

Abstract
Phytoestrogens are plant compounds that structurally mimic the endogenous estrogen 17β-estradiol (E2). Despite intense investigation, the net effect of phytoestrogen exposure on the breast remains unclear. The objective of the current study was to examine the effects of quercetin on E2-induced breast cancer in vivo. Female ACI rats were given quercetin (2.5 g/kg food) for 8 months. Animals were monitored weekly for palpable tumors, and at the end of the experiment, rats were euthanized, breast tumor and different tissues excised so that they could be examined for histopathologic changes, estrogen metabolic activity and oxidant stress. Quercetin alone did not induce mammary tumors in female ACI rats. However, in rats implanted with E2 pellets, co-exposure to quercetin did not protect rats from E2-induced breast tumor development with 100% of the animals developing breast tumors within 8 months of treatment. No changes in serum quercetin levels were observed in quercetin and quercetin + E2-treated groups at the end of the experiment. Tumor latency was significantly decreased among rats from the quercetin + E2 group relative to those in the E2 group. Catechol-O-methyltransferase (COMT) activity was significantly downregulated in quercetin exposed mammary tissue. Analysis of 8-isoprostane F2α (8-iso-PGF2α) levels as a marker of oxidant stress showed that quercetin did not decrease E2-induced oxidant stress. These results indicate that quercetin (2.5 g/kg food) does not confer protection against breast cancer, does not inhibit E2-induced oxidant stress and may exacerbate breast carcinogenesis in E2-treated ACI rats. Inhibition of COMT activity by quercetin may expose breast cells chronically to E2 and catechol estrogens. This would permit longer exposure times to the carcinogenic metabolites of E2 and chronic exposure to oxidant stress as a result of metabolic redox cycling to estrogen metabolites, and thus quercetin may exacerbate E2-induced breast tumors in female ACI rats.
Keywords: Breast Cancer, Quercetin, Phytoestrogen, Estrogen, ACI Rat
Introduction
Phytoestrogens are biologically-active, plant-derived, phenolic compounds that structurally mimic the mammalian steroid hormone 17β-estradiol (E2) (Hodek et al., 2002; Sirtori et al., 2005; Stopper et al., 2005; Limer et al., 2006). These plant compounds exhibit a wide array of pharmacological properties, and in recent years, interest in their potential benefits has increased dramatically. The most highly investigated properties of the phytoestrogens are their possible chemoprotective characteristics, especially those relevant to breast cancer (Yang et al., 2001; Peeters et al., 2003; Galati and O'Brien, 2004; Sirtori et al., 2005; Messina et al., 2006). Epidemiological evidence suggests that diet and nutrition can influence cancer development, and Asian women report fewer post-menopausal symptoms and experience fewer breast cancers than women in Western countries (Le Corre et al., 2005; Nichenametla et al., 2006; Usui, 2006). More specifically, Asian women have a three-fold lower breast cancer risk than women in the United States, independent of body weight (Ursin et al., 1994). However, second and third generation descendants of women who migrated from Asia to Western countries have breast cancer risks similar to those of women in the host country, suggesting that lifestyle and dietary habits, and not genetic factors explain the low breast cancer risk of women in Asia (Probst-Hensch et al., 2000; Pike et al., 2002; Usui, 2006). Furthermore, epidemiologic studies have detected associations between phytoestrogen consumption, phytoestrogen levels in plasma and urine, and reduced risk of breast cancer (Peeters et al., 2003; Brooks and Thompson, 2005).
In recent years, various phytoestrogens have been evaluated for their ability to prevent chemically-induced mammary carcinogenesis. However, the net effect of phytoestrogen exposure on breast carcinogenesis remains unclear. Quercetin has been shown to inhibit nitrosomethylurea (NMU)- and 7,12-dimethylbenzanthracene (DMBA)-induced mammary carcinogenesis in rats (Verma et al., 1988). Resveratrol decreased NMU- and DMBA-induced tumor incidence and multiplicity by 50% in Sprague Dawley rats (Bhat et al., 2001; Banerjee et al., 2002). Similarly, resveratrol blocked the formation of pre-neoplastic lesions, suppressed mammary carcinogenesis, reduced tumor incidence and increased tumor latency in Sprague Dawley rats treated with DMBA (Whitsett et al., 2006). Daidzein effectively inhibited DMBA-induced mammary tumors in rats and significantly increased latency among mammary tumor virus-Neu mice (Constantinou et al., 2001; Jin and MacDonald, 2002). Despite intense investigation, considerable doubt remains as to whether phytoestrogen exposure imparts protection against breast cancer, and some investigations have yielded results indicating that phytoestrogens may exacerbate tumor development. For example, quercetin increased the severity of E2-induced kidney tumorigenesis in male Syrian hamsters (Zhu and Liehr, 1994). Genistein increased tumor cross-sectional area, increased tumor multiplicity, elevated the percentage of proliferative cells in tumors and increased the weight of estrogen-dependent mammary adenocarcinomas in rat models of mammary cancer (Allred et al., 2004; Kijkuokool et al., 2006).
Phytoestrogens have been evaluated for their ability to alter breast development. Resveratrol-exposed female rats displayed more differentiated lobular structures and decreased proliferation in the mammary terminal ductal structures, making them less vulnerable to damage by carcinogens (Whitsett et al., 2006). In addition, exposure to genistein during breast development altered breast morphology, and decreased the formation of terminal ducts (Hilakivi-Clarke et al., 1999). Thus, the relationship between phytoestrogens and tumor development may rely on age at the time of exposure and the hormonal environment. Several studies have demonstrated that rats exposed to genistein early in life have a decreased incidence of DMBA-induced mammary tumors in adulthood (Murrill et al., 1996; Fritz et al., 1998; Lamartiniere et al., 1998; Hilakivi-Clarke et al., 1999). Conversely, prepubescent rats treated with resveratrol showed accelerated NMU-induced mammary carcinogenesis and elevated tumor incidence and multiplicity (Sato et al., 2003).
Taken together, these studies indicate that certain phytoestrogens might reduce the risk of chemically-induced mammary cancers in animal models, particularly if exposure is early in life. However, very few studies have focused on the effect of phytoestrogens on breast cancers using an animal model of estrogen-induced breast tumor, which represents the most relevant model system for gaining insight into the relationship between phytoestrogens and human mammary carcinogenesis. The female ACI rat model of estrogen-induced mammary cancer represents a relevant biological system in which to study the relationship between quercetin exposure and human breast cancer. Continuous treatment of ovary-intact female ACI rats with E2 results in about 80-100% mammary tumor incidence within six to seven months, and tumors have not been observed in ovary-intact female ACI rats not exposed to exogenous E2 (Shull et al., 1997; Harvell et al., 2000; Mense et al., 2008b; Mense et al., 2009; Singh et al., 2009). All mammary tumors observed in this model have been classified as carcinomas, and invasive features have been observed in some (Shull et al., 1997; Harvell et al., 2000; Mense et al., 2008b; Mense et al., 2009; Singh et al., 2009). E2-induced mammary gland tumors in ACI rats and human sporadic breast cancers share many pertinent histopathologic and molecular features, both in early pre-malignant lesions as well as in primary tumors (Makris et al., 1997; Arnerlov et al., 2001; Li et al., 2002a; Li et al., 2002b; Li et al., 2004; Weroha et al., 2006). For example, mammary cancers induced by E2 in ACI rats are estrogen-dependent and exhibit genomic instability, characteristics commonly observed in human breast cancers (Makris et al., 1997; Arnerlov et al., 2001; Li et al., 2002a; Li et al., 2002b; Li et al., 2004; Weroha et al., 2006).
The objective of the present study was to examine the effects of quercetin on estrogen-induced breast cancer in vivo. The ACI rat model of estrogen-induced breast cancer was used. Female ACI rats (4-6 weeks of age) were given quercetin via the diet (2.5 g/kg food) for a period of 240 days. Since no other investigations of phytoestrogen exposure have been performed in the ACI rat model, the experimental dose of quercetin was chosen based on previous studies of phytoestrogens in animal models of chemically-induced breast cancer (Verma et al., 1988; Murrill et al., 1996; Fritz et al., 1998; Lamartiniere et al., 1998; Hilakivi-Clarke et al., 1999; Bhat et al., 2001; Constantinou et al., 2001; Banerjee et al., 2002; Jin and MacDonald, 2002; Whitsett et al., 2006). During the experiment, animals were monitored weekly for palpable tumors, and at the end of the experiments rats were euthanized, mammary tumor and different tissues quickly excised so that they could be used for histochemical and biochemical evaluations. Our findings indicate that, at the experimental dose (2.5 g/kg food) used, quercetin inhibited COMT activity in the mammary, did not prevent oxidative changes in the breast and did not confer protection against breast cancer.
