

INTRODUCTION
Over the past two decades, there has been tremendous progress in molecular genetics of heritable skin diseases, and as many as 500 distinct human genes are now known to harbor mutations in these diseases (Feramisco et al., 2009). The clinical implications of this progress are evident in the diagnosis and management of these diseases. For example, (a) identification of the specific mutations can be used for confirmation of diagnosis with prognostic implications; (b) identification of mutations has facilitated assessment of the precise mode of inheritance, particularly in cases with no previous family history of the disease; and (c) identification of candidate genes and mutations has formed the basis for DNA-based prenatal testing and pre-implantation genetic diagnosis in families at risk for recurrence (Uitto, 2009). However, in spite of significant progress in identification of the molecular basis of heritable skin diseases, there has been relatively little progress until very recently in developing effective and specific treatments. This synopsis will highlight some of the milestones in progress towards treatment and cure of heritable skin diseases, primarily focusing on epidermolysis bullosa (EB) as a paradigm of such conditions, with emphasis on development of gene-,protein-, and cell-based molecular strategies just entering the clinical arena (Uitto et al., 2012).
RECLINICAL MODEL SYSTEMS
EB is a heterogeneous group of heritable blistering disorders due to fragility of the cutaneous basement membrane zone (link to Milestone article no. 1; Fine et al., 2008; Intong and Murrell, 2012). The development of molecular therapies for skin diseases has been largely predicated upon development of animal model systems, particularly transgenic mice that recapitulate their clinical, histopathological, and ultra-structural features (Bruckner-Tuderman et al., 2010; Natsuga et al., 2010). In the case of EB, these include knockout mice with targeted ablation of the corresponding gene, such as those encoding type VII collagen, type XVII collagen, or the subunit polypeptides of laminin 332. Also, identification of naturally occurring spontaneous mutations in mice and other vertebrates has been helpful for development of preclinical approaches. In addition to transgenic animals, human xenograft transplants onto immunocompromised mice have provided useful model systems (Siprashvili et al., 2010). Finally, to our knowledge, previously unreported vertebrate model systems, such as zebrafish, have been explored as potential models for heritable skin diseases (Li et al., 2011).
PROSPECTS OF GENE THERAPY
Information gleaned from the experiments utilizing preclinical animal models has been critical for the development of gene therapy approaches for EB. The first Milestone for applying gene therapy for EB took place in 2005, when cultured keratinocyte stem cells with holoclone potential were cultured from the skin of a patient with a junctional form of EB (Mavilio et al., 2006). This patient was shown to have mutations in the LAMB3 gene, which encodes the β3-subunit polypeptide of laminin 332. The patient, a 36-year-old man, had a form of non-Herlitz junctional EB caused by compound heterozygosity for a LAMB3 null mutation and a missense mutation (E210K) that, in addition to the amino acid substitution, disrupts intron splicing. Holoclone-forming cells were not present in the blistering area of skin, most likely because the continuous proliferative stimulus associated with the chronic wound-healing process had depleted the stem cell pool. However, biopsy from the palm area, which blistered less, allowed a sufficient number of holoclones to be cultured. The cultured cells were transduced with a Maloney leukemia virus–derived retroviral vector expressing the LAMB3 complementary DNA, and these cells were then used to prepare genetically corrected epidermal grafts in culture (Figure 1). The grafts were transplanted onto selected areas of the patient’s legs, which were prepared for acceptance of the graft by laser ablation of the deficient epidermis. Engraftment was shown to result in synthesis and proper assembly of normal levels of functional laminin 332, which clinically resulted in development of adherent epidermis that remained stable and did not demonstrate blistering (Mavilio et al., 2006). A published 3.5 year follow-up noted that on clinical examination, no blisters were observed in the transplanted area of skin, and the regenerated skin was stable, normal looking, and functionally resilient to mechanical trauma (De Luca et al., 2009). There was no evidence of inflammation, and specific tests carried out 3 and 6 months after the transplantation procedure indicated absence of both humoral and T cell–mediated cytotoxic immune responses against the transgene product. It should be noted that one of the mutant alleles contained a single point mutation (E210K), which allowed residual expression of some laminin 332. Thus, the introduction of the complementary DNA–derived laminin β3 polypeptide was not recognized as a new protein by the patient’s immune system, and no evidence of rejection has been noted.
