

Abstract
Fragment screens have successfully identified new scaffolds in drug discovery, often with relatively high hit rates (5%) using small screening libraries (1,000–10,000 compounds). This raises two questions: would other noteworthy chemotypes be found were one to screen all commercially available fragments (> 300,000), and does the success rate imply low specificity of fragments? We used molecular docking to screen large libraries of fragments against CTX-M β-lactamase. We identified ten millimolar-range inhibitors from the 69 compounds tested. The docking poses corresponded closely to the crystallographic structures subsequently determined. Notably, these initial low-affinity hits showed little specificity between CTX-M and an unrelated β-lactamase, AmpC, which is unusual among β-lactamase inhibitors. This is consistent with the idea that the high hit rates among fragments correlate to a low initial specificity. As the inhibitors were progressed, both specificity and affinity rose together, yielding to our knowledge the first micromolar-range noncovalent inhibitors against a class A β-lactamase.
Fragment-based drug discovery focuses on low-molecular-weight compounds (<250 AMU in this study) that target subpockets within the overall active site1. It samples chemical space more efficiently because the number of fragments needed to cover all reasonable chemotypes is many orders of magnitude smaller than that for more drug-like compounds, where multiple chemotypes are combined. In the past decade, this strategy has led to the discovery of new scaffolds that were later combined or grown into high-affinity inhibitors2–8. However, the small size of fragments also makes them difficult to screen. Their few functional groups result in low-affinity inhibitors and concomitantly high concentrations required for experimental testing, making high-throughput methods infeasible. Instead, biophysical methods such as NMR9,10, protein crystallography11 and surface plasmon resonance (SPR)12 are used to test up to hundreds or several thousands of fragment molecules. This necessarily leaves unscreened most of the over 300,000 fragments that are readily available commercially.
The low-throughput nature of fragment testing makes computational docking potentially attractive as a way to prioritize fragments from the much larger commercially available set. Indeed, several groups have used docking to prioritize fragments for testing3,13. Nevertheless, docking of fragment molecules is often regarded as particularly challenging. This reflects two concerns in the community: first, fragments may be more promiscuous in their binding modes than larger ‘drug-like’ molecules, and this is difficult to predict; second, docking scoring functions are inaccurate even for the larger molecules against which they have often been parameterized, and are likely to be still less accurate for fragments14–16. Docking is thus not widely accepted as a reliable method to prioritize fragments, and there are few if any studies that compare docking-predicted fragment geometries to subsequent structural results.
Compared with high-throughput screening of drug-like molecules, fragment screening has frequently yielded high hit rates in different targets, despite the smaller size of the libraries17. This calls into question the specificity of fragment inhibitors. Whereas the small size of fragments may allow them to bind to protein surfaces more easily by reducing the chances of steric clashes with the protein, the same advantage may enable one fragment to interact with binding sites in unrelated proteins, leading to low specificity.
We were interested in exploring these questions in the class A β-lactamase CTX-M. This extended-spectrum β-lactamase confers bacterial resistance to third-generation cephalosporins and has recently emerged as among the most important mechanisms of resistance to the penicillin and cephalosporin antibiotics18,19. It is also an enzyme for which efforts to develop noncovalent inhibitors have been unsuccessful, including our own with compounds in the ‘lead-like’ or ‘drug-like’ size ranges20. Consistent with the idea that CTX-M presents a difficult target, its active site is large and open to solvent, as one might expect from an enzyme that is thought to have evolved from a cell-wall transpeptidase21. Compounding this structural challenge, our molecular libraries undersample the chemotypes suited for antibiotic targets22,23. These factors support the use of a fragment-based approach for inhibitor discovery against CTX-M. Mechanically, CTX-M is well suited to such an approach owing to its sensitive and quantitative activity assay and protein crystals that diffract to high resolution.
Here we investigate several questions. Can molecular docking be used to prioritize fragments and predict their binding modes? If it can do so reliably, it would open up a much larger set of fragments than are usually tested. How specific are the inhibitors that emerge? Can they be optimized for specificity and affinity? How does fragment screening compare to lead-like screening against the same target? We undertook a series of molecular docking screens of both fragment and lead-like compound libraries, followed by experimental assays and crystallography. Whereas docking failed to identify any lead-like inhibitors against CTX-M, it led to the discovery of a series of low-affinity fragment inhibitors. The series was progressed to larger and more potent inhibitors, including several with Ki values in the 10 μM range. Using AmpC β-lactamase, an enzyme that shares some active site features with CTX-M but typically recognizes very different inhibitors, we studied the specificity of the new molecules as they progressed from fragments to more potent, larger inhibitors. The implications of these results are discussed.