Materials and methods
Chemicals
17β-estradiol, cholesterol, quercetin, morin, sodium acetate, acetic acid, ascorbic acid, β-glucuroronidase and sulfatase were purchased from Sigma-Aldrich (St. Louis, MO). Water and acetonitrile of HPLC grade were purchased from Fisher Scientific (Springfield, NJ). LC-MS/MS was performed on an Applied Biosystem/MSD Sciex (Foster City, CA) API 2000 QTrap mass spectrometer equipped with an Agilent 1100 HPLC system. All chromatographic separations were performed on a Nucleodur 100-3 C8 column (125 × 2.0 mm, Macherey-Nagel, Bethlehem, PA). AIN76A phytroestrogen-free diet was purchased from Dyets Inc., Bethlehem, PA. Quercetin in AIN76A diet as pellets was prepared by Dytes Inc.
Quercetin Treatment of ACI Rats and Tumor Induction
Female ACI rats (Harlan Sprague Dawley, Indianapolis, IN) were obtained at 4 weeks of age and housed in the Columbia University animal facility under controlled temperature, humidity, and lighting conditions. Animals were fed AIN76A phytoestrogen-free diet (Dyets Inc., Bethlehem, PA) and water was given ad libitum. After a one week acclimatization period, rats were randomly divided into two groups. One group (E2 + quercetin) was implanted with E2 pellets (3 mg E2 + 17 mg cholesterol, s.c.) as reported previously (Bhat et al., 2003; Mense et al., 2008b; Mense et al., 2009; Singh et al., 2009) and fed quercetin enriched AIN76A diet (2.5 g/kg food), while the control group was implanted with cholesterol pellets (17 mg cholesterol, s.c.) and fed quercetin enriched AIN76A diet. The dose of quercetin was chosen based on previous studies of phytoestrogen exposure in animal models of chemically-induced cancer (Verma et al., 1988; Zhu and Liehr, 1994). E2 and cholesterol pellets were prepared using a pellet press as described previously (Han and Liehr, 1994; Wang and Liehr, 1995; Bhat et al., 2003; Mense et al., 2008b; Mense et al., 2009; Singh et al., 2009). Quercetin treatment began 7 days prior to pellet implantation. The duration of the experiment was 240 days, during which animals were monitored for tumor development weekly. At the end of the experiment, animals were anesthetized using isoflurane and euthanized. Mammary (both tumor and normal), uterus, ovary, lung, brain, kidney and liver tissues were removed and snap-frozen in liquid nitrogen for future analyses. Frozen tissues were stored at -70 °C. A portion of the excised mammary and other tissues was stored in 10% buffered formalin for histopathologic analyses. Tumor incidence and the number of tumor nodules per rat were counted at the time of dissection. The formalin-fixed tissue was embedded in paraffin, and sections of 4 to 5 μm thickness were cut. Paraffin-embedded sections of the mammary, liver, brain, uterus, kidney, lung and ovary were stained with hematoxylin and eosin for histopathologic evaluation by a pathologist. Histopathologic analyses for tumor development and morphologic changes were performed on mammary tissue from all rats from the experimental and control groups.
LC-MS/MS Analysis of Quercetin in Serum Samples
Tuning to maximize sensitivity for precursor and product ions was performed using a syringe pump to infuse standard stock solution into an Applied Biosystem/MSD Sciex API 2000 QTRAP mass spectrometer equipped with a turbo ion electrospray source. Data acquisition and processing were performed using the Analyst 1.4.2 software package (Applied Biosystems). The m/z 303.0 --> 153.1 precursor ion/product ion transition (positive mode) was used for both quercetin and morin. Morin was used as an internal standard for quercetin quantitation (Wang et al., 2005). The standard Analyst quantitative optimization algorithms were run to optimize for LC-MS/MS sensitivity for both quercetin and morin. The optimal declustering potential (DP), entrance potential (EP), and collision energy (CE) were 65 V, 8 V and 30 V respectively for the detection of both quercetin and morin, using an ion spray voltage of 5,500 V and an ion spray temperature of 150 °C.
Chromatographic separation was performed on a Nucleodur 100-3 C8 column (125 × 2.0 mm) at a flow rate of 300 μL/min, with a gradient of 80% solvent A (0.1% formic acid in water) and 20% solvent B (0.1% formic acid in 30:70 water:acetonitrile) for 1 min, then 80% to 30% of solvent A and 20% to 70% of solvent B for next 6 min, then 0 to 100% of solvent C (0.095% formic acid in acetonitrile) for next 2 min, then 0 to 80% of solvent A and 0 to 20% of solvent B for next 1 min, and finally 80% solvent A and 20% solvent B for 4 min. Using these chromatographic conditions quercetin (analyte) and morin (internal standard) were well separated (retention times of 9.7 and 9.1 min respectively, with close to baseline separation).
This assay was then assessed for linearity, matrix (serum) effects, and lower limit of quantitation. A standard curve was prepared over a concentration range of 0.05 – 5 μg/mL using 7 different concentrations of quercetin in both rat serum and water (50 μL for each sample). To each of these samples 100 μL of a 2:1 mixture of acetonitrile:water + 0.1% formic acid was added. Samples were then centrifuged for 10 min at 13,500g and 4 °C to remove precipitated protein in the serum samples, and 100 μL of the clear supernatant removed to autosampler vials for LC-MS/MS analysis with an injection of 15 μL. The lower limit of quantitation was 0.05 μg/mL of quercetin in the original serum or water sample. Good linearity was observed over the range of 0.05 – 5 μg/mL (R2 > 0.99), and no substantial matrix effect was observed by comparison of the serum and water based standards. A similar study with morin, the internal standard in this study, also demonstrated good linearity and a lack of a substantial matrix effect. The percent coefficient of variation (%CV) for the morin internal standard was 7%, and for quercetin replicates in the calibration curve it was 4%.
Since quercetin is known to be glucuronidated and sulfated in serum, it was necessary to hydrolyze these conjugates to allow for quantitation (Hou et al., 2003). Briefly, 50 μL of a serum sample was added to 25 μL β-glucuroronidase (β-glucuroronidase 130.2 U/mL, sulfatase 4.3 U/mL in pH 5 sodium acetate buffer) and 5 μL ascorbic acid (300 mg/mL) in an eppendorf tube. Incubation was then conducted in a shaking water bath at 37 °C for 4 hours. After incubation, 100 μL of 150 nM of morin was added to each tube as an internal standard (in a 2:1 mixture of acetonitrile:water + 0.1% formic acid). The mixture was vortexed for 30 s and then centrifuged at 13,500g for 10 min at 4 °C, and a sample of the supernatant analyzed for quercetin as described above.