Figure 1. Strategy for an ex vivo patient-specific keratinocyte gene therapy.

Keratinocytes cultured from the patient’s skin biopsy are transduced with a vector expressing the transgene, and the transgenic cells are selected and grown into epithelial sheets that can be grafted back to the patient (adapted from Tamai et al., 2009).
Although this study clearly provided proof-of-principle of ex vivo keratinocyte therapy for EB, there have been significant concerns regarding this approach. One of them revolves around the safety of the retroviral vectors used to integrate the transgene into the genome, and specifically, the concern highlights the possibility that the random insertion of the transgene results in activation of proto-oncogenes or inactivation of tumor suppressive genes (De Luca et al., 2009). These concerns were raised in response to clinical trials in which introduction of the curative transgene into hematopoietic progenitor cells of patients with X-linked severe combined immuno-deficiency caused activation of a T cell proto-oncogene, resulting in leukemia (Hacein-Bey-Abina et al., 2003). Although it has been suggested that, on statistical grounds, the probability of such an event to occur is extremely low, and that the retroviral integration into the genome may not be random, these concerns have resulted in the conclusion that the Maloney leukemia virus LTR-based vectors are not acceptable for genetic modification of stem cells, and such clinical trials are now banned by the regulatory agencies in many countries in Europe (see De Luca et al., 2009). Nevertheless, there have been significant improvements in vector design, and many of them have potentially a more favorable safety profile due to targeted integration sites and self-inactivating properties (Titeux et al., 2010). On the basis of these considerations, clinical gene therapy trials focusing on the recessive dystrophic forms of EB have been recently initiated (Dr. Alfred Lane, Stanford University, personal communication).
In the context of ex vivo keratinocyte gene therapy, the phenomenon known as revertant mosaicism is of considerable interest (Lai-Cheong et al., 2011; Pasmooij and Jonkman, 2012). Revertant mosaicism in skin diseases was originally noted in a patient with a junctional form of EB, where areas of skin were shown to reacquire the wild-type phenotype through naturally occurring mitotic gene conversion (Jonkman et al., 1997). Thus, revertant mosaicism reflects “natural gene therapy,” which could provide corrected, patient-specific cells for grafting purposes (Gostynski et al., 2009).
RNA INTERFERENCE TECHNOLOGY
Another gene therapy approach for treatment of inherited skin diseases utilizes silencing of the mutant RNA by short-interfering RNA (siRNA; Figure 2). Specifically, the safety and efficacy of a siRNA targeting mutant keratin 6A (N171K) has been recently reported in a patient with pachyonychia congenita (PC). The Milestone paper by Leachman et al. (2010) describes a patient with PC, a disabling keratinization disorder affecting the skin, nails, oral mucosa, hair, and teeth. This disease is inherited in an autosomal dominant fashion and often leads to painful lesions in the feet characterized by blistering and callus formation on the soles. PC is caused by mutations in either keratin 6, 16, or 17 genes, and the mutant polypeptides cause the clinical manifestation by dominant-negative interference (McLean et al., 2011). The approach of this study was to elicit a selective depletion of the mutant keratin by injection of siRNA using a siRNA molecule carefully selected to be mutant-specific and not interfere with the wild-type K6a mRNA.
Figure 2. Small interfering RNA (siRNA) strategies for autosomal dominant keratin 6a disorders by targeting either mutant or both mutant and wild-type alleles.