RESULTS
Fragment inhibitor discovery through molecular docking
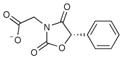
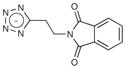
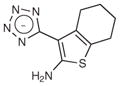
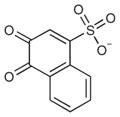
A fragment subset (67,489 compounds) and the lead-like subset (1,147,326 compounds) of the ZINC small-molecule library were docked into the active site of CTX-M-9, and compounds were selected from the top ranking list. In the lead-like database screening, five β-lactam substrates were ranked among the top 200 compounds (top 0.02%) in a pose similar to that observed crystallographically and consistent with proposed catalytic mechanisms for class A β-lactamases24. These are effectively positive controls, suggesting that the sampling and scoring parameters for docking were reasonable. Nevertheless, of the 37 lead-like molecules tested, none showed any inhibition at the limit of solubility. In contrast, ten inhibitors with half-maximal inhibitory concentration (IC50) values in the millimolar range emerged from 69 fragment compounds investigated (Table 1; Supplementary Figs. 1–3 online). These ten inhibitors were chemically distinct from most other fragments; typically the most similar fragment to any of them in ZINC had Tanimoto similarities less than 0.45 using ECFP4 fingerprints, and often lacked the anionic group that is crucial for binding to CTX-M. Clustering the 317,000 ZINC fragments available as of this writing into 2,500 representative clusters and comparing cluster centroids also led to compounds that often differed substantially, and often fatally, from the ten docking hits (Supplementary Table 1 online). These observations are consistent with a punctate chemical space at the fragment level.
Table 1.
Fragment inhibitors discovered by docking
| # | Structure | Docking rank | MW (Da) | CTX-M LEa | CTX-M Ki (mM) | AmpC Ki (mM) |
|---|---|---|---|---|---|---|
| 1 |
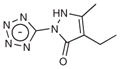
|
950 | 193.2 | – | >10 | 3.1 |
| 2 |
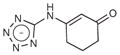
|
102 | 178.2 | 0.25 | 4.4 | 3.9 |
| 3 |
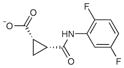
|
8 | 240.2 | 0.20 | 3.1 | 3.1 |
| 4 |
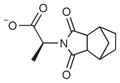
|
457 | 236.3 | 0.25 | 1.3 | 2.2 |
| 5 |
|
77 | 234.2 | 0.27 | 0.7b | 3.5 |
| 6 |
|
25 | 242.2 | 0.28 | 0.194 | 3.3 |
| 7 |
|
623 | 220.3 | 0.23 | 3.0b | N/Ac |
| 8 |
|
262 | 237.2 | 0.26 | 1.0b | N/A |
| 9 |
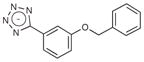
|
202 | 251.3 | 0.22 | 1.0b | N/A |
| 10 |
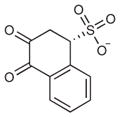
|
430 | 239.2 | 0.27 | 0.7b | N/A |
Compounds 1–6 were tested against both CTX-M and AmpC, and are ordered based on the affinity against CTX-M.
LE, ligand efficiency; ΔGbind(kcal)/(Number of heavy atoms).
Ki estimated from IC50 experiments.
N/A, not tested against AmpC.
Whereas the fragment hit rate was much higher than that from the lead-like library screen (14.5% versus 0), the full reliability of the docking could not be established, and the compounds could not be confidently progressed for affinity, without determining their structures experimentally. Five of these fragment hits were thus crystallized with the enzyme and their structures determined (Supplementary Table 2 online). The electron density for the inhibitors established their position in the active site unambiguously (Fig. 1). For several, the inhibitor density overlaps with those of phosphate and water molecules that were previously observed in the apo structure; half occupancy was used for both the inhibitor and phosphate/water in these cases.
Figure 1.

Crystal structures of fragment inhibitors and optimized derivatives against CTX-M. Five compounds have molecular weight below 250 Da, with two additional ones below 300. The surface of the protein active site is colored white (carbon), red (oxygen) and blue (nitrogen). The crystal pose of the ligand is colored yellow. The docking prediction is shown in cyan. The 2Fo − Fc electron density of the ligands is shown in white at 1σ. For compound 4, the active site is shared by the inhibitor and a phosphate molecule, both modeled at half occupancy. For compound 11, there is a partial-occupancy phosphate in the active site that is not shown here. Waters with half occupancy are also modeled in the structures of compounds 3 and 6.