Western blot analysis
Approximately 50 mg ACI rat mammary and mammary tumor tissues were homogenized in a tissue protein extraction buffer (T-PER, Thermo Scientific, IL) with protease inhibitor cocktail (Sigma Chemicals, MO) using a PRO 200 rotor stator homogenizer (PRO Scientific, CT). After homogenization, samples were centrifuged at 10,000g and the clear supernatant was saved. The Pierce BCA Protein Assay kit was used to determine protein concentration of each sample (Pierce, Rockford IL). Eighty microgram total protein was size fractionated on a 12% SDS-polyacrylamide gel, and transferred onto a PVDF membrane (Millipore Corp., Billerica, MA) under standard conditions (Bhat and Epelboym, 2004; Mense et al., 2008a). PVDF membranes were blocked in 5% dry non-fat milk/PBS/0.5% Tween-20 at room temperature for 2 hours. Affinity purified rabbit polyclonal antibody against proliferating cell nuclear antigen (PCNA) (Santa Cruz Biotechnology, Santa Cruz, CA, sc-7907) was diluted 1:1500 in PBS/0.5% Tween-20 and used for immunodetection. After incubation overnight at room temperature with the primary antibody, membranes were washed four times for 10 min per wash using PBS/0.5% Tween-20. Horse radish peroxidase-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA, sc-2004) was diluted 1:2000 in PBS/0.5% Tween-20 and used as a secondary antibody. After incubation with secondary antibody for 2 hours at room temperature, the membranes were washed again as described above. Chemiluminescent detection was performed using the BM Chemiluminescence Detection kit (Roche, Indianapolis, IN) and Alpha Innotech FluorChem HD2 (Alpha Innotech, San Leandro, CA) gel documentation system with AlphaEase FC StandAlone software (version 6.0.0.14; Alpha Innotech). Membranes probed for PCNA were washed in PBS/0.5% Tween-20 and reprobed with alpha-tubulin monoclonal rabbit antibody (Cell Signaling Technology, Danvers, MA, cat # 2144) using the method described above.
Quantification of COMT activity
Microsomal proteins were prepared from ACI rat mammary and liver tissues and used for quantification of COMT activity using method as described previously (Bock and Matern, 1973; Bai et al., 2007). Briefly, the reaction mixture for quantification of COMT activity consisted of 1 mM 1,4-dithiothreitol, 1.2 mM MgCl2 and 100 μM S-Adenosyl methionine (AdoMet) (containing 1 μCi [methyl-3H] AdoMet) in Tris-HCl buffer (50 mM, pH 7.4). Fourty micomole 4-hydroxy estradiol (4-OHE2) was used as substrate in the reaction. The reaction was initiated by the addition of 100 μg of microsomal protein to the reaction mixture and the reaction carried out at 37 °C for 30 min. The final volume of the reaction mixture was 300 μL. The reaction was arrested by immediately placing the tubes on ice, followed by the addition of 500 μL ice-cold double distilled water. The reaction mixtures were extracted with 2 mL n-heptane for the methylated catechol products. After centrifugation at 2000 rpm for 5 min, 1 mL of aqueous phase was transferred into scintillation tube containing 9 mL of scintillation fluid (PerkinElmer, Waltham, MA) and radioactivity content was measured with liquid scintillation analyzer (Packard Tri-CARB 2900TR; Downers Grove, IL). The rate of methylation of 4-OHE2 was expressed as nmol of methylated product formed/mg of microsomal protein/min.
8-iso-PGF2α Quantification
Total 8-iso-PGF2α levels in the mammary tissue of female ACI rats were quantified using a direct 8-iso-PGF2α enzyme immunoassay kit obtained from Assay Designs (Ann Arbor, MI) according to the supplier's instructions as described previously (Bhat et al., 2003). Fifty to 100 mg rat mammary tissue was homogenized in cold PBS (pH 7.4) containing 0.005% butylated hydroxytolulene. Tissue homogenates were prepared in 2 mL round-bottom microcentrifuge tubes by using a TissueLyser (Qiagen, Valencia CA). Homogenization was carried out in the TissueLyser at 29 cycles per second for 4 min. Protein concentrations from the neutralized homogenates were determined using the Pierce BCA protein assay kit (Pierce, Rockford IL). 8-iso-PGF2α esters in 100 μL of the total mammary homogenate were hydrolyzed by incubation with 25 μL of 10 N NaOH at 45 °C for 2 hours. The reaction mixture was cooled on ice for 5 min, neutralized with 25 μL of 12 N HCl and centrifuged in a microcentrifuge for 5 min at 4 °C. The clear neutralized supernatant was transferred into a new microcentrifuge tube and 50 μL of the hydrolyzed/neutralized sample was used for the 96-well format 8-iso-PGF2α assay. Samples were incubated with the 8-iso-PGF2α antibody for 18 hours at 4 °C. After incubation, the contents of the wells were emptied and washed with wash buffer. Wash buffer was then removed from the wells and the color was developed by incubation with 200 μL of p-nitrophenyl phosphate for 45 min at room temperature. The reaction was stopped by the addition of 50 μL of stop solution and the plate was read at 405 nm. Standard curves were run on each plate and were generated by measuring the optical density of 160-100,000 pg/mL of 8-iso-PGF2α standards that were processed simultaneously with the unknown samples. Data are expressed as mean 8-iso-PGF2α pg/mg protein ± standard error of the mean. Fold changes were calculated by comparing 8-iso-PGF2α levels detected in the mammary tissue of E2-exposed animal tissues to levels in the mammary tissue of age-matched control animals.
Statistics and Data Analysis
All data were analyzed using Sigma Plot 8.0 software (Systat Software, San Jose CA). Fisher's exact test was used to compare tumor incidences between the E2 and E2 + quercetin treatment groups, or between a treatment group and its control group. The average number of tumor nodules per tumor-bearing animal was calculated by dividing the sum of the tumor nodules in all tumor-bearing animals by the total number of tumor-bearing animals. The average number of tumor nodules per rat is expressed as the mean ± the standard error of the mean. After calculating Kaplan-Meier survival curve of tumor occurrence for latency, we used log-rank test to detect differences in the survival curves between the E2 and E2 + quercetin treatment groups (Mense et al., 2008b; Mense et al., 2009; Singh et al., 2009). The two sample T-test was used to detect group differences in quantitative variables, including tumor multiplicity (defined as number of tumor nodules per tumor-bearing rat), specific fold changes in 8-iso-PGF2α levels, serum quercetin levels, COMT activity levels and PCNA protein levels. p values ≤0.05 were considered significant.
Results
Breast Morphology and Proliferation in Quercetin-Treated ACI Rats
No morphologic changes were detected in the mammary tissue of quercetin-treated ACI rats relative to those of controls (Figure 1). Mammary tissue from all animals in the quercetin group displayed normal lobular architecture, consisting of ducts surrounded by small lobules (Figure 1). Histopathologic analysis of mammary tissue from all rats in the quercetin + E2 experimental group revealed hyperplastic lobular units and intraductal proliferation (Figure 1), both of which are consistent with morphologic changes present in rats treated with E2 (Mense et al., 2008b). Both ductal carcinoma in situ (DCIS) and micro-invasive cancers were present in the mammary tissue of animals from the quercetin + E2 group (Figure 2). These tumor characteristics are similar to E2-induced breast tumors (Makris et al., 1997; Arnerlov et al., 2001; Li et al., 2002a; Li et al., 2002b; Li et al., 2004; Weroha et al., 2006). Breast tissue from ACI rats exposed to dietary quercetin for 8 months was similar to that of controls and did not exhibit increased proliferation (Figure 1). This observation was further confirmed with PCNA immunoblot that showed no significant change in PCNA levels between control and quercetin exposed breast (Figure 3). However, PCNA expression was significantly upregulated in E2 and quercetin + E2 exposed breast and in breast tumors compared to control breast (Figure 3). PCNA is an established marker for proliferating cells (Connolly and Bogdanffy, 1993). No significant morphological differences were observed between ovary, kidney, uterus or liver tissues from rats in the quercetin and quercetin + E2 groups (data not shown). In one instance, squamous metaplasia was observed in the uterus of a quercetin + E2-treated rat (data not shown).
Figure 1. Mammary Tissue Morphology in Control, E2 and Quercetin-Treated ACI Rats.
