

Abstract
Recent data on DNA sequencing of human tumours have established that cancer cells contain thousands of mutations. These data support the concept that cancer cells express a mutator phenotype. This Perspective considers the evidence supporting the mutator phenotype hypothesis, the origin and consequences of a mutator phenotype, the implications for personalized medicine and the feasibility of ablating tumours by error catastrophe.
Many years ago, I put forth the concept that the mutation rate of nonmalignant cells is insufficient to generate the large numbers of mutations that are present in human cancers1. Instead, it was hypothesized that cancers express a mutator phenotype, and as a result progressively accumulate2 large numbers of mutations during tumour progression.
The human genome is dynamic; it is estimated that each cell undergoes >20,000 DNA damaging events3–5 and >10,000 replication errors per cell per day6. As a result, mutations occur throughout the genome, including in genes that maintain genetic stability. DNA damage that escapes correction by base excision repair (BER) or nucleotide excision repair (NER)4 can generate misincorporations during DNA replication7. Misincorporations by mutant DNA polymerases5–7 that escape mismatch repair (MMR)8 result in single-base substitutions. Unrepaired DNA alterations and crosslinks that block DNA replication can result in chromosome rearrangements, amplifications and deletions9. The number of proteins that function in DNA replicative processes in human cells is not known. However, studies in yeast indicate that >100 genes are required for the maintenance of genetic stability10. Among these are genes that encode error-prone DNA polymerases that can replicate past bulky lesions on DNA11. Mutations or misregulation of any of these genes could increase the probability that subsequent mutations will occur in oncogenes (resulting in driver mutations that confer a growth advantage). Such repetitive cycles of mutagenesis and selection mimic Darwinian evolution. Most mutations are ‘passengers’ that do not confer a growth advantage. The concept of cancer being initiated by DNA damage and the generation of large numbers of driver, mutator and passenger mutations after each round of selection is illustrated in FIG. 1. In addition to driver mutations, there are subclonal mutations that are present in a large proportion of cells as well as random mutations that are generated during the last round of clonal selection. By the time a solid tumour is detected, it frequently measures 1 cm3 and encompasses 108–109 cells, each cell containing tens of thousands of clonal, subclonal and random mutations12.
Figure 1. Cascade of mutations during tumour progression.

In the case of solid tumours, epidemiological evidence indicates that as many as 20 years pass between the time an individual is exposed to a carcinogen to the clinical appearance of a tumour. Various barriers to tumour progression exist, including DNA repair processes, the availability of nutrition, the requirement of angiogenesis to allow the tumour to increase in size and responses to hypoxia. Circles represent mutations in genes that result in enhanced mutagenesis, triangles indicate driver mutations that are selected on the basis of changes in the tumour microenvironment and white rectangles represent passenger mutations.
In order for environmental agents to introduce mutations that cause cancer, the mutations would need to be in excess of those produced by normal cellular processes. The major source of endogenous DNA damage is likely to be reactive oxygen species (ROS) and related reactive molecules13. The principal alteration produced by ROS is 8-oxo-deoxyguanosine (8-oxo-dG)13, and mice harbouring mutations in genes that encode proteins that repair oxygen-damaged DNA are cancer-prone4. DNA damage by ROS14 as well as errors by replicative DNA polymerases in vitro2,15 and methylcytidine deamination16 can result in a disproportionally high frequency of single GC→AT transitions. These are also the most frequent mutations that accumulate in human tumours17. Thus, it is tempting to speculate that these processes are a major source of mutations in spontaneous tumours.
Evidence for the expression of a mutator phenotype in human cancer has been presented18,19. Recent studies in mice have shown that if the genes encoding the replicative DNA polymerases Pol δ or Pol ε are replaced with genes harbouring mutations that render them error-prone, tumours occur in various tissues20; this lends further credence to the mutator phenotype concept (BOX 1). The efficiency by which cancers arise with and without mutator mutations has also been modelled by varying all clinically relevant parameters21. The importance of a mutator mutation is greatest when more oncogenic mutations are required for the commitment to cancer. For cancers that require only one or two mutations, such as inherited retinoblastoma, a mutator phenotype may not be necessary. However, for most cancers that require three or more driver mutations, a mutator phenotype may be inevitable21. Based on age of onset, it is postulated that prostate cancer, for example, requires as many as 12 driver mutations117,118. By contrast, I have argued that human cancers contain thousands of mutations, many of which are random, and that mutations in multiple pathways can result in a malignant phenotype.
Box 1. Mouse models of a mutator phenotype.
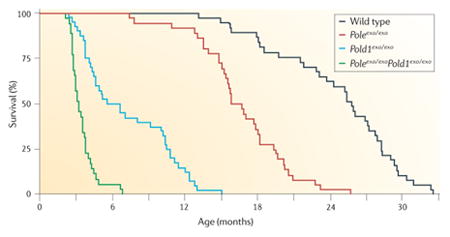
Mice with mutations in DNA damage repair genes and DNA polymerases have an increased incidence of cancer. In pioneering studies, Preston and colleagues20,95 created mutant mice (Poleexo/exo mice and Pold1exo/exo mice) in which the endogenous genes encoding the catalytic subunits of the replicative DNA polymerases Pol δ and Pol ε were replaced with genes that encoded mutants of the catalytic subunits that lacked 3′–5′ proofreading exonucleolytic activity. Poleexo/exo mice and Pold1exo/exo mice had strong spontaneous mutator and cancer phenotypes (see the figure) in the absence of environmental stress. The heterozygotes were normal. The homozygotes had reduced survival that correlated with increased tumour incidence. In Poleexo/exoPold1exo/exo mice or mice expressing either mutant DNA polymerase in the presence of inactive mismatch repair (MMR) genes96, the incidence of different cancers was further increased.
Mice have also been generated with substitutions in the polymerase domain of Pol δ97. Mice heterozygous for a Pol δ -L604K substitution did not exhibit an increased incidence of cancer but tumours progressed more rapidly. Cells from these mutant mice had a decreased rate of fork progression and an increased number of chromosome rearrangements92, and the purified corresponding human mutant polymerase was defective in copying past altered template nucleotides92. Translesion DNA polymerases11,98 have an expanded active site99, which enables them to copy past altered template bases. Some of these polymerases are overexpressed in mouse and human tumours, and it has been suggested that they can copy undamaged DNA, incorporate non-complementary nucleosides and contribute to a mutator phenotype100. Therefore, in addition to deficits in DNA repair, alterations in either replicative or translesion DNA polymerases can generate and/or accelerate tumorigenesis in mice.