The crystallographic geometries of the five fragments resemble those predicted by docking, with r.m.s. errors ranging from 1.12 Å to 2.86 Å (Fig. 1). These r.m.s. errors can in some cases overemphasize the differences between the predicted and observed geometries, as they typically made most of the same polar and nonpolar contacts, notwithstanding the structural differences. Thus, even the compound with the largest r.m.s. error, compound 1, nevertheless made many of the same interactions in the docked and crystallographic structure, including similar hydrogen bonds formed with the protein by the tetrazole group and the pyrazole nitrogen (Fig. 1). For compound 3, when the complex structure was determined, we found that the species in the active site was a diastereoisomer of the one picked by docking from the fragment library. Still, the docked geometry captures most of the key interactions observed in the X-ray structure (Fig. 1). Additionally, although the discrepancy between the experimental and docking structures undoubtedly owed to approximations and inaccuracies in the calculations, it was also partly due to the artifacts of crystal packing. For crystals in the P21 space group (Supplementary Table 2), the active site is close to the crystal packing interface. In three cases, inhibitors interacted with a threonine residue from the neighboring monomer, distorting their experimental geometries compared to that expected in solution and modeled by docking.
A notable aspect of many of the fragment inhibitors was that they were tetrazole derivatives placing the anionic ring in the canonical carboxylic acid binding site of CTX-M (this site would ordinarily bind the C(3)4′ carboxylate of penicillins and cephalosporins). This is a rarely used chemotype among β-lactamase inhibitors, though tetrazole is well known as a carboxylate bioisostere for other targets25. The dominance of this chemotype among the docking hits, its favorable biological properties and its extensive interactions in the X-ray structures drew us to this series. In the fragment structures, the tetrazole often hydrogen bonds with Ser130, Thr235 and Ser237. Ser237 is alanine in many class A β-lactamases, including TEM-1, that do not have extended-spectrum β-lactamase (ESBL) activity. In CTX-M, Ser237 interacts with the oxyimino side chains of third-generation cephalosporins and is partly responsible for the ESBL activity of these enzymes26,27. Here, the same substitution makes CTX-M more susceptible to the tetrazole compounds, which suggests that these small molecules may be particularly useful for targeting related ESBL enzymes.
Based on the favorable complementarity between the tetrazole groups and the binding site, we looked for larger analogs with which to progress this new series. Returning to the docking screen of the lead-like database, from which our initial selection had been so unfavorable, we selected four compounds with relatively high docking ranks (Table 2; Supplementary Fig. 1). All showed inhibition when tested at 5 mM concentration, and one inhibited CTX-M with a Ki of 21 μM (compound 12, Table 2). This compound ranked 982 from the lead-like docking screen, which was a low enough ranking that we had simply overlooked it initially. Here too we determined the structure of the CTX-M–12 complex, to 1.41- Å resolution, and the experimental structure showed high fidelity to the docking-predicted pose (Fig. 1) and high complementarity to the enzyme (Fig. 2). Particularly noteworthy was the role of the added amide linkage, which is recognized by the canonical site in CTX-M that typically interacts with the conserved R1 amide side chain of β-lactam drugs (that is, Asn132 and Asn104). Subsequent similarity searches of the ZINC databases returned analogs of 12, compounds 13 and 14, which when tested had Ki values of 10 and 12 μM, respectively. To our knowledge, these compounds represent the most potent noncovalent inhibitors that have been identified against CTX-M or any other class A β-lactamase.
Table 2.
Lead-like inhibitors developed from the fragments
| # | Structure | MW (Da) | CTX-M LE | CTX-M Ki (mM) | AmpC Ki (mM) |
|---|---|---|---|---|---|
| 11 |
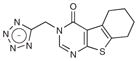
|
287.3 | 0.21 | 1.1 | 1.5 |
| 12 |
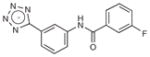
|
282.3 | 0.32 | 0.021 | 2.8 |
| 13 |
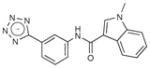
|
318.3 | 0.30 | 0.010 | N/A |
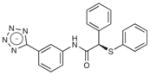
| 14 |
|
386.4 | 0.25 | 0.012 | N/A |
Figure 2.
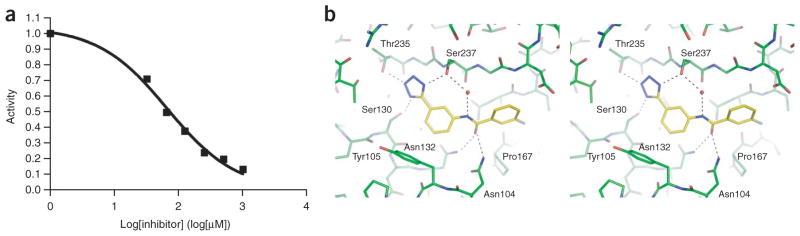
Inhibition and binding of compound 12. (a) Dose-response curve for CTX-M. The error for any point is 2% activity or less. (b) CTX-M complex structure (stereoview). Potential hydrogen bonds are represented by black dash lines. A mediating water molecule is shown as a red sphere.