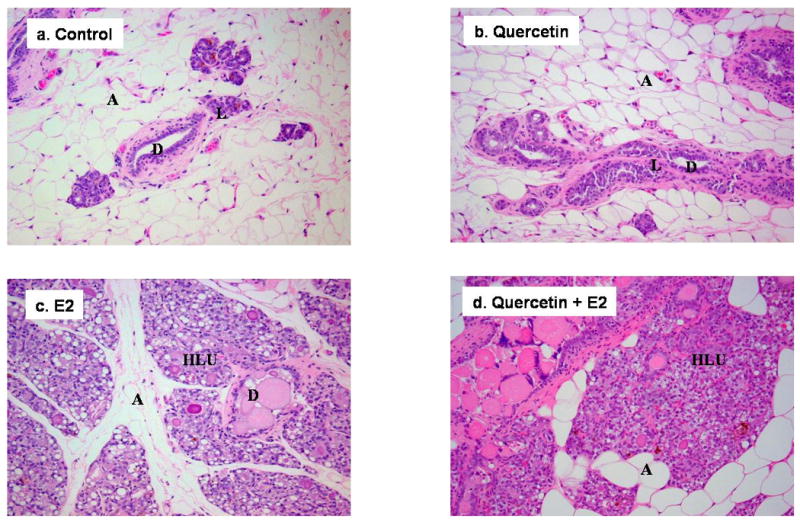
Female ACI rats were treated with E2, quercetin, E2 + quercetin for 240 days. Animals in the E2 and E2 + quercetin groups were implanted with E2 pellets (s.c., 3 mg E2 + 17 mg cholesterol). Control and quercetin-treated rats were implanted with pellets containing 17 mg cholesterol only. Quercetin and quercetin + E2-treated rats were given quercetin via diet (2.5 g/kg food). a-b) The mammary glands of ACI rats in control or quercetin group show normal lobular architecture (L) with branched ducts (D) and normal distribution of fat/adipose tissue (A) (all panels 100×). c-d) Mammary glands from rats in the E2 and quercetin + E2 groups show increased proliferation with dilated ducts containing inspissated secretions (D) and increased proliferation and expansion of terminal lobular units (HLU) accompanied by compression of and expansion into the surrounding fat/adipose tissue (A) (all panels 100×).
Figure 2. Mammary Tumors in Quercetin + E2-Treated ACI Rats.
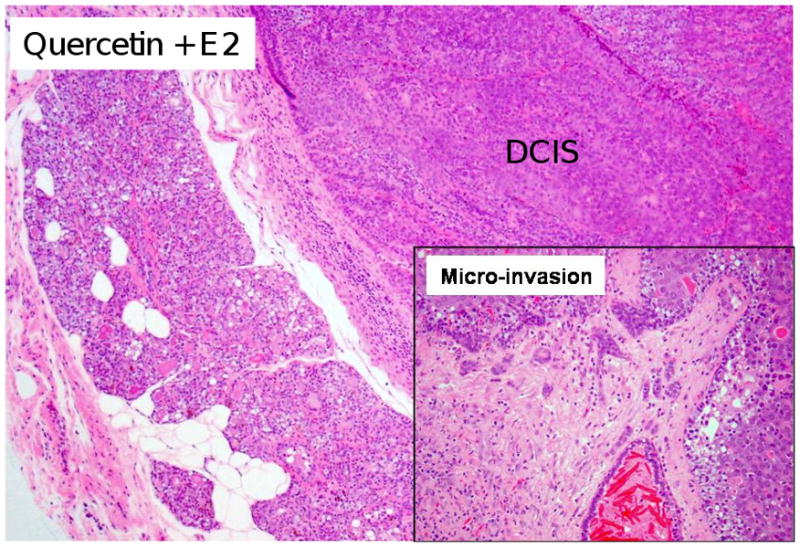
Female ACI rats were treated with E2 + quercetin for 240 days as described in figure 1. The mammary tissue from rats treated with quercetin + E2 show ductal carcinoma in situ (DCIS) (40×). Inset) Invasive adenocarcinoma was observed in the mammary tissue of quercetin + E2-treated rats (100×).
Figure 3. Western blot analysis of PCNA in ACI rat mammary and mammary tumors.
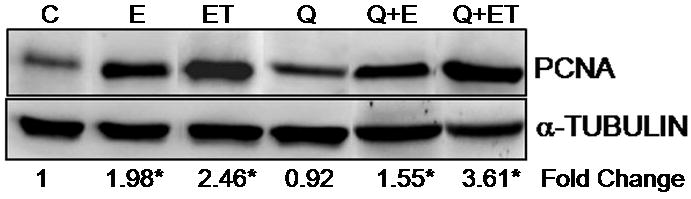
Female ACI rats were treated with E2, quercetin or E2 + quercetin for 240 days as detailed in figure 1. At the end of the experiment, mammary and mammary tumor tissues were collected. Tissues were homogenized and approximately 80 μg protein was used for western blot analysis. Membrane was probed with antibody against PCNA, stripped and re-probed for alpha-tubulin. Fold change compared to control mammary was calculated for atleast 3 different animals in each group. * indicates a p value <0.05 compared to control mammary (C, E, ET, Q, Q+E and Q+ET represent control mammary, E2 mammary, E2 tumor, quercetin mammary, quercetin + E2 mammary and quercetin + E2 tumor respectively).
Serum Quercetin Levels did not Change in Quercetin and Quercetin + E2-treated ACI rats
Rats were fed a quercetin enriched AIN76A diet (2.5g/kg food) for the entire duration of experiment. Food intake in quercetin and quercetin + E2-treated group did not differ significantly. Combined mean food intake in each group was ∼ 20 g/rat per day. The LC-MS/MS analysis was used to determine total quercetin (parent quercetin and the quercetin after enzymatic hydrolysis with β-glucuronidase/sulfatase) in rat serum samples following the ingestion of quercetin enriched diet. The serum quercetin levels in quercetin + E2-treated rats did not differ significantly from the serum levels in quercetin group. The serum quercetin levels were 2.01 ± 0.21 μg/mL or 6.68 ± 0.70 μmol/L and 1.88 ± 0.24 μg/mL or 6.24 ± 0.78 μmol/L (mean ± SEM; n = 5, p = 0.706) respectively in quercetin and quercetin + E2-treated group. Quercetin levels were undetected in control group rats fed with phytroestrogen-free AIN76A diet.
Quercetin Exacerbates Breast Cancer Development in ACI Rats
At the dose used in the experimental conditions (2.5 g/kg food), quercetin did not confer protection against estrogen-induced breast cancer development. No tumors were observed in control animals treated with quercetin alone; however, tumor incidence reached 100% in rats co-treated with quercetin and E2 (Table 1). Rats in the quercetin + E2 group displayed an increased mammary tumor incidence relative to animals from the E2 treatment group, where tumor incidence was equal to 82% (Table 1). However, the increase in tumor incidence in the quercetin + E2 group compared to the E2 group was not statistically significant. Average tumor multiplicity was equal to 3.4 ± 0.7 in rats from the quercetin + E2 group (Table 1). This represents a non-significant increase relative to rats in the E2 group, where a tumor multiplicity of 3.1 ± 0.7 was observed. According to Kaplan-Meier survival curve analysis, average tumor latency was significantly shorter for animals in the quercetin + E2 group versus those in the E2 group, indicating that breast tumors appeared earlier in quercetin + E2-treated animals relative to animals exposed to E2 only (Figure 4).
Table 1. Breast Tumor Incidence and Multiplicity by Treatment Group.
| Treatment | n | Tumor Incidence (%) | Tumor Multiplicity |
|---|---|---|---|
| Control | 10 | 0 | N.A |
| E2 | 11 | 82 | 3.1 ± 0.7 |
| Quercetin | 6 | 0 | N.A |
| Quercetin + E2 | 10 | 100 | 3.4 ± 0.7 |
Columns one lists the different treatments each group of animals received and column two lists the number of animals in each group. The percentage of animals that developed tumors during the 240 day treatment period (tumor incidence) is listed in column three. Column four shows the number of tumors per tumor bearing rat (tumor multiplicity ± SEM).
Figure 4. Quercetin Decreases the Latency of E2-Induced Mammary Tumors in ACI Rats.