(a) In normal keratinocytes, synthesis of K6a (blue), K6b (red), and K6c (green) occurs; (b) in pachyonychia congenita keratinocytes with a heterozygous missense mutation in KRT6A, there is a dominant-negative interference between the wild-type and mutant K6a protein that perturbs the entire keratin network and compromises cell integrity, leading to skin blistering as a result of minor trauma; (c) one siRNA approach is to target the mutant KRT6A allele to leave only residual wild-type KRT6A allele expression; (d) an alternative siRNA strategy is to silence all KRT6A, both mutant and wild-type—blistering does not occur in the absence of K6a because of functional redundancy with K6b and K6c, allowing normal intermediate filament network integrity (reproduced from Uitto et al., 2012).
This double-blind study consisted of injection of the siRNA to one foot while the other foot received a vehicle–control solution (Leachman et al., 2010). The treatment consisted of 17 weeks of two times weekly injections and was followed by a 3-month washout. Careful assessment of the clinical signs and symptoms revealed definitive improvement, and the size of the callus in siRNA-injected sites was getting smaller. Thus, this first-in-human double-blinded phase 1b clinical trial suggested the proof-of-principle of RNA interference technology for patients with keratinization disorders.
This study also highlighted some of the drawbacks of this approach. First, the siRNA was delivered by direct injection to the lesional skin, resulting in intense pain (Leachman et al., 2010). Consequently, significant efforts are now focusing on improved delivery methods, such as pharmaceutical formulations for topical, non-invasive delivery for PC, and other keratin disorders (Kaspar et al., 2009; McLean and Moore, 2011). Another concern is that although there was a dramatic and specific response in the treated area of skin, the improvement appeared to be temporary, and upon discontinuation of the treatment, the lesions returned to their original size. Thus, continuous treatment by improved topical delivery system might be required for this approach (Smith et al., 2008). Finally, it has been pointed out that one of the disadvantages of the allele-specific gene silencing approach is that the US Food and Drug Administration and corresponding international agencies may consider each mutation-specific siRNA as a separate entity requiring individual toxicity studies and regulatory approval (see Uitto et al., 2012). Thus, an alternative approach to the mutant allele–specific ablation, for example, in case of PC, would be total silencing of all KRT6A alleles, both mutant and wild type (Figure 2). It should be noted that the blistering phenotype does not occur in the complete absence of K6a because of functional redundancy with K6b and K6c, allowing normal intermediate filament network integrity. Clinical trials are now being contemplated using these approaches for PC and other keratinization disorders (Dr Irwin McLean, University of Dundee, personal communication).
ALLOGENEIC FIBROBLAST THERAPY
The Milestone article by Wong et al. (2008) reported on a clinical trial in which the potential for intradermal injections of allogeneic fibroblasts were assessed in individuals with recessive dystrophic EB (RDEB). These studies were based on the premise that human skin fibroblasts, in addition to keratinocytes, express type VII collagen (Stanley et al., 1985; Chen et al., 1994). Furthermore, previous animal studies have suggested that COL7A1 gene–corrected human RDEB fibroblasts overexpressing type VII collagen, when injected intradermally into immunodeficient mouse skin or into a transplanted human RDEB skin xenograft, allowed sustained human type VII collagen deposition at the dermal epidermal junction, accompanied by anchoring fibril formation (Woodley et al., 2003). These observations lead the authors to postulate that injection of a sufficient number of normal fibroblasts could lead to amelioration of the skin phenotype in patients with RDEB.
Five subjects were injected with cultured autologous fibroblasts into the edge of unblistered skin, and the expression of type VII collagen was evaluated by immunofluorescence in biopsies at 2 weeks and 3 months after a single injection (Wong et al., 2008). Significant, up to ~2-fold increase in type VII collagen immunostaining was noted at sites injected with parental fibroblasts or from those of unrelated donors. The increased type VII collagen at the dermal–epidermal junction was accompanied with an increase in anchoring fibrils, although these were not fully developed. Nevertheless, this and complementary studies demonstrated the potential of allogeneic fibroblast therapy for treatment of RDEB (Petrova et al., 2010; Yan and Murrell, 2010).