Figure is modified, with permission, from REF. 20 © (2009) National Academy of Sciences.
This Perspective focuses on results obtained from DNA sequencing, and the implications of a mutator phenotype in cancer. The feasibility of modifying the growth of cancers by altering the rate of accumulation of mutations is considered. Undoubtedly, changes at the level of transcription and translation also contribute to a mutator phenotype in cancer. However, these epigenetic changes will not be considered owing to limited direct evidence that they are stably transferred from one generation to another in cancer cells, their reversible nature and the lack of knowledge about the functional significance of specific alterations.
Sequencing of human tumour DNA
Ironically, the most significant evidence for the existence of a mutator phenotype comes from The Cancer Genome Atlas, which was designed to catalogue mutations in human cancers with the expectation of identifying new targets for chemotherapy. The results indicate that each tumour is unique and contains tens to hundreds of thousands of mutations17,18.
It is important to emphasize that the current methods of DNA sequencing detect only the most frequent substitutions at each position; a single base change is detected only if present in >10% of the molecules. To detect less frequent substitutions, it is necessary to sequence either the same region multiple times (deep sequencing) or to sequence single molecules without errors. Thus, mutations catalogued in The Cancer Genome Atlas and similar cancer databanks are not derived from deep sequencing and are therefore predominately clonal; sub-clonal22,23 and random23,24 mutations have not been extensively characterized. Moreover, the nucleotide sequences of repeats and telomeres have not yet been determined owing to slippage of DNA polymerase during PCR amplification, the lack of fixed primer sites and complexities in aligning reads. Repetitive DNA sequences often assume non-B DNA conformations that are frequently mutated25,26. Also, deletions, insertions and rearrangements are frequently not reported. As a result, no human genome has yet been completely sequenced.
Exon sequencing
A compilation of the numbers and types of mutations found in exons from a variety of tumours is presented in TABLE 1. More than 1,000 different genes have been reported to be mutated in human tumours, and many tumours contain as many as 100 non-synonymous mutations. In any tumour type, none of the genes is invariably mutated nor is there a set of mutated genes that are diagnostic of a specific tumour17. Next-generation sequencing has identified many known cancer genes, including TP53, KRAS and epidermal growth factor receptor (EGFR)27, as well as genes that previously were not known to be involved in carcinogenesis. Many genes contain multiple mutations. A single breast tumour, for example, contained 70 mutant protein kinases; some with three separate substitutions28. The most frequently reported mutant gene was TP53, and in lung tumours TP53 mutations correlated with tumour grade: for example, somatic mutations in TP53 were reported in 13%, 24% and 52% of tumours of grades 1, 2 and 5, respectively29. So far, only a few new genes have been shown to be commonly mutated, and these are neither highly prevalent nor in multiple tumour types.
Table 1. Cancer genome sequencing studies.
| Tumour type | Genes sequenced | Mutations | Mutated genes | Non-syn. mutations per tumour | Known cancer genes (%)* | New cancer-associated genes (%)* | Predominant mutation type | Refs |
|---|---|---|---|---|---|---|---|---|
| Exon sequencing | ||||||||
| Breast (n = 11) | 18,191 | 1,243 | 1,137 | 101 | TP53 (33%), PIK3CA (17%) | CHD5 (3%) | CG→TA (35%), CG→GC (29%) | 101,102 |
| Colorectal (n = 11) | 18,191 | 942 | 848 | 77 | APC (67%), TP53 (50%), KRAS (46%) | TGM3 (8.3%) | CG→TA (59%) | 101,102 |
| Colorectal (MSI n = 1, MSS n = 1) | Exome capture | MSI = 1,304, MSS = 198 | NA | MSI = 359, MSS = 45 | BRAF (∼10%), TP53 (∼50%) | NA | CG→TA (∼35%) | 103 |
| Pancreatic (n = 24) | 20,661 | 1,163 | 1,007 | 48 | KRAS (99%), TP53 (86%), SMAD4 (26%), CDKN2A (24%) | NA | CG→TA (54%) | 104 |
| Pancreatic neuroendocrine (n = 10) | ∼18,000 | 157 | 149 | 16 | MEN1 (45%) | DAXX (24%), ATRX (19%) | CG→TA (42%), CG→GC (18%) | 105 |
| Glioblastoma (n = 21) | 20,661 | 748 | 685 | 47 | CDKN2A (50%), TP53 (40%), EGFR (37%) | IDH1 (12%) | CG→TA (64%) | 106 |
| Lung (n = 188) | 623 | 1,013 | 348‡ | NA | TP53 (35%), KRAS (32%), EGFR (18%) | LRP1B (9%), PTPRD (5%) | CG→AT (43% versus 13%)§ | 29 |
| Clear cell RCC (n = 101) | 3,544 | 515 | 319 | NA | VHL (55%) | SETD2 (3%), KDM5C (3%) | CG→TA (∼40%) | 107 |
| Clear cell RCC (n = 7) | 20,921 | 156 | 108 | 16 | NA | PBRM1 (41%) | CG→TA (31%), AT→GC (22%) | 108 |
| Ovarian clear cell (n = 8) | ∼18,000 | 258 | 253 | 20‖ | PIK3CA (40%), PPP2R1A (7%), KRAS (5%) | ARID1A (57%) | CG→TA (49%) | 109 |
| Medulloblastoma (childhood) (n = 21)¶ | 21,039 | 202 | 196 | 11 | PTCH1 (30%), TP53 (7.6%) | MLL2 (10.6%), MLL3 (2%) | CG→TA (55%), CG→AT (21%) | 110 |
| Whole-genome sequencing | ||||||||
| Acute myeloid leukaemia (n = 1) | NA | 500–1,000 | 10 | 10 | NPM1 (28%), FLT3 (24%) | DNMT3A (22%) | NA | 41,111 |
| Acute myeloid leukaemia (n = 1) | NA | ∼750 | 64# | 12 | FLT3 (27%), NPM1 (23%), NRAS (11%) | IDH1 (16%) | NA | 112 |
| Lobular breast (n = 1) | NA | NA | NA | 32 | CDH1 (∼60%) | HAUS3 (∼1%) | CG→TA (54%) | 113 |
| Basal-like breast (n = 3) | NA | 27,173 | ∼200 | NA | TP53 (65%) | CHGB (NA) | CG→TA (55%) | 35,114 |
| Non-small-cell lung (n = 1) | NA | >50,000 | NA | >302 | KRAS (∼30%) | MIR598 (NA) | CG→AT (46%) | 35 |
| Small-cell lung (n = 1) | NA | 22,910 | 105 | NA | TP53 (85%), RB1 (80%) | CHD7 (NA) | CG→AT (∼36%) | 36 |
| Melanoma (n = 1) | NA | 33,345 | 195 | NA | BRAF (∼70%), TP53 (∼50%) | SPDEF (NA) | CG→TA (∼66%) | 31 |