Specificity
Because we were interested in how hit rate and affinity relate to the specificity of fragments, the CTX-M inhibitors were tested against AmpC, a class C β-lactamase. Whereas AmpC also hydrolyzes β-lactams, it is mechanistically distinct from class A β-lactamases such as CTX-M and, more importantly, has different inhibition patterns. Molecules that inhibit AmpC rarely inhibit CTX-M, and the converse is also true. For instance, the two purely noncovalent inhibitor series against AmpC, the phthalimides and the thiophene sulfonamides, are inactive against CTX-M28,29. Conversely, clavulanate, a covalent inhibitor against CTX-M, does not inhibit AmpC at relevant concentrations. We tested eight of the CTX-M inhibitors against AmpC (compounds 1–6, 11 and 12; Tables 1 and 2). All eight compounds inhibited AmpC with IC50 values in the low millimolar range. For fragments 1–5, this affinity closely resembled that for CTX-M, and there was essentially no specificity between the two enzymes for these fragments. As the potency of the inhibitors increased against CTX-M (for instance, for compound 6 and especially for the optimized inhibitor 12), activity against AmpC remained unaltered, so specificity rose substantially. Thus, the 21 μM CTX-M inhibitor 12 has a Ki against AmpC of 2,800 μM, giving a specificity of over 100-fold.
DISCUSSION
Fragment-based drug design has become established as an effective approach for developing new inhibitors against challenging protein targets in the past decade. Still, important questions remain about both fundamental and practical aspects of this technique. This study investigated two of them: the utility of molecular docking for prioritizing fragments and the specificity of fragment inhibitors. The ability to prioritize fragments by docking for CTX-M is consistent with earlier studies using docking to find fragments. What stands out here is the case-by-case comparison of the docking structure predictions with those determined by crystallography. Seven docking predictions are compared to subsequent X-ray structures with what turns out to be good geometric fidelity between them. This addresses widely debated concerns that fragments are too small to be treated well by docking, not only for hit rates but also for geometry. On the other hand, the specificity of fragments for CTX-M was inversely correlated with their affinities. At low affinities, the fragments showed little specificity between CTX-M and AmpC. As the inhibitors were progressed, both specificity and affinity rose together. This is consistent with the idea that the high hit rates among fragments partly arise from their initial low specificity, and that specificity can be designed into these molecules as they are advanced.
With well-known challenges when docking even large inhibitors, a key question was how the scoring functions and sampling algorithms would perform when docking small fragments. Two potential problems have been previously raised against fragment-based docking: first, the lack of restraints in fragment binding can lead to a high rate of false binding modes; second, many docking scoring functions are empirically derived for drug-like ligands and may be unsuitable for fragments15.
Whereas the relatively few fragment-protein interactions do present a challenge, the results from this study suggest that molecular docking can correctly predict binding poses of fragment inhibitors. There are several reasons for this, some general and some specific to CTX-M. First, DOCK3.5.54 uses a physics-based scoring function, which has no explicit bias against fragment compounds. Second, fragments are typically more rigid than larger lead-like and drug-like molecules, making them easier to treat by docking. Finally, the high quality of the ultra-high-resolution CTX-M structure, and the overall rigidity of the active site, undoubtedly reduced several docking challenges, including the ambiguity of hydrogen atom positions and the potential for protein flexibility27,30. Notwithstanding these caveats, the high relative hit rate of the fragment screen and the high correspondence of the docked and crystallographic poses support the reliability of the docking calculation. Indeed, there are few studies that we know of outside of toy cavity sites31 where so many docking predictions correspond with such high fidelity to subsequent X-ray crystallographic results. Judged by this criterion alone, one might be tempted to conclude that docking works better for fragments than for larger, more classical molecules.
A crucial contributor to the success of the fragment screen, especially as compared to the lead-like docking screens, is the better coverage of chemical space among fragments than among drug-like or even lead-like molecules. Chemotypes are seen among high-scoring fragments that are rarely if ever observed among high-scoring lead-like molecules. The tetrazole-containing molecules are a good example of this—though a few are present among the lead-like docking hits, their ranks are low enough that they are not selected for testing. From an inhibitor design perspective, tetrazoles are noteworthy bioisosteres, and once one knows to look for them they may be found among the docked lead-like molecules as well. Thus, in the fragment docking screen there were eight tetrazoles among the top 500 ranking molecules, which is the slice of the high-scoring molecules from which we usually select compounds to test. Conversely, there was only one tetrazole compound in the top 500 ranked molecules from the lead-like screen. The highest affinity compound from docking, compound 12, ranked 982 in the lead-like virtual screening and would not have been picked had we not had the information provided by the tetrazole fragments.