One of the observations in this study was that the allogeneic cells, as determined by Y chromosome positivity in female patients injected with male fibroblasts, were not detectable 2 weeks after the initial injection, yet type VII collagen deposition continued up to 3 months (Wong et al., 2008). Although initial observations of the injection sites did not reveal evidence of significant inflammation, subsequent studies suggested a subclinical immunological mechanism by which the allogeneic fibroblasts elicit increased type VII collagen expression in the basal keratinocytes of the recipient’s skin (Nagy et al., 2011) (Figure 3). This conclusion was supported by the observation that increased type VII collagen deposition at the dermal–epidermal junction was particularly pronounced in those patients who expressed some type VII collagen at the baseline, and who, therefore, had an enhanced capacity to increase synthesis of their own mutant type VII collagen. These studies identified heparin-binding epidermal growth factor–like growth factor as a novel putative mediator induced in the recipient cells by allogeneic fibroblast injection (Figure 3).
Figure 3. Postulated mechanism by which fibroblast therapy may ameliorate the blistering tendency in recessive dystrophic epidermolysis bullosa (RDEB).

(a) In normal skin, keratinocytes synthesize type VII collagen molecules (red), which assemble into anchoring fibrils. These fibrils entrap the interstitial collagen fibers in the dermis, securing the stable association at the dermal–epidermal junction. (b) In some patients with RDEB, there are only a few rudimentary anchoring fibrils, allowing formation of blisters below the lamina densa as a result of minor trauma. (c) Allogeneic fibroblasts injected directly into dermis elicit a subclinical immune reaction that leads to synthesis of heparin-binding epidermal growth factor–like growth factor (HB–EGF), which upregulates the synthesis and assembly of patient’s own mutated type VII collagen. The increase in the rudimentary anchoring fibrils, which are partially functional, stabilizes the association of epidermis to the underlying dermis and ameliorates the blistering tendency (adapted from Uitto, 2011a).
Although these initial studies utilized a single injection of allogeneic fibroblasts to the skin, the increase in type VII collagen persisted for several months. This can be explained, in part, by the observation that type VII collagen, once deposited to the cutaneous basement membrane zone, has a relatively long half-life as judged from mouse studies (Kern et al., 2009). Clinical trials utilizing multiple injections at regular intervals are now being contemplated to counteract the development of chronic wounds in patients with RDEB (Dr John McGrath, St John’s Institute of Dermatology, personal communication).
BONE MARROW STEM CELL THERAPY
Cell-based therapy for EB and potentially other heritable skin diseases has recently been extended to include bone marrow–derived adult stem cells. Although these cells are known to have a critical role in skin homeostasis, it has also become clear that the plasticity of these cells enables their differentiation into cell types responsible for skin regeneration after injury (Badiavas et al., 2003; Tamai et al., 2009). The Milestone article describing the first clinical trial of allogeneic bone marrow transplantation for EB was reported by Wagner et al. (2010). In this study seven children with severe RDEB were enrolled to be recipients of bone marrow transplantation, using standard myeloablative approach (Wagner et al., 2010). This study was predicated on previous animal studies, which have been conducted to evaluate the potential of bone marrow–derived stem cells to treat EB. For example, bone marrow transfer into the fetal circulation of mice that are deficient in type VII collagen resulted in deposition of type VII collagen in the skin, associated with reduction in the severity of the blistering in neonatal animals (Chino et al., 2008). Also, hematopoietic and nonhematopoetic cell populations were infused into type VII collagen knockout mice at birth, and this treatment was shown to extend the survival of the recipient mice by several weeks or months (Tolar et al., 2009). The EB phenotype was also rescued in a type XVII collagen knockout model by bone marrow transplantation (Fujita et al., 2010). Although type VII collagen is synthesized both by keratinocytes and fibroblasts, type XVII collagen, a component of hemidesmosomes, is synthesized exclusively by keratinocytes. Collectively, these studies demonstrated that different bone marrow–derived stem cells, including mesenchymal stem cells, can ameliorate the clinical symptoms and increase the survival rate of EB mice, thus paving a way to clinical trials in patients with different forms of EB.
