google Scholar]; (b) O’Hara F, Blackmond DG, Baran PS. J Am Chem Soc. 2013 doi: 10.1021/ja406223k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.