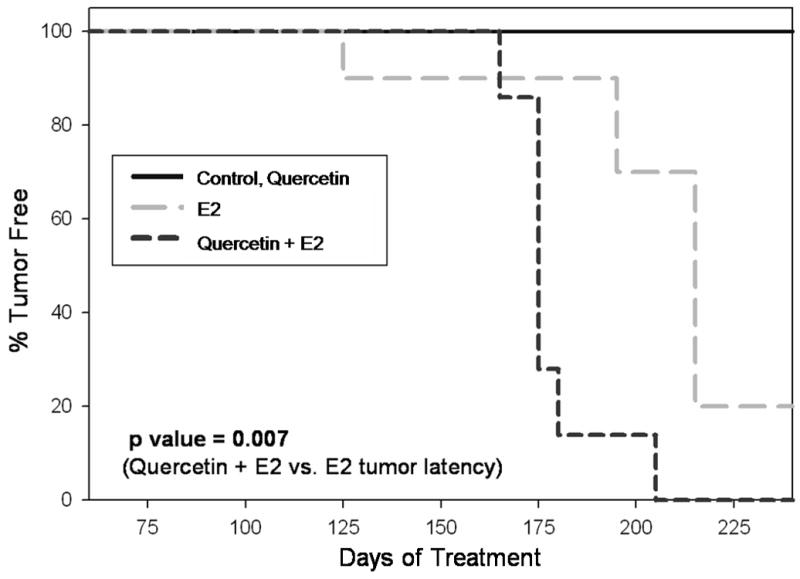
Female ACI rats were treated with E2, quercetin and E2 + quercetin as described in figure 1. Kaplan-Meier survival curves for tumor occurrence were plotted for each treatment group, and the log rank test was used to detect differences in the tumor latency curves between groups. Animals in the control and quercetin group did not develop any tumors and are represented by the same line on the graph. Tumor latency was significantly shorter in the E2 + quercetin group compared to the E2 group (p = 0.007).
Quercetin inhibits COMT activity in ACI Rats
COMT enzyme activity was evaluated in microsomes of the mammary and liver tissue of control, E2-, quercetin- and quercetin + E2-treated ACI rats (Table 2). In breast, 2 and 4-hydroxylation of E2 are the main pathways of estrogen metabolism. COMT detoxifies the catechols (2- and 4-hydroxy estradiol) to form 2- and 4-methoxyestradiol (2-MeOE2 and 4-MeOE2), which are hormonally inactive and more quickly excreted. COMT enzyme activity was increased in mammary tissue from rats in the E2 group relative to control group (Table 2) but significantly downregulated in quercetin and quercetin + E2-treated group mammary compared to rats in the control group (Table 2). Furthermore, the downregulation of COMT activity was mammary specific. COMT activity did not change significantly in liver tissue of any treatment group compared to control liver (Table 2).
Table 2. COMT enzyme activity in ACI rat mammary and liver tissues.
| Treatment and Tissue | COMT Activity (nmol/mg protein/min) |
Fold Change |
|---|---|---|
| Control-mammary | 0.095 ± 0.009 | 1 |
| E2-treated mammary | 0.22 ± 0.029 | 2.31* |
| Quercetin-treated mammary | 0.043 ± 0.015 | 0.46* |
| Quercetin + E2-treated mammary | 0.038 ± 0.015 | 0.39* |
| Control-liver | 5.48 ± 0.751 | 1 |
| E2-treated liver | 4.59 ± 0.285 | 0.84 |
| Quercetin-treated liver | 3.37 ± 0.176 | 0.61 |
| Quercetin + E2-treated liver | 4.36 ± 0.463 | 0.79 |
Activity of the COMT enzyme was measured in mammary and liver tissues from control, E2, quercetin and quercetin + E2-treated rats after 240 days of treatment. Column one lists the treatment each group of animals received and organs in which COMT enzyme activity was quantified. Column two shows the COMT activity as an average of values obtained for at least 4 different animals ± SEM. Column three shows the fold change in COMT enzyme activity compared to the respective control tissue.
Indicates a p value <0.05 compared to respective control tissue.
8-iso-PGF2α Formation in Quercetin-Treated ACI Rats
Oxidative stress in the mammary tissue of ACI rats was evaluated by quantification of 8-iso-PGF2α, an established marker of oxidative stress in vivo (Morrow et al., 1990; Pratico et al., 1995; Bhat et al., 2003; Mense et al., 2008b; Mense et al., 2009; Singh et al., 2009). 8-iso-PGF2α levels were significantly increased (approximately 8-fold) in the breast tissue of rats co-treated with quercetin + E2 for 240 days relative to age-matched quercetin-treated rats (Figure 5). In rats treated with E2 only, a 5-fold increase in 8-iso-PGF2α levels was observed relative to age-matched cholesterol-treated controls. While the fold increase in 8-iso-PGF2α was larger for animals in the quercetin + E2 group compared to the E2 group, the difference was not statistically significant (Figure 5).
Figure 5. 8-iso-PGF2α Formation in E2 and Quercetin + E2-Treated ACI Rats.
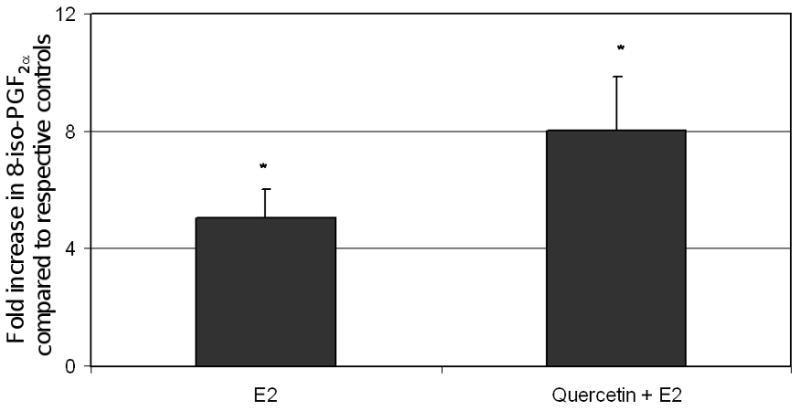
Female ACI rats were treated with E2, quercetin or E2 + quercetin as described in figure 1. 8-iso-PGF2α levels were measured in mammary tissue from animals in each of these groups. Fold changes were calculated for E2-treated animals relative to age-matched controls. Fold changes were calculated for E2 + quercetin -treated animals relative to age-matched quercetin-treated controls. These data are reported as an average of values obtained for at least 6 different animals ± SEM. * Indicates a p value <0.05 compared to age-matched controls.
Discussion
Dietary quercetin (2.5 g/kg food) did not induce mammary tumors in ovary-intact female ACI rats (Table 1). Histopathologic analysis and PCNA expression suggest no changes in cell proliferation and morphology in the mammary tissue of quercetin-treated ACI rats relative to those of controls (Figures 1 and 3). However, in ACI rats implanted with E2 pellets, co-exposure to dietary quercetin enhanced cell proliferation and breast tumor development (Table 1, Figures 2 and 3). One hundred percent of animals co-treated with quercetin + E2 developed mammary tumors during the 240 day experiment (Table 1). Compared to the 82% tumor incidence observed in animals exposed to E2, animals from the quercetin + E2 treatment group displayed an increased tumor incidence, however the difference in tumor incidences was not statistically significant (Table 1, p = 0.15). The trend was similar for tumor multiplicity, where animals from the quercetin + E2 group had more tumors on average than animals exposed to E2, but the difference between tumor multiplicities was not statistically significant (Table 1). However, tumor latency was significantly decreased among rats from the quercetin + E2 group relative to those in the E2 treatment group, indicating that quercetin treatment causes estrogen-induced breast tumors to appear more rapidly (Figure 4). COMT activity in mammary was significantly decreased among rats from quercetin and quercetin + E2 group relative to those in the control group, indicating that quercetin treatment downregulated COMT activity (Table 2). E2 co-treatment did not have any effect on quercetin metabolism as serum quercetin levels in quercetin + E2-treated group were same as in quercetin alone treated group. Previous study has also reported the equivalent levels of plasma quercetin (14 μmol/L vs. 6.7 μmol/L in our study) in rats fed with nearly same amount of quercetin (58.5 mg or 194 μmol vs. 50 mg or 166 μmol in our study) as daily ingested dose (Graf et al., 2006). Moreover, quercetin administration did not decrease lipid peroxidation in ACI rats. In fact, fold increases in 8-iso-PGF2α in the quercetin + E2 group were larger than those in the E2 group, suggesting that quercetin does not prevent lipid peroxidation (Figure 5). Taken together, these data indicate that dietary quercetin (2.5 g/kg food) does not confer protection against estrogen-induced breast cancer in female ACI rats.