Bone marrow transplantation in the children with RDEB was noted to result in synthesis of new type VII collagen and clinical improvement that was sustained for at least 1 year after the transplantation (Wagner et al., 2010; Tolar et al, 2011). Although these preliminary studies were promising and generated cautious optimism, it should be noted that two of the seven children died as a result of complications of the bone marrow transplant procedure, which utilized traditional chemoablative preconditioning of the recipients. A second clinical bone marrow transplantation trial has been initiated with the approach to use reduced-intensity chemotherapy before transplantation, perhaps having lesser side effects with reduced morbidity and mortality (Kiuru et al., 2010).
In addition to bone marrow transplantation, a pilot study on two patients with severe RDEB has tested the efficacy of intradermal injection of allogeneic mesenchymal stem cells into chronic ulcerative sites in these patients (Conget et al., 2010). Improved wound healing lasting up to 4 months was attributed to replenishment of type VII collagen, which was undetectable before the procedure. Similar studies are now being developed to examine whether intradermal or intravenous injection of bone marrow–derived mesenchymal stem cells can improve the clinical outcome. Collectively, these early observations support the usefulness of bone marrow stem cell populations in the treatment of heritable skin diseases, such as RDEB.
NOVEL THERAPEUTIC APPROACHES
A number of new technologies are currently being developed for potential treatment of EB, but these have not reached the clinical trial stage as yet. One of such approaches focuses on induced pluripotent stem cells (iPSCs), which allow patient-specific cells to be corrected for the gene defect, followed by introduction of differentiated fibroblast and/or keratinocytes to the skin (Uitto et al., 2012) (Figure 4). This approach would circumvent the difficulties in obtaining sufficient numbers of patient-specific cells and avoid the problem of immune rejection. Recently, patient-specific iPSCs have been generated from several human diseases to investigate the disease mechanisms, test potential drugs, and develop cell-based therapies. In the case of heritable skin diseases, patient-specific iPSCs have been generated from patients with dyskeratosis congenita as well as RDEB (Agarwal et al., 2010; Itoh et al., 2011; Tolar et al., 2011). Although a number of technological issues still need to be resolved before iPSC-based therapy can be moved to the clinic, there is rapid technological progress in this area, and the first report of gene correction utilizing patient-specific iPSCs has already been reported in a case with gyrate atrophy (Howden et al., 2011), providing a proof-of-concept for this technology.
Figure 4. Schematic steps of reprogramming somatic cells, such as fibroblasts, to induced pluripotent stem (iPS) cells, and their differentiation into epidermal keratinocytes capable of forming skin-like structures.
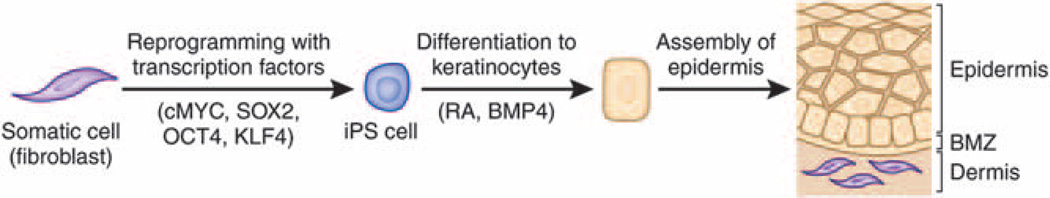
The reprogramming process is initiated by introduction of transcription factors (cMYC, SOX2, OCT4, and KLF4) into the somatic cells by transduction of expression vectors, synthetic mRNA, or recombinant protein. The iPS cells have characteristic features that allow their identification and enrichment. The iPS cells can then be differentiated into keratinocytes under specific culture conditions, e.g., medium supplemented with retinoic acid (RA) and bone morphogenic protein-4 (BMP4). BMZ, basement membrane zone (adapted from Uitto, 2011b).