| Prostate (n = 7) | NA | 3,866 | NA | 20 | SPOP (28%) | SPTA1 (28%) | AT→GC (32%) | 115 |
| Multiple myeloma (n = 38) | NA | 7,450 | NA | 35 | KRAS (26%), NRAS (24%) | FAM46C (13%) | CG→TA (47%), AT→TA (13%) | 116 |
APC, adenomatous polyposis coli; ARID1A, AT-rich interactive domain 1A; ATRX, alpha thalassaemia/mental retardation syndrome X-linked; CDH1, E-cadherin; CDKN2A, cyclin-dependent kinase inhibitor 2A; CHD, chromodomain helicase DNA binding protein; CHGB, chromogranin B; DAXX, death-domain associated protein; DNMT3A, DNA methyltransferase 3α; EGFR, epidermal growth factor receptor; FLT3, FMS-related tyrosine kinase 3; HAUS3, HAUS augmin-like complex, subunit 3; IDH1, isocitrate dehydrogenase 1; KDM5C, lysine (K)-specific demethylase 5C (which encodes JARID1C); LRP1B, low density lipoprotein receptor-related protein 1B; MEN1, multiple endocrine neoplasia 1; MIR598, microRNA-598; MLL, mixed-lineage leukaemia; MSI, microsatellite instability; MSS, microsatellite stable; NA, not applicable; non-syn., non-synonymous; NPM1, nucleophosmin; PBRM1, polybromo 1; PIK3CA, PI3K, catalytic, α-polypeptide; PPP2R1A, protein phosphatase 2, regulatory subunit A, α; PTCH1, patched 1; PTPRD, protein tyrosine phosphatase, receptor type, D; RB1, retinoblastoma 1; RCC, renal cell carcinoma; SETD2, SET domain containing 2; SPDEF, SAM pointed domain containing ETS transcription factor; SPOP, speckle-type POZ protein; SPTA1, spectrin, α, erythrocytic 1; TGM3, transglutaminase 3; VHL, von Hippel-Lindau tumour suppressor.
Where possible, values are taken from follow-up studies or are prevalence frequencies of the same study (excludes discovery samples).
Number of genes containing at least one non-synonymous point mutation.
Values for tumours from smokers versus never-smokers, respectively.
One treated sample with 125 mutations is excluded from this calculation, as done by the original authors.
Excluding cell line sample.
Somatic point mutations in conserved or regulatory portions of the genome.
Whole-genome sequencing
The types of somatic mutations in normal human tissue have been difficult to establish. However, DNA sequences of family members, generations apart, indicate that single-base transitions are the most common mutations detected30. Most mutations reported in tumours (TABLE 1) are also single-base substitutions; CG→TA transitions predominate. In lung tumour cell lines23,31 and melanoma cell lines31, the mutation frequency on the transcribed strand is lower than that on the non-transcribed strand, which affirms the concept of preferential removal of endogenous DNA damage by transcription-coupled NER32. In some tumours, the range of mutations is unique and is indicative of exposure to environmental agents. Tobacco smoke contains large amounts of polycyclic hydrocarbons and aromatic amines33 that form bulky adducts in DNA; when bypassed by a translesion DNA polymerase (Pol κ)34, they result in predominantly G→T transversions, which are precisely the most frequent errors reported in lung cancers29,35,36. In skin cancer, the most frequent mutations are found at potential sites of ultraviolet-radiation-induced pyrimidine dimer formation23,27. Thus, DNA sequencing will increasingly yield important clues about environmental exposures to mutagens that could enhance a mutator phenotype.
Even in haematopoietic malignancies in which morphological homogeneity is frequently diagnostic, genetic heterogeneity is extensive. For example, the most successful demonstration of targeted cancer therapy is the treatment of myelogenous leukaemia with imatinib37,38, a specific inhibitor of the breakpoint cluster region (BCR)–ABL1 fusion kinase. Resistance emerges in more than 30% of patients and is most frequently mediated by a point mutation in the ATP binding site of ABL1 (REF. 39). The emergence of resistance is associated with pre-existing mutations40. In one study, whole-genome sequencing revealed only ten non-synonymous mutations41, of which eight were de novo mutations; however, none of these mutations was found in 187 other patients.
Heterogeneity within tumours
An important hallmark of cancer42 is cellular heterogeneity. Although subtypes of normal cells within an organ are morphologically similar, cancer cells within a tumour are strikingly different. There may be small cells, large cells, cells with multiple nuclei and cells with different shapes and staining properties within a tumour. Indeed, morphological heterogeneity is an important criterion for grading tumours. It seems reasonable to suggest that this morphological heterogeneity derives from functional heterogeneity encoded by multiple mutations in the cancer genome. The heterogeneity of nuclear DNA in tumours was first established by cytological studies. Chromosome alterations are found in many types of neoplastic cells43, and it has been proposed that aneuploidy (an abnormal number of chromosomes) is sufficient to explain genetic instability in tumours without requiring gene mutations44. Changes in chromosomes can encompass millions of nucleotides and are thus very difficult to investigate. The simple observation that deletions involving large numbers of genes are compatible with cell viability indicates the enormous genetic redundancy in human cells.
Historically, genetic instability in cancer was hypothesized by Boveri45 on the basis of the effects of aneuploidy on the growth of sea urchin embryos. Foulds46 further observed that malignant characteristics appeared to occur in a stepwise fashion, and Nowell47 suggested that genomic instability, governed by the generation and selection of mutations, contributes to a stepwise progression of tumorigenesis — a recapitulation of Darwinian selection. The processes of enhanced mutagenesis and Darwinian evolution are not mutually exclusive.