The high hit rate of fragment-based screening raises the question of whether fragments typically have low specificities. It has been suggested that fragments are more likely to bind to protein surfaces compared with larger compounds, whose multiple functional groups often demand compromises throughout the molecule to fit the protein, thereby reducing ligand efficiency. Conversely, the multiple functional groups of larger molecules also reduce the likelihood of binding to off-targets or off-sites, as the compromises they demand are often so substantial as to eliminate binding entirely17. The results from our studies supported this idea. Most of the fragments we tested inhibited not only CTX-M but also AmpC, an enzyme with what is typically a wholly different inhibition pattern. As the complementarity increased between the ligand and a given enzyme, specificity versus the second enzyme was improved by over 100-fold, exactly tracking the affinity increase. Thus, compound 12 has a Ki of 21 μM against CTX-M but only 2.8 mM against AmpC, whereas a developed thiophene-sulfonamide inhibitor has a Ki against AmpC of 1 μM but no measurable affinity against CTX-M.
From where, then, does the low specificity of the fragments derive? The crystal structures of the same fragment inhibitors bound to CTX-M and AmpC suggest that fragment promiscuity arises not because they target the same local binding motif in different proteins, but rather because of the ability of fragments to adapt to the different environments in these proteins, despite their limited flexibility. In X-ray structures determined with both enzymes, compound 1 for instance targeted two non-overlapping regions in the superimposed active sites of CTX-M and AmpC (D.G. Teotico and B.K.S., unpublished data). For compound 4, although the carboxylate group binds close to the oxyanion holes of both CTX-M and AmpC, it forms different hydrogen bonds with surrounding residues and makes hydrophobic contacts with different parts of the active site in the two enzymes. In all of these cases, the electron density for the ligand is well defined, which suggests that the energy minima for ligand binding are at least closely clustered.
Perhaps the most compelling observation to emerge from this study is the close correspondence of the docking predictions to the subsequent X-ray results for the fragment inhibitors. Few if any studies have directly compared docking predictions to experimental fragment complexes, and this has opened the door for concerns about our ability to predict fragment geometries. This study, though far from assuring that docking will always work with fragments, does suggest enough reliability for docking to be pragmatic. This enables one to probe libraries of available fragments that are one to two orders of magnitude larger than those that are typically screened empirically. Given an increasingly punctate chemical space as one moves to the fragment size scale, it is unlikely that any single library of 1,000 or even 10,000 molecules can adequately represent the chemotypes present among the over 300,000 fragments that are commercially available. Indeed, an implication of this study is that high fragment hit rates reflect the lower specificity of fragments compared to larger, more classical molecules; the high hit rate cannot be taken to mean that one has adequately represented all possible chemical space in a library of 10,000 fragments. Rather, the converse is true—for fragments in particular, missing out on 300,000 available molecules represents an unusually large gap in chemical space coverage. Admittedly, one cannot hope to capture by docking the full diversity of hits likely to result from empirically screening a well-constructed fragment library; the two approaches are likely to be complementary. Still, the ability to prioritize fragments by docking, one of the main results of this work, will allow investigators to access a much larger pool of fragments than is addressable empirically, thus substantially expanding the chemo-types sampled in fragment-based screens.
METHODS
Molecular docking
A fragment subset (67,489 compounds) and the lead-like subset (1,147,326 compounds) of the ZINC small-molecule database (http://zinc.docking.org/) were docked into the active site of the 0.88- Å CTX-M structure using DOCK 3.5.54 (refs. 30,32,33). To prepare the protein for docking, all waters and ions were removed from the structure except for the conserved catalytic water. The positions of polar group hydrogen atoms on Ser70, Ser130, Thr235 and the catalytic water were placed based on the electron density of the ultra-high-resolution structure30. The proton position for Ser237 was more ambiguous; two positions were initially sampled in separate docking screens and one was chosen for later experiments based on performance.