Another approach to counteract the clinical manifestations of EB, which has not reached the clinical trial stage yet, is potential protein-replacement therapy. This concept is again predicated on the use of Col7a1 knockout mice as a platform, which demonstrated that infusion of a purified type VII collagen results in significantly reduced blistering and extends the lifespan of these mice (Remington et al., 2009). Again, once the type VII collagen has been properly deposited on the cutaneous basement membrane zone, the protein remains stably assembled for several months. Clinical trials utilizing GMP-purified type VII collagen are currently contemplated for patients with severe RDEB (Dr David Woodley, University of Southern California, personal communication).
Collectively, regenerative medicine for heritable skin diseases is moving very rapidly, and with novel technological innovations the field is making progress towards treatment and cure of these, currently intractable disorders.
ACKNOWLEDGMENTS
Author thank Angela Christiano, Irwin McLean, and John McGrath for helpful discussions. This study was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
Footnotes
CONFLICT OF INTEREST
The author states no conflict of interest.
REFERENCES
- Agarwal S, Loh YH, McLoughlin EM, et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature. 2010;464:292–296. doi: 10.1038/nature08792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badiavas EV, Abedi M, Butmarc J, et al. Participation of bone marrow derived cells in cutaneous wound healing. J Cell Physiol. 2003;196:245–250. doi: 10.1002/jcp.10260. [DOI] [PubMed] [Google Scholar]
- Bruckner-Tuderman L, McGrath JA, Robinson EC, et al. Animal models of epidermolysis bullosa: update 2010. J Invest Dermatol. 2010;130:1485–1488. doi: 10.1038/jid.2010.75. [DOI] [PubMed] [Google Scholar]
- Chen YQ, Mauviel A, Ryynanen J, et al. Type VII collagen gene expression by human skin fibroblasts and keratinocytes in culture: influence of donor age and cytokine responses. J Invest Dermatol. 1994;102:205–209. doi: 10.1111/1523-1747.ep12371763. [DOI] [PubMed] [Google Scholar]
- Chino T, Tamai K, Yamazaki T, et al. Bone marrow cell transfer into fetal circulation can ameliorate genetic skin diseases by providing fibroblasts to the skin and inducing immune tolerance. Am J Pathol. 2008;173:803–814. doi: 10.2353/ajpath.2008.070977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conget P, Rodriguez F, Kramer S, et al. Replenishment of type VII collagen and reepithelialization of chronically ulcerated skin after intradermal administration of allogeneic mesenchymal stromal cells in two patients with recessive dystrophic epidermolysis bullosa. Cytotherapy. 2010;12:429–431. doi: 10.3109/14653241003587637. [DOI] [PubMed] [Google Scholar]
- De Luca M, Pellegrini G, Mavilio F. Gene therapy of inherited skin adhesion disorders: a critical overview. Br J Dermatol. 2009;161:19–24. doi: 10.1111/j.1365-2133.2009.09243.x. [DOI] [PubMed] [Google Scholar]
- Feramisco JD, Sadreyev RI, Murray ML, et al. Phenotypic and genotypic analyses of genetic skin disease through the Online Mendelian Inheritance in Man (OMIM) database. J Invest Dermatol. 2009;129:2628–2636. doi: 10.1038/jid.2009.108. [DOI] [PubMed] [Google Scholar]
- Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58:931–950. doi: 10.1016/j.jaad.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Abe R, Inokuma D, et al. Bone marrow transplantation restores epidermal basement membrane protein expression and rescues epidermolysis bullosa model mice. Proc Natl Acad Sci USA. 2010;107:14345–14350. doi: 10.1073/pnas.1000044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gostynski A, Deviaene FC, Pasmooij AM, et al. Adhesive stripping to remove epidermis in junctional epidermolysis bullosa for revertant cell therapy. Br J Dermatol. 2009;161:444–447. doi: 10.1111/j.1365-2133.2009.09118.x. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- Howden SE, Gore A, Li Z, et al. Genetic correction and analysis of induced pluripotent stem cells from a patient with gyrate atrophy. Proc Natl Acad Sci USA. 2011;108:6537–6542. doi: 10.1073/pnas.1103388108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intong LR, Murrell DF. Inherited epidermolysis bullosa: new diagnostic criteria and classification. Clin Dermatol. 2012;30:70–77. doi: 10.1016/j.clindermatol.2011.03.012. [DOI] [PubMed] [Google Scholar]
- Itoh M, Kiuru M, Cairo MS, et al. Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa-induced pluripotent stem cells. Proc Natl Acad Sci USA. 2011;108:8797–8702. doi: 10.1073/pnas.1100332108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkman MF, Scheffer H, Stulp R, et al. Revertant mosaicism in epidermolysis bullosa caused by mitotic gene conversion. Cell. 1997;88:543–551. doi: 10.1016/s0092-8674(00)81894-2. [DOI] [PubMed] [Google Scholar]
- Kaspar RL, McLean WH, Schwartz ME. Achieving successful delivery of nucleic acids to skin: 6th Annual Meeting of the International Pachyonychia Congenita Consortium. J Invest Dermatol. 2009;129:2085–2087. doi: 10.1038/jid.2009.220. [DOI] [PubMed] [Google Scholar]
- Kern JS, Loeckermann S, Fritsch A, et al. Mechanisms of fibroblast cell therapy for dystrophic epidermolysis bullosa: high stability of collagen VII favors long-term skin integrity. Mol Ther. 2009;17:1605–1615. doi: 10.1038/mt.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiuru M, Itoh M, Cairo MS, et al. Bone marrow stem cell therapy for recessive dystrophic epidermolysis bullosa. Dermatol Clin. 2010;28:371–382. doi: 10.1016/j.det.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Lai-Cheong JE, McGrath JA, Uitto J. Revertant mosaicism in skin: natural gene therapy. Trends Mol Med. 2011;7:140–148. doi: 10.1016/j.molmed.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leachman SA, Hickerson RP, Schwartz ME, et al. First-in-human mutation-targeted siRNA phase Ib trial of an inherited skin disorder. Mol Ther. 2010;18:442–446. doi: 10.1038/mt.2009.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Frank M, Thisse CI, et al. Zebrafish: a model system to study heritable skin diseases. J Invest Dermatol. 2011;131:565–571. doi: 10.1038/jid.2010.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavilio F, Pellegrini G, Ferrari S, et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med. 2006;12:1397–1402. doi: 10.1038/nm1504. [DOI] [PubMed] [Google Scholar]
- McLean WH, Hansen CD, Eliason MJ, et al. The phenotypic and molecular genetic features of pachyonychia congenita. J Invest Dermatol. 2011;131:1015–1017. doi: 10.1038/jid.2011.59. [DOI] [PubMed] [Google Scholar]
- McLean WH, Moore CB. Keratin disorders: from gene to therapy. Hum Mol Genet. 2011;20:R189–R197. doi: 10.1093/hmg/ddr379. [DOI] [PubMed] [Google Scholar]
- Nagy N, Almaani N, Tanaka A, et al. HBEGF induces COL7A1 expression in keratinocytes and fibroblasts: possible mechanism underlying allogeneic fibroblast therapy in recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2011;131:1771–1774. doi: 10.1038/jid.2011.85. [DOI] [PubMed] [Google Scholar]
- Natsuga K, Shinkuma S, Nishie W, et al. Animal models of epidermolysis bullosa. Dermatol Clin. 2010;28:137–142. doi: 10.1016/j.det.2009.10.016. [DOI] [PubMed] [Google Scholar]
- Pasmooij AM, Jonkman MF. First symposium on natural gene therapy of the skin. Exp Dermatol. 2012;21:236–239. doi: 10.1111/j.1600-0625.2011.01426.x. [DOI] [PubMed] [Google Scholar]
- Petrova A, Ilic D, McGrath JA. Stem cell therapies for recessive dystrophic epidermolysis bullosa. Br J Dermatol. 2010;163:1149–1156. doi: 10.1111/j.1365-2133.2010.09981.x. [DOI] [PubMed] [Google Scholar]