Gene rearrangements
The heterogeneity and complexity of the cancer genome was documented initially by techniques that analysed gene rearrangements. Rearrangements and large deletions occur at a much lower frequency in cancer genomes than do single-base substitutions, but they have the potential to inactivate multiple contiguous genes. Using comparative genomic hybridization (CGH), oligonucleotide arrays or PCR amplification, it is now feasible to detect changes involving as few as 250 nucleotides48. Although these approaches are of relatively low resolution, they have been important in demonstrating the multiplicity of rearrangements that occur within tumours. Fifty-nine recurrent copy number changes have been identified in lung adenocarcinoma49, and most of these did not encompass known cancer genes. Massively parallel sequencing has been used to identify various somatic and germline rearrangements, and multiple genomic rearrangements at specific loci have been attributed to single catastrophic events50.
Microsatellites
Repetitive nucleotide sequences are present throughout the genome and are hot spots for mutagenesis. Changes in the length of repetitive sequences in DNA51–54 are characteristic of colon cancer in patients with inherited mutations in MMR genes55,56. Microsatellites are also frequently mutated in various sporadic human malignancies, often when MMR genes are silenced57. Because microsatellites are mutated at a high frequency, they have served to establish the heterogeneity of cells within tumours and to delineate cell lineages during tumour evolution58. Maley et al.59 used microsatellites as prognostic markers for progression of a pre-malignant condition, Barrett's oesophagus, to adenocarcinoma of the oesophagus. Changes in the lengths of poly(dG) tracts in chronic ulcerative colitis correlate with the presence of occult adenocarcinoma of the colon60. Some expanded tracts were found in biopsies located tens of centimetres from the primary tumours, suggesting that the fields of mutated premalignant cells from which cancer may arise could be very extensive.
Subclonal mutations
In addition to clonal mutations within a tumour, there are likely to be hundreds of thousands of subclonal mutations that would not be detected by routine DNA sequencing. These mutations could be the remnants of mutant genes that offered an increased growth advantage but were outcompeted by other driver mutations. However, as the tumour microenvironment changes, cells harbouring subclonal mutations could be rejuvenated and drive the malignant process. Campbell et al.23 used pyrosequencing to identify subclonal mutations among V(D)J rearrangements at the immunoglobulin G (IgG) locus in chronic lymphocytic leukaemia. Subclonal mutations could be spatially clustered within a tumour, as indicated in FIG. 1. Heppner61 proposed that tumours consist of dynamically evolving subpopulations. Regions of diversity within tumours62 have been verified by karyotype analysis63, DNA fingerprinting64 and the identification of microsatellite alterations65. Furthermore, DNA sequencing of pancreatic tumours demonstrated a submicroscopic distribution of subclonal populations within tumours66.
Random mutations
Single-base substitutions are the most prevalent mutations found in human cancers (TABLE 1). They most frequently arise from DNA polymerase errors that escape correction by MMR or by small alterations in template bases that are copied by translesion DNA polymerases67. Most single-base substitutions do not alter protein function68 and are unlikely to confer a selective growth advantage. A method to detect and identify mutations at predetermined sites in human cells at a frequency of 1 per 108 base pairs has been established19. By comparing the frequency of random mutations in cancer and adjacent normal tissues, the mutation frequency in normal tissues was found to be less than one mutation in 108 base pairs. By contrast, the first six tumours analysed exhibited large numbers of mutations, the mean being 205 × 10−8 mutations per base pair19. Sequence analysis indicated that all were single-base substitutions and that most were not clonally expanded. The high number of random mutations in tumours that was recorded by this technology has been verified by studies of lung adenocarcinomas in both mice and humans69.
Metastasis
Most cancer deaths result from metastasis. Understanding the complexities of this process may be fundamental to effective cancer therapy. Riethmuller and Johnson70 observed single metastatic cells in bone marrow by staining cells for epithelial cytokeratins. This opened the path for key observations that were contrary to the predominant dogma that classified metastasis as a late-stage event in tumour progression. The concept of metastasis arising late was fuelled by the expectation that early detection of primary tumours and their subsequent removal would prevent metastatic spread. However, Klein and colleagues quantified chromosome changes in single metastatic cells in mice and humans bearing primary tumours for different lengths of time71–73. They found that chromosome changes in metastatic lesions were different from those in the primary tumours and, moreover, that discrete metastatic lesions in the same individual had different chromosome alterations73. These results provide strong evidence that metastasis occurs early, and perhaps continuously, during tumour progression. By contrast, from DNA sequencing studies of pancreatic tumours it was concluded that metastasis occurs late during tumour evolution66.
It is important to note that no single genetic alteration or group of genetic alterations has been shown to drive the metastatic process. All studies report that metastases contain many clonal mutations that are present in the primary tumours and thereafter acquire additional mutations during expansion at distant sites23. The findings that metastatic lesions differ from the primary tumour and from one another is in accord with the expression of a mutator phenotype in metastatic lesions21. An issue that needs to be addressed is whether metastatic lesions harbour mutations that are generated late in the evolution of the primary tumour (FIG. 2). It should be noted that the most frequent chromosome alterations in primary breast tumours and in prostate tumours are rarely present in metastatic lesions73. The persistence of independently evolving metastatic lesions has important implications for cancer therapy. If metastatic spread occurs early during tumour progression and metastases accumulate mutations that are different from those in the primary lesions, the response of the primary lesions to chemotherapy may not be indicative of the response of the metastatic lesions.
Figure 2. Dissemination of metastasis early and late during tumour progression.

Metastatic spread may occur throughout tumour progression, both early in tumour evolution and as the tumour evolves more genetic alterations. The metastases may then evolve to develop metastasis-specific mutations that are different to those found in the primary tumour.
The impact of mutators on cancer therapy
The expression of a mutator phenotype in human cancers and in particular the accumulation of subclonal and random mutations has important implications. From the data so far presented, I estimate that each cancer cell within most tumours contains >10,000 mutations. By the time a tumour is clinically detected it is likely to contain 108–109 cells and could, in principle, harbour >1011 different mutations. Therefore, every tumour could contain mutant genes that will render some cells resistant to any single chemotherapeutic agent. This is a major impediment to personalized medicine. It can be argued that, although a tumour will contain cells that are resistant to any single drug, it is unlikely to contain single cells that are resistant to multiple drugs that target different pathways. In fact, this provides a strong theoretical argument for the increased efficacy of combination chemotherapy. The efficacy of combination therapy can also be based on synthetic lethality74,75; this phenomenon describes the loss of cell viability when the function of multiple genes is simultaneously lost, even though the loss of each gene individually is compatible with cell viability. Synthetic lethality is probably the basis of the efficacy of poly(ADP-ribose) polymerase (PARP) inhibitors in patients with mutations in BRCA1 (REF. 76) or BRCA2 (REFS 77,78). Unfortunately, we lack a sufficient number of effective cancer chemotherapeutic agents that have limited toxicity and that precisely target a particular range of cellular molecules. Perhaps the most important immediate therapeutic results from DNA sequencing will be the identification and quantification of cancer cells with pre-existing resistant mutations. These cells will selectively proliferate on administration of a drug to which the surrounding cancer cells are sensitive. Thus, it is important to determine the frequency and types of drug-resistant mutations in tumours to avoid the use of toxic and ineffective chemotherapeutic agents.