The matching sphere set used for placing ligands in the binding pocket was generated by combining the ligand atoms in the complex structures determined for CTX-M27,30. A total of 60 atom-derived spheres were used. Spheres were labeled for chemical matching based on the hydrogen bonding properties and ionization states of nearby protein atoms34. Guided by the matching spheres, pre-generated ligand conformations (1 to 4,224 with an average of 7.5 per ligand for fragment compounds) were placed in the binding pocket, and up to 1,148,899 poses (averaging approximately 90,000) per ligand were sampled. Ligand bin size/overlap and receptor bin size/overlap were 0.4/0.3 and 0.4/0.3, respectively. The energy potential (scoring) grids were calculated as outlined previously35. These include an excluded volume grid calculated using DISTMAP, a van der Waals potential grid calculated by CHEMGRID, an electrostatic potential grid from DelPhi and a desolvation grid36,37. Ligand configurations were scored by summing the energies of van der Waals and electrostatic interactions, corrected by ligand desolvation energy. Ligand desolvation energies and partial atomic charges were precalculated for the library using AMSOL (University of Minnesota, Minneapolis), and the desolvation energy was corrected for the degree of burial of each ligand atom at the time of docking. Burial was based on the volume of high dielectric solvent displaced by the protein, which differed from ligand atom to ligand atom and from pose to pose; it is this displaced volume that is precalculated on the desolvation grid (this method is included in the DOCK3.5.54 distribution). The absolute magnitude of the partial charges of polar protein atoms involved in hydrogen bonds with the ligand in the complex structures was increased by 0.4 electrons to favor such electrostatic interactions. The net charges of the residues were unaltered. The atoms with altered partial charges included the Oγ, HOγ, HN and O atoms of Ser70, Ser130 and Ser237, and the Oδ1 and HNδ2 atoms of Asn132 and Asn104.
After completing the docking screens, compounds were chosen from among the top-ranked 500 (occasionally the top 1,000) molecules for testing. Whereas the primary criterion for prioritizing fragments for testing was thus the docking rank directly, compounds were also visually inspected for favorable interactions with the CTX-M structure. Emphasis was given to those compounds that placed negatively charged moieties in the oxyanion hole (formed by the backbone amide groups of Ser70 and Ser237) or the pocket that recognizes the C(3)4′ carboxylate of penicillins and cephalosporins. Compounds that appeared too flexible or left too many polar groups uncomplemented by CTX-M were de-prioritized. Of the ten fragments that inhibited CTX-M, eight ranked in the top 500 (0.7% of the database) and two ranked in the top 1,000 (1.5%) by docking.
Inhibition assays
The activities of CTX-M and AmpC were measured based on hydrolysis of the β-lactam substrate nitrocefin in 100 mM Tris-HCl (pH 7.0, with 0.01% v/v Triton X-100 to control for aggregation-based artifacts) and monitored at 480 nM wavelength on a Hewlett-Packard HP-8453 spectrometer38. Final protein concentrations were 0.1 nM for CTX-M and 0.75 nM for AmpC β-lactamase. Compounds were purchased from Chemical Block, Enamine and Chembridge and assayed without further purification. They were dissolved in DMSO and tested at final concentrations of up to 5–15 mM (depending on their solubility) in IC50 experiments, and at varied concentrations for Ki determination. Nitrocefin concentration was 25 μM for CTX-M and 200 μM for AmpC in the inhibition assays. The Km of nitrocefin for CTX-M was determined to be 11.6 μM, whereas that for AmpC was 400 μM. Protein was typically added to the reaction buffer first, and the reaction was initiated by the addition of nitrocefin. To control for incubation effects, this order was reversed (enzyme added last) for characteristic inhibitors; no effect on inhibition was observed.
Crystallization and structure determination
CTX-M-9, a clinically isolated CTX-M mutant that we have studied extensively, was used as a representative enzyme of the CTX-M family. It was purified as previously described19 and crystallized in 1.4 M potassium phosphate (pH 8.7) from hanging drops at 19 °C. Final concentration of protein in the drop was 9 mg ml−1 or 4.5 mg ml−1. Often, higher occupancy of the inhibitor in the active site was achieved with lower concentrations of protein, probably because of the higher inhibitor/protein ratio in the drop. Complex crystals were obtained either by cocrystallization or by soaking. DMSO stocks of inhibitors were added to the phosphate buffer to a final concentration of 25 mM (final DMSO concentration 2.5% v/v), and then mixed with equal volumes of protein solution in the drop. For a few tetrazole compounds, the inhibitor concentration in the phosphate buffer was reduced to 5 mM or 2.5 mM owing to low solubility. Cocrystallization was attempted first. In the case of failures, preformed crystals were soaked for 24 h with the same phosphate buffer containing the inhibitor. In two cases where crystals did not survive soaking after the transfer, the phosphate buffer containing the inhibitor was added directly to the original drop containing the apo crystals, and crystals remained stable after 24 h. Diffraction was measured at beamline 8.3.1 of the Advanced Light Source, Berkeley, California, and processed with HKL200039. Initial models were obtained by one step of rigid-body refinement using Refmac in CCP4, with an apo CTX-M-9 structure. Refinement and model rebuilding were carried out in CCP4 and Coot40,41. Ligands were modeled after the refinement of the protein and most waters was nearly completed. The Fo − Fc map at this stage clearly identified the ligand configuration, particularly for structures determined to 1.5 Å or higher resolutions. The 2Fo − Fc density for the ligand in the final structure closely matched the initial Fo − Fc density. For the two structures determined to 1.7 and 1.8 Å, alternative ligand configurations were tested in parallel refinement experiments to compare with the conformation chosen in the final structure. Simulated annealing omit maps were also used to check and prevent model bias in the final stages of refinement.