The observation that administration of quercetin alone was not sufficient to induce breast cancers in ACI rats indicates that quercetin does not directly initiate carcinogenesis in this animal model. However, quercetin substantially decreased the latency of estrogen-induced breast cancers in female ACI rats, illustrating that quercetin potentiates the effect of E2 on breast carcinogenesis. One possible explanation for the effect of quercetin on tumor latency may be that quercetin promotes growth of malignant cells transformed by estrogen. While quercetin, like many other phytoestrogens, has a higher affinity for estrogen receptor β (ERβ) than ERα, this phytoestrogen exerts estrogenic effects on cell proliferation if concentrations are sufficient (Kuiper et al., 1998; Rodgers and Grant, 1998; Balabhadrapathruni et al., 2000; Harris et al., 2005; van der Woude et al., 2005). If malignant clones are capable of faster growth in the presence of quercetin, tumor formation would occur more rapidly, thus decreasing the time needed for appearance of palpable breast tumors. However, no proliferative changes were detected in mammary tissue from animals treated with only quercetin based on histopathologic evaluation and PCNA expression (Figures 1 and 3). These results suggest that quercetin probably has only a weak effect on proliferation in normal breast tissue, but may potentiate the mitogenic effects of E2.
Increased severity of estrogen-induced tumors in quercetin-treated rats may be related to the effect of quercetin on COMT enzyme activity. COMT catalyzes the methylation of hydroxylated estrogen metabolites to hormonally-inactive methylated products, including 4-MeOE2 and 2-MeOE2 (Zhu, 2002). Methylation of catechol estrogens by COMT is thought to be a detoxification reaction, reducing exposure to highly carcinogenic estrogen metabolites such as 4- hydroxyl estradiol (4-OHE2) (Harris et al., 2005). COMT enzyme activity in quercetin and quercetin + E2-treated mammary was significantly decreased compared to control group mammary, indicating that quercetin treatment downregulated COMT activity (Table 2). Moreover, quercetin co-treatment with E2 overcomes E2-mediated increase in COMT enzyme activity (Table 2), thereby increased exposure to catechol estrogens and reduced detoxification to their methylated products. Quercetin mediated COMT inhibition has been previously shown in the kidneys of estrogen-treated Syrian hamsters, resulting in accumulation of 4-OHE2 and more severe kidney tumors than those present in rats exposed to only E2 (Zhu and Liehr, 1994; Zhu and Liehr, 1996). Similarly, other studies have found that quercetin treatment of cytosolic proteins from human mammary tissues decreased COMT activity, and inhibited COMT-mediated detoxification of 4-OHE2 in MCF7 breast cancer cells (Nagai et al., 2004; van Duursen et al., 2004; Lehmann et al., 2008). The effect of COMT inhibition by quercetin may be two-fold, as protective effects have been ascribed to some methylated estrogen metabolites (Zhu and Conney, 1998; Brueggemeier et al., 2001; LaVallee et al., 2002; Schumacher and Neuhaus, 2006). 2-MeOE2, the methylated product of 2-hydroxy estradiol (2-OHE2), has been shown to inhibit angiogenesis and increase apoptosis both in vitro and in vivo (Zhu and Liehr, 1996; Nagai et al., 2004; van Duursen et al., 2004; Lehmann et al., 2008). Thus, by decreasing methylation of catechol estrogens by COMT, quercetin may increase concentrations of the highly carcinogenic 4-OHE2 while simultaneously minimizing levels of 2-MeOE2, an estrogen metabolite with anti-cancer properties. The alteration in COMT activity may thus result in the buildup of estrogen and its carcinogenic metabolites in the target organ and thus enhances the possibility of cancer development through DNA damage and the associated processes. It is interesting to note that quercetin does not alter COMT activity in liver (table 2), thus this effect on COMT seems to be target-organ specific.
Our results provide evidence that exposure to dietary quercetin (2.5 g/kg food), even when consumption begins early in life, does not confer any protective effect against breast carcinogenesis. In contrast, it appears that quercetin may actually exacerbate E2-associated mammary carcinogenesis. Quercetin exposure dramatically decreased the latency of E2-induced breast tumors, and shows trends toward increasing tumor incidence and multiplicity. Inhibition of COMT activity by quercetin may be one of the mechanisms responsible for promotion of estrogen-induced carcinogenesis by quercetin.
Acknowledgments
This work was supported by the National Institutes of Health Grants ES009089 and CA 109551 (HKB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allred CD, Allred KF, Ju YH, Clausen LM, Doerge DR, Schantz SL, Korol DL, Wallig MA, Helferich WG. Dietary genistein results in larger MNU-induced, estrogen-dependent mammary tumors following ovariectomy of Sprague-Dawley rats. Carcinogenesis. 2004;25:211–218. doi: 10.1093/carcin/bgg198. [DOI] [PubMed] [Google Scholar]
- Arnerlov C, Emdin SO, Cajander S, Bengtsson NO, Tavelin B, Roos G. Intratumoral variations in DNA ploidy and s-phase fraction in human breast cancer. Anal Cell Pathol. 2001;23:21–28. doi: 10.1155/2001/430674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai HW, Shim JY, Yu J, Zhu BT. Biochemical and molecular modeling studies of the O-methylation of various endogenous and exogenous catechol substrates catalyzed by recombinant human soluble and membrane-bound catechol-O-methyltransferases. Chem Res Toxicol. 2007;20:1409–1425. doi: 10.1021/tx700174w. [DOI] [PubMed] [Google Scholar]
- Balabhadrapathruni S, Thomas TJ, Yurkow EJ, Amenta PS, Thomas T. Effects of genistein and structurally related phytoestrogens on cell cycle kinetics and apoptosis in MDA-MB-468 human breast cancer cells. Oncol Rep. 2000;7:3–12. [PubMed] [Google Scholar]
- Banerjee S, Bueso-Ramos C, Aggarwal BB. Suppression of 7,12-dimethylbenz(a)anthracene-induced mammary carcinogenesis in rats by resveratrol: role of nuclear factor-kappaB, cyclooxygenase 2, and matrix metalloprotease 9. Cancer Res. 2002;62:4945–4954. [PubMed] [Google Scholar]
- Bhat HK, Calaf G, Hei TK, Loya T, Vadgama JV. Critical role of oxidative stress in estrogen-induced carcinogenesis. Proc Natl Acad Sci U S A. 2003;100:3913–3918. doi: 10.1073/pnas.0437929100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat HK, Epelboym I. Suppression of calbindin D28K in estrogen-induced hamster renal tumors. J Steroid Biochem Mol Biol. 2004;92:391–398. doi: 10.1016/j.jsbmb.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Bhat KP, Lantvit D, Christov K, Mehta RG, Moon RC, Pezzuto JM. Estrogenic and antiestrogenic properties of resveratrol in mammary tumor models. Cancer Res. 2001;61:7456–7463. [PubMed] [Google Scholar]
- Bock KW, Matern S. Immunochemical studies on rat-liver microsomal NAD glycohydrolase. Eur J Biochem. 1973;38:20–24. doi: 10.1111/j.1432-1033.1973.tb03027.x. [DOI] [PubMed] [Google Scholar]