Targeting a mutator phenotype
It might be more effective to target aberrant processes in tumours than to target specific genes or pathways. Mathematical models indicate that a mutator phenotype would be most efficient if expressed early during tumour progression21. The large numbers of clonal mutations in tumour cells may not render it feasible to target multiple pathways. I envision that a decrease in mutation rate would slow down tumour progression, whereas an increase might facilitate clonal evolution. However, there is probably a maximum mutation frequency that a tumour can tolerate: a further increase would be detrimental, reducing cell fitness and enhancing cell killing.
Lethal mutagenesis
The multiple genotypes in viral populations and subclonal mutations in tumours enable them to overcome environmental diversity. Culturing virally infected cells with mutagenic nucleoside analogues has been shown to cause a small increase in the mutation frequencies of RNA tumour viruses79 and HIV80 that obliterates viral infections. The concept of lethal mutagenesis is counter-intuitive for cancer; an increase in mutations would be expected to increase mutations in genes that enhance proliferation as well as increase the generation of resistance mutations. However, this increase in mutation rate might be preferentially detrimental for cancers that have already accumulated many mutations owing to a mutator phenotype and prior chemotherapy81. A tumour can be considered as a cooperative society of cells, and thus might be analogous to a viral quasi-species82. This approach is currently being tested using human cancer cells.
Arguments against a mutator phenotype
The concept that cancers exhibit a mutator phenotype is not universally accepted for several reasons66,83,84. First, many mutations are detrimental68 and would be unlikely to increase proliferation. To address this, a panel of 66 DNA polymerase I mutants with differences in fidelity spanning >10,000-fold was established in Escherichia coli85. All mutants were cultured together for 350 generations; no wild-type bacteria, antimutators (mutants with increased replicative fidelity) or high-frequency mutators were recovered. At the end of the competition, all bacteria exhibited a mutation rate that was 10- to 47-fold greater than that of wild-type bacteria. This suggests that mutators, over a narrow range in fidelity, can confer a growth advantage. In addition, mathematical models indicate that the acquisition of mutators with reduced fitness would not eliminate premalignant mutator clones during tumour evolution86.
Second, it is argued that tumours undergo thousands of rounds of replication in vivo, and as a result a sufficient number of mutations might accumulate normally during replication83. The estimate of thousands of rounds of replication is based on measurements of the length of each cell division cycle and assumes that all cells are actively dividing throughout the lifespan of a tumour. However, this assumption ignores the radiographic evidence for the wide disparities in growth rates of human tumours87, the evidence for extensive periods of dormancy during tumour progression88, and the presence of disseminated cancer cells more then 10 years after the removal of primary tumours89. If tumours grow exponentially for thousands of doublings, extensive cell death would be required to restrict their size to that observed in patients. Skin and gastrointestinal tumours can dispose of large numbers of dead cells; other solid tumours would have greater difficulty.
Third, with the possible exception of somatic mutations in TP53 in most tumours90 and mutations in POLB (which encodes Pol β) in stomach and prostate cancer91, there seem to be few reported mutations in DNA repair or DNA replication genes in human tumours. However, many of the genes that are required for the maintenance of genetic stability are essential, and mutations that drastically alter their activity are likely to result in reduced fitness. As a result, cells within a tumour may have mutations in multiple genes that increase mutation rates, and these would only be apparent by either deep sequencing or sequencing multiple copies of the same gene.
Perspective
The thousands of clonal mutations that have been identified in tumours and the lack of common mutations show that the cancer phenotype is very complex. Each tumour has evolved differently as a result of stochastic and environmental processes. Tumour cells contain thousands of mutations, and it seems reasonable to suggest that mutations in many pathways can confer similar growth advantages and result in the same malignant properties. Therefore, it is unlikely that there will be rate-limiting steps or pathways that can be uniquely targeted to prevent progression of a tumour. Considering the numbers of mutations in tumours and the paucity of effective drugs, the most important immediate goal for personalized chemotherapy may be to identify resistant mutations that are already present in tumours and to avoid toxicity to the patient from the use of ineffective chemotherapeutic agents.
The Cancer Genome Atlas has identified thousands of mutations in human tumours. Of greater importance than accumulating additional data on mutations in large numbers of human cancers would be studies that are designed to use the power of DNA sequencing to answer mechanistic questions about the origins of these mutations. Many of these mutations are in non-coding regions and are presumed to have no selective growth advantage. However, we are continually learning that these non-coding regions have important regulatory functions. The most frequent mutations are single-base substitutions; these could be indicative of errors generated by replicative DNA polymerases. This hypothesis can be tested by sequentially downregulating each of the different DNA polymerases in cancer cells. Most recent studies have identified large numbers of deletions and amplifications in the cancer genome. These could result from stalled replication forks, including those caused by mutations in DNA polymerases2,92. Although the reported mutations appear to be evenly distributed throughout the genome, they may be localized to mutational hot spots at alternative DNA structures93,94. The identification of subclones by deep DNA sequencing may allow us to identify alternative driver mutations and additional mutations in genetic stability genes, as well as to determine the presence of drug-resistant mutations before chemotherapy. Some of the important questions that massively parallel DNA sequencing is poised to address are listed in BOX 2. Shallow sequencing of tumours from many people may identify a few additional driver mutations, but it might be more important to sequence a few tumours or specific genes at great depth to identify the range and frequency of all types of mutations, including those that do not increase cell growth but are nevertheless revealing of carcinogenic mechanisms that generate genetic instability.
Box 2. Questions to be addressed with the aid of next-generation DNA sequencing.
DNA polymerases
Are mutant DNA polymerases selected for during tumour progression?
Are known mutations that reduce the fidelity of DNA polymerases present in tumour cells?
Are translesion DNA polymerases upregulated in tumours?
Are translesion DNA polymerases error-prone in copying unaltered DNA templates?
DNA repair
Are mutations in DNA repair genes selectively enriched during tumour progression?