Cheminformatics
Compound similarity comparison and search was performed using ECFP-4 fingerprint and Tanimoto coefficient in SciTegic Pipeline pilot. To search for compounds similar to compound 12 from the whole ZINC database, approximately 5 million purchasable small molecules were compared with compound 12, and those with a similarity score better than 0.5 were retained, ranked and sorted. Compounds were selected from the top 250 molecules that shared the core scaffold with compound 12.
Supplementary Material
Acknowledgments
This work was supported by US National Institutes of Health grants GM63813 and GM59957 (to B.K.S.). We thank D.G. Teotico (University of California, San Francisco) for providing the AmpC inhibitors and for insightful discussions, and J. Hert and C. Laggner for assistance with similarity search. We thank D.G. Teotico, S. Boyce, M. Mysinger and J. Irwin for reading the manuscript. We also thank R. Bonnet.
Footnotes
Accession codes. Protein Data Bank: The coordinates and structure factors of the described structures have been deposited with accession codes 3G2Y (1), 3G2Z (2), 3G30 (3), 3G31 (4), 3G32 (6), 3G34 (11) and 3G35 (12).
Note: Supplementary information and chemical compound information is available on the Nature Chemical Biology website.
AUTHOR CONTRIBUTIONS
This project was designed by Y.C. and B.K.S. together; all the experiments were undertaken by Y.C. Both authors contributed to interpreting the results and writing the paper.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Rees DC, Congreve M, Murray CW, Carr R. Fragment-based lead discovery. Nat Rev Drug Discov. 2004;3:660–672. doi: 10.1038/nrd1467. [DOI] [PubMed] [Google Scholar]
- 2.Congreve M, Chessari G, Tisi D, Woodhead AJ. Recent developments in fragment-based drug discovery. J Med Chem. 2008;51:3661–3680. doi: 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]
- 3.Murray CW, et al. Application of fragment screening by X-ray crystallography to beta-secretase. J Med Chem. 2007;50:1116–1123. doi: 10.1021/jm0611962. [DOI] [PubMed] [Google Scholar]
- 4.Haydon DJ, et al. An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science. 2008;321:1673–1675. doi: 10.1126/science.1159961. [DOI] [PubMed] [Google Scholar]
- 5.Card GL, et al. A family of phosphodiesterase inhibitors discovered by cocrystallography and scaffold-based drug design. Nat Biotechnol. 2005;23:201–207. doi: 10.1038/nbt1059. [DOI] [PubMed] [Google Scholar]
- 6.Edwards PD, et al. Application of fragment-based lead generation to the discovery of novel, cyclic amidine beta-secretase inhibitors with nanomolar potency, cellular activity, and high ligand efficiency. J Med Chem. 2007;50:5912–5925. doi: 10.1021/jm070829p. [DOI] [PubMed] [Google Scholar]
- 7.Fahr BT, et al. Tethering identifies fragment that yields potent inhibitors of human caspase-1. Bioorg Med Chem Lett. 2006;16:559–562. doi: 10.1016/j.bmcl.2005.10.048. [DOI] [PubMed] [Google Scholar]
- 8.Oltersdorf T, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 9.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 10.Pellecchia M, et al. Perspectives on NMR in drug discovery: a technique comes of age. Nat Rev Drug Discov. 2008;7:738–745. doi: 10.1038/nrd2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartshorn MJ, et al. Fragment-based lead discovery using X-ray crystallography. J Med Chem. 2005;48:403–413. doi: 10.1021/jm0495778. [DOI] [PubMed] [Google Scholar]
- 12.Sweeney ZK, et al. Discovery of triazolinone non-nucleoside inhibitors of HIV reverse transcriptase. Bioorg Med Chem Lett. 2008;18:4348–4351. doi: 10.1016/j.bmcl.2008.06.080. [DOI] [PubMed] [Google Scholar]
- 13.Huang JW, et al. Fragment-based design of small molecule X-linked inhibitor of apoptosis protein inhibitors. J Med Chem. 2008;51:7111–7118. doi: 10.1021/jm8006992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marcou G, Rognan D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J Chem Inf Model. 2007;47:195–207. doi: 10.1021/ci600342e. [DOI] [PubMed] [Google Scholar]
- 15.Hubbard RE, Chen I, Davis B. Informatics and modeling challenges in fragment-based drug discovery. Curr Opin Drug Discov Devel. 2007;10:289–297. [PubMed] [Google Scholar]
- 16.Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug Discov Today. 2006;11:580–594. doi: 10.1016/j.drudis.2006.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hann MM, Leach AR, Harper G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J Chem Inf Comput Sci. 2001;41:856–864. doi: 10.1021/ci000403i. [DOI] [PubMed] [Google Scholar]