- Brooks JD, Thompson LU. Mammalian lignans and genistein decrease the activities of aromatase and 17beta-hydroxysteroid dehydrogenase in MCF-7 cells. J Steroid Biochem Mol Biol. 2005;94:461–467. doi: 10.1016/j.jsbmb.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Brueggemeier RW, Bhat AS, Lovely CJ, Coughenour HD, Joomprabutra S, Weitzel DH, Vandre DD, Yusuf F, Burak WE., Jr 2-Methoxymethylestradiol: a new 2-methoxy estrogen analog that exhibits antiproliferative activity and alters tubulin dynamics. J Steroid Biochem Mol Biol. 2001;78:145–156. doi: 10.1016/s0960-0760(01)00090-5. [DOI] [PubMed] [Google Scholar]
- Connolly KM, Bogdanffy MS. Evaluation of proliferating cell nuclear antigen (PCNA) as an endogenous marker of cell proliferation in rat liver: a dual-stain comparison with 5-bromo-2′-deoxyuridine. J Histochem Cytochem. 1993;41:1–6. doi: 10.1177/41.1.7678022. [DOI] [PubMed] [Google Scholar]
- Constantinou AI, Lantvit D, Hawthorne M, Xu X, van Breemen RB, Pezzuto JM. Chemopreventive effects of soy protein and purified soy isoflavones on DMBA-induced mammary tumors in female Sprague-Dawley rats. Nutr Cancer. 2001;41:75–81. doi: 10.1080/01635581.2001.9680615. [DOI] [PubMed] [Google Scholar]
- Fritz WA, Coward L, Wang J, Lamartiniere CA. Dietary genistein: perinatal mammary cancer prevention, bioavailability and toxicity testing in the rat. Carcinogenesis. 1998;19:2151–2158. doi: 10.1093/carcin/19.12.2151. [DOI] [PubMed] [Google Scholar]
- Galati G, O'Brien PJ. Potential toxicity of flavonoids and other dietary phenolics: significance for their chemopreventive and anticancer properties. Free Radic Biol Med. 2004;37:287–303. doi: 10.1016/j.freeradbiomed.2004.04.034. [DOI] [PubMed] [Google Scholar]
- Graf BA, Ameho C, Dolnikowski GG, Milbury PE, Chen CY, Blumberg JB. Rat gastrointestinal tissues metabolize quercetin. J Nutr. 2006;136:39–44. doi: 10.1093/jn/136.1.39. [DOI] [PubMed] [Google Scholar]
- Han X, Liehr JG. DNA single-strand breaks in kidneys of Syrian hamsters treated with steroidal estrogens: hormone-induced free radical damage preceding renal malignancy. Carcinogenesis. 1994;15:997–1000. doi: 10.1093/carcin/15.5.997. [DOI] [PubMed] [Google Scholar]
- Harris DM, Besselink E, Henning SM, Go VL, Heber D. Phytoestrogens induce differential estrogen receptor alpha- or Beta-mediated responses in transfected breast cancer cells. Exp Biol Med (Maywood) 2005;230:558–568. doi: 10.1177/153537020523000807. [DOI] [PubMed] [Google Scholar]
- Harvell DM, Strecker TE, Tochacek M, Xie B, Pennington KL, McComb RD, Roy SK, Shull JD. Rat strain-specific actions of 17beta-estradiol in the mammary gland: correlation between estrogen-induced lobuloalveolar hyperplasia and susceptibility to estrogen-induced mammary cancers. Proc Natl Acad Sci U S A. 2000;97:2779–2784. doi: 10.1073/pnas.050569097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilakivi-Clarke L, Onojafe I, Raygada M, Cho E, Skaar T, Russo I, Clarke R. Prepubertal exposure to zearalenone or genistein reduces mammary tumorigenesis. Br J Cancer. 1999;80:1682–1688. doi: 10.1038/sj.bjc.6690584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodek P, Trefil P, Stiborova M. Flavonoids-potent and versatile biologically active compounds interacting with cytochromes P450. Chem Biol Interact. 2002;139:1–21. doi: 10.1016/s0009-2797(01)00285-x. [DOI] [PubMed] [Google Scholar]
- Hou YC, Chao PD, Ho HJ, Wen CC, Hsiu SL. Profound difference in pharmacokinetics between morin and its isomer quercetin in rats. J Pharm Pharmacol. 2003;55:199–203. doi: 10.1211/002235702487. [DOI] [PubMed] [Google Scholar]
- Jin Z, MacDonald RS. Soy isoflavones increase latency of spontaneous mammary tumors in mice. J Nutr. 2002;132:3186–3190. doi: 10.1093/jn/131.10.3186. [DOI] [PubMed] [Google Scholar]
- Kijkuokool P, Parhar IS, Malaivijitnond S. Genistein enhances N-nitrosomethylurea-induced rat mammary tumorigenesis. Cancer Lett. 2006;242:53–59. doi: 10.1016/j.canlet.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B, Gustafsson JA. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology. 1998;139:4252–4263. doi: 10.1210/endo.139.10.6216. [DOI] [PubMed] [Google Scholar]
- Lamartiniere CA, Zhang JX, Cotroneo MS. Genistein studies in rats: potential for breast cancer prevention and reproductive and developmental toxicity. Am J Clin Nutr. 1998;68:1400S–1405S. doi: 10.1093/ajcn/68.6.1400S. [DOI] [PubMed] [Google Scholar]
- LaVallee TM, Zhan XH, Herbstritt CJ, Kough EC, Green SJ, Pribluda VS. 2-Methoxyestradiol inhibits proliferation and induces apoptosis independently of estrogen receptors alpha and beta. Cancer Res. 2002;62:3691–3697. [PubMed] [Google Scholar]
- Le Corre L, Chalabi N, Delort L, Bignon YJ, Bernard-Gallon DJ. Resveratrol and breast cancer chemoprevention: molecular mechanisms. Mol Nutr Food Res. 2005;49:462–471. doi: 10.1002/mnfr.200400094. [DOI] [PubMed] [Google Scholar]
- Lehmann L, Jiang L, Wagner J. Soy isoflavones decrease the catechol-O-methyltransferase-mediated inactivation of 4-hydroxyestradiol in cultured MCF-7 cells. Carcinogenesis. 2008;29:363–370. doi: 10.1093/carcin/bgm235. [DOI] [PubMed] [Google Scholar]
- Li JJ, Papa D, Davis MF, Weroha SJ, Aldaz CM, El-Bayoumy K, Ballenger J, Tawfik O, Li SA. Ploidy differences between hormone- and chemical carcinogen-induced rat mammary neoplasms: comparison to invasive human ductal breast cancer. Mol Carcinog. 2002a;33:56–65. doi: 10.1002/mc.10022. [DOI] [PubMed] [Google Scholar]
- Li JJ, Weroha SJ, Lingle WL, Papa D, Salisbury JL, Li SA. Estrogen mediates Aurora-A overexpression, centrosome amplification, chromosomal instability, and breast cancer in female ACI rats. Proc Natl Acad Sci U S A. 2004;101:18123–18128. doi: 10.1073/pnas.0408273101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SA, Weroha SJ, Tawfik O, Li JJ. Prevention of solely estrogen-induced mammary tumors in female aci rats by tamoxifen: evidence for estrogen receptor mediation. J Endocrinol. 2002b;175:297–305. doi: 10.1677/joe.0.1750297. [DOI] [PubMed] [Google Scholar]
- Limer JL, Parkes AT, Speirs V. Differential response to phytoestrogens in endocrine sensitive and resistant breast cancer cells in vitro. Int J Cancer. 2006;119:515–521. doi: 10.1002/ijc.21863. [DOI] [PubMed] [Google Scholar]
- Makris A, Allred DC, Powles TJ, Dowsett M, Fernando IN, Trott PA, Ashley SE, Ormerod MG, Titley JC, Osborne CK. Cytological evaluation of biological prognostic markers from primary breast carcinomas. Breast Cancer Res Treat. 1997;44:65–74. doi: 10.1023/a:1005717924761. [DOI] [PubMed] [Google Scholar]
- Mense SM, Chhabra J, Bhat HK. Preferential induction of cytochrome P450 1A1 over cytochrome P450 1B1 in human breast epithelial cells following exposure to quercetin. J Steroid Biochem Mol Biol. 2008a;110:157–162. doi: 10.1016/j.jsbmb.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mense SM, Remotti F, Bhan A, Singh B, El-Tamer M, Hei TK, Bhat HK. Estrogen-induced breast cancer: alterations in breast morphology and oxidative stress as a function of estrogen exposure. Toxicol Appl Pharmacol. 2008b;232:78–85. doi: 10.1016/j.taap.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mense SM, Singh B, Remotti F, Liu X, Bhat HK. Vitamin C and alpha-naphthoflavone prevent estrogen-induced mammary tumors and decrease oxidative stress in female ACI rats. Carcinogenesis. 2009;30:1202–1208. doi: 10.1093/carcin/bgp093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina M, McCaskill-Stevens W, Lampe JW. Addressing the soy and breast cancer relationship: review, commentary, and workshop proceedings. J Natl Cancer Inst. 2006;98:1275–1284. doi: 10.1093/jnci/djj356. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ., 2nd A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci U S A. 1990;87:9383–9387. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrill WB, Brown NM, Zhang JX, Manzolillo PA, Barnes S, Lamartiniere CA. Prepubertal genistein exposure suppresses mammary cancer and enhances gland differentiation in rats. Carcinogenesis. 1996;17:1451–1457. doi: 10.1093/carcin/17.7.1451. [DOI] [PubMed] [Google Scholar]