Is there coordinate regulation of DNA repair pathways in response to DNA damage?
Does DNA repair capacity foretell the effectiveness of some chemotherapeutic drugs?
Metastasis
When are metastases seeded?
When do metastases proliferate?
Are there metastasis genes?
Are there regions of primary tumours that generate metastasis?
Are metastases inhibited by the same drugs as primary tumours?
Other questions
Do cancers arise in fields of premalignant cells?
Are there mutant genes that render tumours more susceptible to chemotherapy?
Do tumours or metastases have periods of dormancy?
Are there genes that specify dormancy?
Are passenger mutations neutral?
Do passenger mutations occur more frequently at hot spots for mutagenesis in nonmalignant cells?
Will the quantification of mutations that render cells resistant to a drug correlate with lack of therapeutic response?
As an alternative to targeting drugs against individual enzymes or pathways, it might be feasible to affect the rate of production of mutations in cancer cells. I have postulated that the progressive accumulation of mutations is one of the underlying processes that characterize the cancer phenotype. A decrease in the accumulation of mutations could delay cancer progression and serve as a preventative measure. Conversely, increasing the mutation rate might exceed the threshold of viability. This option would be targeted to cancers that have a high frequency of mutations as a result of prior treatment with mutagenic chemotherapeutic agents. Chemicals that induce lethal mutagenesis in tumours should be sought and explored therapeutically in individuals who lack other treatment options. Although many chemotherapeutic agents are already mutagenic, evaluating the therapeutic potential of highly mutagenic nucleoside analogues may present an additional approach to cancer therapy.
Acknowledgments
I thank E. Fox, B. Preston, A. Kamath, M. Horwitz and S. Nishimura for critical reading and D. Lim for constructing the figures. I am indebted to present and former members of our laboratory for establishing some of the evidence presented and for daily discussions. This work was supported by Grants from the US National Cancer Institute (CA-102029, CA-105802 and CA-77852) and the US National Institute of Aging (AG-033061).
Footnotes
Competing interests statement: The author declares no competing financial interests.
References
- 1.Loeb LA, Springgate CF, Battula N. Errors in DNA replication as a basis of malignant change. Cancer Res. 1974;34:2311–2321. [PubMed] [Google Scholar]
- 2.Loeb L, Monnat R. DNA polymerases and human disease. Nature Rev Genet. 2008;9:594–604. doi: 10.1038/nrg2345. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 4.Friedberg EC, et al. In: DNA Repair and Mutagenesis. Friedberg EC, editor. Ch. 2. ASM Publishing; Washington, DC: 2006. [Google Scholar]
- 5.Lange SS, Takata K, Wood RD. DNA polymerases and cancer. Nature Rev Cancer. 2011;11:96–110. doi: 10.1038/nrc2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Preston RJ. Mechanistic data and cancer risk assessment: the need for quantitative molecular endpoints. Environ Mol Mutagen. 2005;45:214–221. doi: 10.1002/em.20093. [DOI] [PubMed] [Google Scholar]
- 7.Schmitt MW, et al. Active site mutations in mammalian DNA polymerase delta alter accuracy and replication fork progression. J Biol Chem. 2010;285:32264–32272. doi: 10.1074/jbc.M110.147017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination and cancer biology. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 9.Tlsty TD, et al. Loss of chromosomal integrity in neoplasia. Cold Spring Harbor Symp Quant Biol. 1993;58:645–654. doi: 10.1101/sqb.1993.058.01.072. [DOI] [PubMed] [Google Scholar]
- 10.Kolodner RD, Putnam CD, Myung K. Maintenance of genome stability in Saccharomyces cerevisiae. Science. 2002;297:552–557. doi: 10.1126/science.1075277. [DOI] [PubMed] [Google Scholar]
- 11.Masutani C, et al. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 12.Loeb KR, Loeb LA. Significance of multiple mutations in cancer. Carcinogenesis. 2000;21:379–385. doi: 10.1093/carcin/21.3.379. [DOI] [PubMed] [Google Scholar]
- 13.Nishimura S. 8-Hydroxyguanine: from its discovery in 1983 to the present status. Proc Jpn Acad. 2006;82:127–141. doi: 10.2183/pjab.82.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McBride TJ, Preston BD, Loeb LA. Mutagenic spectrum resulting from DNA damage by oxygen radicals. Biochemistry. 1991;30:207–213. doi: 10.1021/bi00215a030. [DOI] [PubMed] [Google Scholar]
- 15.Kunkel TA, Patel SS, Johnson KA. Error-prone replication of repeated DNA sequences by T7 DNA polymerase in the absence of its processivity subunit. Proc Natl Acad Sci USA. 1994;91:6830–6834. doi: 10.1073/pnas.91.15.6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen JC, Rideout WM, Jones PA. The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 1994;22:972–976. doi: 10.1093/nar/22.6.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenman C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fox EJ, Salk JJ, Loeb LA. Cancer genome sequencing — an interim analysis. Cancer Res. 2009;69:4948–4950. doi: 10.1158/0008-5472.CAN-09-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bielas JH, Loeb KR, Rubin BP, True LD, Loeb LA. Human cancers express a mutator phenotype. Proc Natl Acad Sci USA. 2006;103:18238–18242. doi: 10.1073/pnas.0607057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Albertson TM, et al. DNA polymerase ε and δ proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci USA. 2009;10617104:17101-. doi: 10.1073/pnas.0907147106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beckman RA, Loeb LA. Efficiency of carcinogenesis with and without a mutator mutation. Proc Natl Acad Sci USA. 2006;103:14140–14145. doi: 10.1073/pnas.0606271103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campbell PJ, et al. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc Natl Acad Sci USA. 2008;105:13081–13086. doi: 10.1073/pnas.0801523105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bielas J, Loeb L. Quantification of random genomic mutations. Nature Methods. 2005;2:285–290. doi: 10.1038/nmeth751. [DOI] [PubMed] [Google Scholar]
- 24.Salk JJ, Fox EJ, Loeb LA. Mutational heterogeneity in human cancers: origins and consequences. Annu Rev Pathol. 2010;5:51–75. doi: 10.1146/annurev-pathol-121808-102113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wells RD, Sinden RR. Genome Analysis, Genome Rearrangement and Stability. Cold Spring Harbor Laboratory Press; Plainview, New York: 1993. pp. 107–138. [Google Scholar]
- 26.Wang G, Carbajal S, Vijg J, DiGiovanni J, Vasquez KM. DNA structure-induced genomic instability in vivo. J Natl Cancer Inst. 2008;100:1815–1817. doi: 10.1093/jnci/djn385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stephens PJ, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stephens P, et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nature Genet. 2005;37:590–592. doi: 10.1038/ng1571. [DOI] [PubMed] [Google Scholar]