- Remington J, Wang X, Hou Y, et al. Injection of recombinant human type VII collagen corrects the disease phenotype in a murine model of dystrophic epidermolysis bullosa. Mol Ther. 2009;17:26–33. doi: 10.1038/mt.2008.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siprashvili Z, Nguyen NT, Bezchinsky MY, et al. Long-term type VII collagen restoration to human epidermolysis bullosa skin tissue. Hum Gene Ther. 2010;21:1299–1310. doi: 10.1089/hum.2010.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FJD, Hickerson RP, Sayers JM, et al. Development of therapeutic siRNAs for pachyonychia congenita. J Invest Dermatol. 2008;128:50–58. doi: 10.1038/sj.jid.5701040. [DOI] [PubMed] [Google Scholar]
- Stanley JR, Rubinstein N, Klaus-Kovtun V. Epidermolysis bullosa acquisita antigen is synthesized by both human keratinocytes and human dermal fibroblasts. J Invest Dermatol. 1985;85:542–545. doi: 10.1111/1523-1747.ep12277377. [DOI] [PubMed] [Google Scholar]
- Tamai K, Kaneda Y, Uitto J. Molecular therapies for heritable blistering diseases. Trends Mol Med. 2009;15:285–292. doi: 10.1016/j.molmed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Titeux M, Pendaries V, Zanta-Boussif MA, et al. SIN retroviral vectors expressing COL7A1 under human promoters for ex vivo gene therapy of recessive dystrophic epidermolysis bullosa. Mol Ther. 2010;18:1509–1518. doi: 10.1038/mt.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolar J, Blazar BR, Wagner JE. Concise review: transplantation of human hematopoietic cells for extracellular matrix protein deficiency in epidermolysis bullosa. Stem Cells. 2011;29:900–906. doi: 10.1002/stem.647. [DOI] [PubMed] [Google Scholar]
- Tolar J, Ishida-Yamamoto A, Riddle M, et al. Amelioration of epidermolysis bullosa by transfer of wild-type bone marrow cells. Blood. 2009;113:1167–1174. doi: 10.1182/blood-2008-06-161299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolar J, Xia L, Riddle MJ, et al. Induced pluripotent stem cells from individuals with recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2011;131:848–856. doi: 10.1038/jid.2010.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uitto J. Progress in heritable skin diseases: translational implications of mutation analysis and prospects of molecular therapies. Acta Derm Venereol. 2009;89:228–235. doi: 10.2340/00015555-0648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uitto J. Cell-based therapy for RDEB: how does it work? J Invest Dermatol. 2011a;131:1597–1599. doi: 10.1038/jid.2011.125. [DOI] [PubMed] [Google Scholar]
- Uitto J. Regenerative medicine for skin diseases: iPS cells to the rescue. J Invest Dermatol. 2011b;131:812–814. doi: 10.1038/jid.2011.2. [DOI] [PubMed] [Google Scholar]
- Uitto J, Christiano AM, McLean WH, et al. Novel molecular therapies for heritable skin disorders. J Invest Dermatol. 2012;132:820–828. doi: 10.1038/jid.2011.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JE, Ishida-Yamamoto A, McGrath JA, et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med. 2010;363:629–639. doi: 10.1056/NEJMoa0910501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong T, Gammon L, Liu L, et al. Potential of fibroblast cell therapy for recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2008;128:2179–2189. doi: 10.1038/jid.2008.78. [DOI] [PubMed] [Google Scholar]
- Woodley DT, Krueger GG, Jorgensen CM, et al. Normal and gene-corrected dystrophic epidermolysis bullosa fibroblasts alone can produce type VII collagen at the basement membrane zone. J Invest Dermatol. 2003;121:1021–1028. doi: 10.1046/j.1523-1747.2003.12571.x. [DOI] [PubMed] [Google Scholar]
- Yan WF, Murrell DF. Fibroblast-based cell therapy strategy for recessive dystrophic epidermolysis bullosa. Dermatol Clin. 2010;28:367–370. xi. doi: 10.1016/j.det.2010.01.015. [DOI] [PubMed] [Google Scholar]