- 18.Bonnet R. Growing group of extended-spectrum beta-lactamases: the CTX-M enzymes. Antimicrob Agents Chemother. 2004;48:1–14. doi: 10.1128/AAC.48.1.1-14.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Y, Delmas J, Sirot J, Shoichet B, Bonnet R. Atomic resolution structures of CTX-M beta-lactamases: extended spectrum activities from increased mobility and decreased stability. J Mol Biol. 2005;348:349–362. doi: 10.1016/j.jmb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 20.Oprea TI, Davis AM, Teague SJ, Leeson PD. Is there a difference between leads and drugs? A historical perspective. J Chem Inf Comput Sci. 2001;41:1308–1315. doi: 10.1021/ci010366a. [DOI] [PubMed] [Google Scholar]
- 21.Massova I, Mobashery S. Kinship and diversification of bacterial penicillin-binding proteins and beta-lactamases. Antimicrob Agents Chemother. 1998;42:1–17. doi: 10.1128/aac.42.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 23.O’Shea R, Moser HE. Physicochemical properties of antibacterial compounds: implications for drug discovery. J Med Chem. 2008;51:2871–2878. doi: 10.1021/jm700967e. [DOI] [PubMed] [Google Scholar]
- 24.Beadle BM, Trehan I, Focia P, Shoichet BK. Structural milestones in the pathway of an amide hydrolase: substrate, acyl, and product complexes of cephalothin with AmpC b-lactamase. Structure. 2002;10:413–424. doi: 10.1016/s0969-2126(02)00725-6. [DOI] [PubMed] [Google Scholar]
- 25.Kumar S, Pearson AL, Pratt RF. Design, synthesis, and evaluation of alpha-ketoheterocycles as class C beta-lactamase inhibitors. Bioorg Med Chem. 2001;9:2035–2044. doi: 10.1016/s0968-0896(01)00107-9. [DOI] [PubMed] [Google Scholar]
- 26.Ibuka AS, et al. Crystal structure of extended-spectrum beta-lactamase Toho-1: insights into the molecular mechanism for catalytic reaction and substrate specificity expansion. Biochemistry. 2003;42:10634–10643. doi: 10.1021/bi0342822. [DOI] [PubMed] [Google Scholar]
- 27.Chen Y, Shoichet B, Bonnet R. Structure, function, and inhibition along the reaction coordinate of CTX-M beta-lactamases. J Am Chem Soc. 2005;127:5423–5434. doi: 10.1021/ja042850a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Babaoglu K, et al. Comprehensive mechanistic analysis of hits from high-throughput and docking screens against beta-lactamase. J Med Chem. 2008;51:2502–2511. doi: 10.1021/jm701500e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Powers RA, Morandi F, Shoichet BK. Structure-based discovery of a novel, noncovalent inhibitor of AmpC beta-lactamase. Structure. 2002;10:1013–1023. doi: 10.1016/s0969-2126(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 30.Chen Y, Bonnet R, Shoichet BK. The acylation mechanism of CTX-M beta-lactamase at 0.88 a resolution. J Am Chem Soc. 2007;129:5378–5380. doi: 10.1021/ja0712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Graves AP, et al. Rescoring docking hit lists for model cavity sites: predictions and experimental testing. J Mol Biol. 2008;377:914–934. doi: 10.1016/j.jmb.2008.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lorber DM, Shoichet BK. Hierarchical docking of databases of multiple ligand conformations. Curr Top Med Chem. 2005;5:739–749. doi: 10.2174/1568026054637683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Irwin JJ, Shoichet BK. ZINC–a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lorber DM, Shoichet BK. Flexible ligand docking using conformational ensembles. Protein Sci. 1998;7:938–950. doi: 10.1002/pro.5560070411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang N, Shoichet BK, Irwin JJ. Benchmarking sets for molecular docking. J Med Chem. 2006;49:6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meng EC, Gschwend DC, Blaney JM, Kuntz ID. Orientational sampling and rigid-body minimization in molecular docking. Proteins. 1993;17:266–278. doi: 10.1002/prot.340170305. [DOI] [PubMed] [Google Scholar]
- 37.Shoichet BK, Leach AR, Kuntz ID. Ligand solvation in molecular docking. Proteins. 1999;34:4–16. doi: 10.1002/(sici)1097-0134(19990101)34:1<4::aid-prot2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 38.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 39.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 40.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 41.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.