- 29.Ding L, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roach JC, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pleasance ED, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2011;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nature Rev Mol Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 33.Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 34.Avkin S, et al. Quantitative analysis of translesion DNA synthesis across a benzo[a]pyrene-guanine adduct in mammalian cells. J Biol Chem. 2004;279:53298–53305. doi: 10.1074/jbc.M409155200. [DOI] [PubMed] [Google Scholar]
- 35.Lee W, et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature. 2011;465:473–477. doi: 10.1038/nature09004. [DOI] [PubMed] [Google Scholar]
- 36.Pleasance ED, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Druker BJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 38.Sawyers CL, et al. Imatinib induces hematologic and cytogenetic reponses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. 2002;99:3530–3539. doi: 10.1182/blood.v99.10.3530. [DOI] [PubMed] [Google Scholar]
- 39.Radich JP, et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci USA. 2006;103:2794–2799. doi: 10.1073/pnas.0510423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soverini S, et al. ABL mutations in late chronic phase chronic myeloid leukemia patients with up-front cytogenetic resistance to imatinib are asociated with a greater likeihood of progression to blast crisis and shorter survival: a study by the GIMEMA Working Party on Chronic Myeloid Leukemia. J Clin Oncol. 2005;23:4100–4109. doi: 10.1200/JCO.2005.05.531. [DOI] [PubMed] [Google Scholar]
- 41.Ley TJ, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 43.Misra A, et al. Clonal mutations in primary human glial tumors: evidence in support of the mutator hypothesis. BMC Cancer. 2007;7:190. doi: 10.1186/1471-2407-7-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li R, Sonik A, Stindl R, Rasnick D, Duesberg P. Aneuploidy vs. gene mutation hypothesis of cancer: recent study claims mutation but is found to support aneuploidy. Proc Natl Acad Sci USA. 2000;97:3236–3241. doi: 10.1073/pnas.040529797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boveri T. Über mehrpolige Mitosen als Mittel zur Analyse des Zellkerns. Verh Phys Med Ges Würzburg. 1902;35:67–90. [Google Scholar]
- 46.Foulds L. The experimental study of tumor progression: a review. Cancer Res. 1954;14:327–339. [PubMed] [Google Scholar]
- 47.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 48.Leary RJ, et al. Integrated analysis of homozygous deletions, focal amplifications, and sequence alterations in breast and colorectal cancers. Proc Natl Acad Sci USA. 2008;105:16224–16229. doi: 10.1073/pnas.0808041105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weir B, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stephens PJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aaltonen LA, et al. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812–816. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 52.Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993;260:816–819. doi: 10.1126/science.8484122. [DOI] [PubMed] [Google Scholar]
- 53.Boland CR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 54.Risinger JL, et al. Genetic instability of microsatellites in endometrial carcinoma. Cancer Res. 1993;53:5100–5103. [PubMed] [Google Scholar]
- 55.Peinado MA, Malkhosyan S, Velazquez A, Perucho M. Isolation and characterization of allelic loss and gains in colorectal tumors by arbitrarily primed polymerase chain reaction. Proc Natl Acad Sci USA. 1992;89:10065–10069. doi: 10.1073/pnas.89.21.10065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–561. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- 57.Wooster R, et al. Instability of short tandem repeats (microsatellites) in human cancers. Nature Genet. 1994;6:152–156. doi: 10.1038/ng0294-152. [DOI] [PubMed] [Google Scholar]
- 58.Shibata D, Navidi W, Salovaara R, Li ZH, Aaltonen LA. Somatic microsatellite mutations as molecular tumor clocks. Nature Med. 1996;2:676–681. doi: 10.1038/nm0696-676. [DOI] [PubMed] [Google Scholar]
- 59.Maley CC, et al. Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett's esophagus. Cancer Res. 2004;64:3414–3427. doi: 10.1158/0008-5472.CAN-03-3249. [DOI] [PubMed] [Google Scholar]
- 60.Salk JJ, et al. Clonal expansions in ulcerative colitis identify patients with neoplasia. Proc Natl Acad Sci USA. 2009;106:20871–20876. doi: 10.1073/pnas.0909428106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heppner GH. Tumor heterogeneity. Cancer Res. 1984;44:2259–2265. [PubMed] [Google Scholar]
- 62.Fidler IJ, Hart IR. Biological and experimental consequences of the zonal composition of solid tumors. Cancer Res. 1981;41:3266–3267. [PubMed] [Google Scholar]
- 63.Mitelman F, Mark J, Levan G, Levan A. Tumor etiology and chromosome pattern. Science. 1972;176:1340–1341. doi: 10.1126/science.176.4041.1340. [DOI] [PubMed] [Google Scholar]
- 64.Misra A, et al. Extensive intra-tumor heterogeneity in primary human glial tumors as a result of locus non- specific genomic alterations. J Neurooncol. 2000;48:1–12. doi: 10.1023/a:1006435201961. [DOI] [PubMed] [Google Scholar]
- 65.Gonzalez-Garcia I, Sole RV, Costa J. Metapopulation dynamics and spatial heterogeneity in cancer. Proc Natl Acad Sci USA. 2002;99:13085–13089. doi: 10.1073/pnas.202139299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yachida S, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Friedberg EC, et al. DNA Repair and Mutagenesis. Ch. 17. ASM Press; Washington, DC: 2006. [Google Scholar]
- 68.Guo HH, Choe J, Loeb LA. Protein tolerance to random amino acid change. Proc Natl Acad Sci USA. 2004;101:9205–9210. doi: 10.1073/pnas.0403255101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zheng L, et al. Fen1 mutations result in autoimmunity, chronic inflammation and cancers. Nature Med. 2007;13:812–819. doi: 10.1038/nm1599. [DOI] [PubMed] [Google Scholar]
- 70.Riethmuller G, Johnson JP. Monoclonal antibodies in the detection and therapy of micrometastatic epithelial cancers. Curr Opin Immunol. 1992;4:647–655. doi: 10.1016/0952-7915(92)90041-c. [DOI] [PubMed] [Google Scholar]
- 71.Schmidt-Kittler O, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci USA. 2003;100:7737–7742. doi: 10.1073/pnas.1331931100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Klein A, et al. Identification of brain- and bone-specific breast cancer metastasis genes. Cancer Lett. 2009;276:212–220. doi: 10.1016/j.canlet.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 73.Klein CA. Parallel progression of primary tumours and metastases. Nature Rev Cancer. 2009;9:302–312. doi: 10.1038/nrc2627. [DOI] [PubMed] [Google Scholar]