- Nagai M, Conney AH, Zhu BT. Strong inhibitory effects of common tea catechins and bioflavonoids on the O-methylation of catechol estrogens catalyzed by human liver cytosolic catechol-O-methyltransferase. Drug Metab Dispos. 2004;32:497–504. doi: 10.1124/dmd.32.5.497. [DOI] [PubMed] [Google Scholar]
- Nichenametla SN, Taruscio TG, Barney DL, Exon JH. A review of the effects and mechanisms of polyphenolics in cancer. Crit Rev Food Sci Nutr. 2006;46:161–183. doi: 10.1080/10408390591000541. [DOI] [PubMed] [Google Scholar]
- Peeters PH, Keinan-Boker L, van der Schouw YT, Grobbee DE. Phytoestrogens and breast cancer risk. Review of the epidemiological evidence. Breast Cancer Res Treat. 2003;77:171–183. doi: 10.1023/a:1021381101632. [DOI] [PubMed] [Google Scholar]
- Pike MC, Kolonel LN, Henderson BE, Wilkens LR, Hankin JH, Feigelson HS, Wan PC, Stram DO, Nomura AM. Breast cancer in a multiethnic cohort in Hawaii and Los Angeles: risk factor-adjusted incidence in Japanese equals and in Hawaiians exceeds that in whites. Cancer Epidemiol Biomarkers Prev. 2002;11:795–800. [PubMed] [Google Scholar]
- Pratico D, Lawson JA, FitzGerald GA. Cyclooxygenase-dependent formation of the isoprostane, 8-epi prostaglandin F2 alpha. J Biol Chem. 1995;270:9800–9808. doi: 10.1074/jbc.270.17.9800. [DOI] [PubMed] [Google Scholar]
- Probst-Hensch NM, Pike MC, McKean-Cowdin R, Stanczyk FZ, Kolonel LN, Henderson BE. Ethnic differences in post-menopausal plasma oestrogen levels: high oestrone levels in Japanese-American women despite low weight. Br J Cancer. 2000;82:1867–1870. doi: 10.1054/bjoc.1999.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers EH, Grant MH. The effect of the flavonoids, quercetin, myricetin and epicatechin on the growth and enzyme activities of MCF7 human breast cancer cells. Chem Biol Interact. 1998;116:213–228. doi: 10.1016/s0009-2797(98)00092-1. [DOI] [PubMed] [Google Scholar]
- Sato M, Pei RJ, Yuri T, Danbara N, Nakane Y, Tsubura A. Prepubertal resveratrol exposure accelerates N-methyl-N-nitrosourea-induced mammary carcinoma in female Sprague-Dawley rats. Cancer Lett. 2003;202:137–145. doi: 10.1016/j.canlet.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Schumacher G, Neuhaus P. 2-Methoxyestradiol -- a new compound for cancer treatment. Dtsch Med Wochenschr. 2006;131:825–830. doi: 10.1055/s-2006-939855. [DOI] [PubMed] [Google Scholar]
- Shull JD, Spady TJ, Snyder MC, Johansson SL, Pennington KL. Ovary-intact, but not ovariectomized female ACI rats treated with 17beta-estradiol rapidly develop mammary carcinoma. Carcinogenesis. 1997;18:1595–1601. doi: 10.1093/carcin/18.8.1595. [DOI] [PubMed] [Google Scholar]
- Singh B, Mense SM, Remotti F, Liu X, Bhat HK. Antioxidant butylated hydroxyanisole inhibits estrogen-induced breast carcinogenesis in female ACI rats. J Biochem Mol Toxicol. 2009;23:202–211. doi: 10.1002/jbt.20281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirtori CR, Arnoldi A, Johnson SK. Phytoestrogens: end of a tale? Ann Med. 2005;37:423–438. doi: 10.1080/07853890510044586. [DOI] [PubMed] [Google Scholar]
- Stopper H, Schmitt E, Kobras K. Genotoxicity of phytoestrogens. Mutat Res. 2005;574:139–155. doi: 10.1016/j.mrfmmm.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Ursin G, Bernstein L, Pike MC. Breast cancer. Cancer Surv. 1994;19-20:241–264. [PubMed] [Google Scholar]
- Usui T. Pharmaceutical prospects of phytoestrogens. Endocr J. 2006;53:7–20. doi: 10.1507/endocrj.53.7. [DOI] [PubMed] [Google Scholar]
- van der Woude H, Ter Veld MG, Jacobs N, van der Saag PT, Murk AJ, Rietjens IM. The stimulation of cell proliferation by quercetin is mediated by the estrogen receptor. Mol Nutr Food Res. 2005;49:763–771. doi: 10.1002/mnfr.200500036. [DOI] [PubMed] [Google Scholar]
- van Duursen MB, Sanderson JT, de Jong PC, Kraaij M, van den Berg M. Phytochemicals inhibit catechol-O-methyltransferase activity in cytosolic fractions from healthy human mammary tissues: implications for catechol estrogen-induced DNA damage. Toxicol Sci. 2004;81:316–324. doi: 10.1093/toxsci/kfh216. [DOI] [PubMed] [Google Scholar]
- Verma AK, Johnson JA, Gould MN, Tanner MA. Inhibition of 7,12-dimethylbenz(a)anthracene- and N-nitrosomethylurea-induced rat mammary cancer by dietary flavonol quercetin. Cancer Res. 1988;48:5754–5758. [PubMed] [Google Scholar]
- Wang MY, Liehr JG. Induction by estrogens of lipid peroxidation and lipid peroxide-derived malonaldehyde-DNA adducts in male Syrian hamsters: role of lipid peroxidation in estrogen-induced kidney carcinogenesis. Carcinogenesis. 1995;16:1941–1945. doi: 10.1093/carcin/16.8.1941. [DOI] [PubMed] [Google Scholar]
- Wang Y, Cao J, Weng JH, Zeng S. Simultaneous determination of quercetin, kaempferol and isorhamnetin accumulated human breast cancer cells, by high-performance liquid chromatography. J Pharm Biomed Anal. 2005;39:328–333. doi: 10.1016/j.jpba.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Weroha SJ, Li SA, Tawfik O, Li JJ. Overexpression of cyclins D1 and D3 during estrogen-induced breast oncogenesis in female ACI rats. Carcinogenesis. 2006;27:491–498. doi: 10.1093/carcin/bgi278. [DOI] [PubMed] [Google Scholar]
- Whitsett T, Carpenter M, Lamartiniere CA. Resveratrol, but not EGCG, in the diet suppresses DMBA-induced mammary cancer in rats. J Carcinog. 2006;5:15. doi: 10.1186/1477-3163-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CS, Landau JM, Huang MT, Newmark HL. Inhibition of carcinogenesis by dietary polyphenolic compounds. Annu Rev Nutr. 2001;21:381–406. doi: 10.1146/annurev.nutr.21.1.381. [DOI] [PubMed] [Google Scholar]
- Zhu BT. Catechol-O-Methyltransferase (COMT)-mediated methylation metabolism of endogenous bioactive catechols and modulation by endobiotics and xenobiotics: importance in pathophysiology and pathogenesis. Curr Drug Metab. 2002;3:321–349. doi: 10.2174/1389200023337586. [DOI] [PubMed] [Google Scholar]
- Zhu BT, Conney AH. Is 2-methoxyestradiol an endogenous estrogen metabolite that inhibits mammary carcinogenesis? Cancer Res. 1998;58:2269–2277. [PubMed] [Google Scholar]
- Zhu BT, Liehr JG. Quercetin increases the severity of estradiol-induced tumorigenesis in hamster kidney. Toxicol Appl Pharmacol. 1994;125:149–158. doi: 10.1006/taap.1994.1059. [DOI] [PubMed] [Google Scholar]
- Zhu BT, Liehr JG. Inhibition of catechol O-methyltransferase-catalyzed O-methylation of 2- and 4-hydroxyestradiol by quercetin. Possible role in estradiol-induced tumorigenesis. J Biol Chem. 1996;271:1357–1363. doi: 10.1074/jbc.271.3.1357. [DOI] [PubMed] [Google Scholar]