- 74.Hartwell LH, Kastan MB. Cell cycle control and cancer. Science. 1994;266:1821–1827. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 75.Paulovich AG, Toczyski DP, Hartwell LH. When checkpoints fail. Cell. 1997;88:315–321. doi: 10.1016/s0092-8674(00)81870-x. [DOI] [PubMed] [Google Scholar]
- 76.King MC, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643–646. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 77.Fong PC, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28:2512–2519. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 78.Fong PC, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 79.Holland JJ, Domingo E, de la Torre JC, Steinhauer DA. Mutation frequencies at defined single codon sites in vesicular stomatitis virus and poliovirus can be increased only slightly by chemical mutagenesis. J Virol. 1990;64:3960–3962. doi: 10.1128/jvi.64.8.3960-3962.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Loeb LA, et al. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc Natl Acad Sci USA. 1999;96:1492–1497. doi: 10.1073/pnas.96.4.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fox EJ, Loeb LA. Lethal mutagenesis: targeting the mutator phenotype in cancer. Semin Cancer Biol. 2010;20:353–359. doi: 10.1016/j.semcancer.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Eigen M. Viral quasispecies. Sci Am. 1993;269:42–49. doi: 10.1038/scientificamerican0793-42. [DOI] [PubMed] [Google Scholar]
- 83.Wang TL, et al. Prevalence of somatic alterations in the colorectal cancer cell genome. Proc Natl Acad Sci USA. 2002;99:3076–3080. doi: 10.1073/pnas.261714699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bodmer W, Bielas JH, Beckman RA. Genetic instability is not a requirement for tumor development. Cancer Res. 2008;68:3558–3560. doi: 10.1158/0008-5472.CAN-07-6544. discussion 3560-3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Loh E, Choe J, Loeb LA. Highly tolerated amino acid substitutions increase the fidelity of E. coli DNA polymerase I. J Biol Chem. 2007;282:12201–12209. doi: 10.1074/jbc.M611294200. [DOI] [PubMed] [Google Scholar]
- 86.Beckman RA, Loeb LA. Negative clonal selection in tumor evolution. Genetics. 2005;171:2123–2131. doi: 10.1534/genetics.105.040840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weedon-Fekjaer H, Lindqvist BH, Vatten LJ, Aalen OO, Tretli S. Breast cancer tumor growth estimated through mammography screening data. Breast Cancer Res. 2008;10:R41. doi: 10.1186/bcr2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Barkan D, Green JE, Chambers AF. Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. Eur J Cancer. 2010;46:1181–1188. doi: 10.1016/j.ejca.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weckermann D, et al. Perioperative activation of disseminated tumor cells in bone marrow of patients with prostate cancer. J Clin Oncol. 2009;27:1549–1556. doi: 10.1200/JCO.2008.17.0563. [DOI] [PubMed] [Google Scholar]
- 90.Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991;351:453–456. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- 91.Starcevic D, Dalal S, Sweasy JB. Is there a link between DNA polymerase beta and cancer? Cell Cycle. 2004;3:998–1001. [PubMed] [Google Scholar]
- 92.Schmitt MW, Matsumoto Y, Loeb LA. High fidelity and lesion bypass capability of human DNA polymerase δ. Biochimie. 2009;91:1163–1172. doi: 10.1016/j.biochi.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fry M, Loeb LA. A DNA polymerase α pause site is a hot spot for nucleotide misinsertion. Proc Natl Acad Sci USA. 1992;89:763–767. doi: 10.1073/pnas.89.2.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vasquez KM, Hanawalt PC. Intrinsic genomic instability from naturally occurring DNA structures: an introduction to the special issue. Mol Carcinog. 2009;48:271–272. doi: 10.1002/mc.20519. [DOI] [PubMed] [Google Scholar]
- 95.Goldsby RE, et al. Defective DNA polymerase-δ proofreading causes cancer susceptibility in mice. Nature Med. 2001;7:638–639. doi: 10.1038/88963. [DOI] [PubMed] [Google Scholar]
- 96.Albertson DG, Collins C, McCormick F, Gray JW. Chromosome aberrations in solid tumors. Nature Genet. 2003;34:369–376. doi: 10.1038/ng1215. [DOI] [PubMed] [Google Scholar]
- 97.Venkatesan RN, Hsu JJ, Lawrence NA, Preston BD, Loeb LA. Mutator phenotypes caused by substitution at a conserved motif A residue in eukaryotic DNA polymerase δ. J Biol Chem. 2006;281:4486–4494. doi: 10.1074/jbc.M510245200. [DOI] [PubMed] [Google Scholar]
- 98.Levine RL, et al. Translesion DNA synthesis catalyzed by human pol η and pol κ across 1,N6-ethenodeoxyadenosine. J Biol Chem. 2001;276:18717–18721. doi: 10.1074/jbc.M102158200. [DOI] [PubMed] [Google Scholar]
- 99.Haracska L, Prakash L, Prakash S. Role of human DNA polymerase κ as an extender in translesion synthesis. Proc Natl Acad Sci USA. 2002;99:16000–16005. doi: 10.1073/pnas.252524999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hoffmann JS, Cazaux C. Aberrant expression of alternative DNA polymerases: a source of mutator phenotype as well as replicative stress in cancer. Semin Cancer Biol. 2011;20:312–319. doi: 10.1016/j.semcancer.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 101.Sjoblom T, et al. The concensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 102.Wood LD, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 103.Timmermann B, et al. Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS ONE. 2011;5:e15661. doi: 10.1371/journal.pone.0015661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jones S, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jiao Y, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011 Jan 20; doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Parsons D, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dalgliesh GL, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2011;463:360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Varela I, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jones S, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2011;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Parsons DW, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435–439. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ley TJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shah SP, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 114.Ding L, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Berger MF, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chapman MA, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 1954;8:1–12. doi: 10.1038/bjc.1954.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Renan MJ. How many mutations are required for tumorigenesis? Implications from human cancer data. Mol Carcinog. 1993;7:139–146. doi: 10.1002/mc.2940070303. [DOI] [PubMed] [Google Scholar]