

Abstract
Licochalcone (LC), a major phenolic retrochalcone from licorice, has anti-inflammatory activity. This study investigated the effects of licochalcone A (LCA) and licochalcone E (LCE) on Liver X receptor-α (LXRα)-mediated lipogenic gene expression and the molecular mechanisms underlying those effects. LCA and LCE antagonized the ability of LXRα agonists (T0901317 or GW3965) to increase sterol regulatory element binding protein-1c (SREBP-1c) expression and thereby inhibited target gene expression (e.g., FAS and ACC) in HepG2 cells. Moreover, treatment with LCA and LCE impaired LXRα/RXRα-induced CYP7A1-LXRE-luciferase (CYP7A1) transactivation. The AMPK-Sirt1 signaling pathway is an important regulator of energy metabolism and, therefore, a potential therapeutic target for metabolic diseases, including hepatic steatosis. We found here that LCE increased AMPK phosphorylation and Sirt1 expression. We conclude that LC inhibits SREBP-1c-mediated hepatic lipogenesis via activation of the AMPK/Sirt1 signaling pathway.
Keywords: Licochalcone A, Licochalcone E, Hepatic steatosis, Sterol regulatory element binding protein-1c, Liver X receptor-α
INTRODUCTION
Liver X receptor-α (LXRα), a member of the nuclear receptor superfamily, binds to the DR-4 motif known as the LXR response element (LXRE) in its target genes and acts as an important regulator of cholesterol, fatty acids, and bile acids (1). LXRα increased the efflux of free cholesterol as well as nascent and mature HDL through upregulation of the ATP binding cassette (ABC) sterol transporters, such as ABCA1 and ABCG1 (2). Activation of LXRα, however, is associated with increased lipogenesis, hypertriglycemia, and fat accumulation through de novo fatty acid synthesis in the liver due to the LXRα-induced increase in the expression of lipogenic genes, such as fatty acid synthase (FAS), acetyl-CoA carboxylase (ACC), and stearoyl-CoA desaturase-1 (SCD-1) (3). Sterol regulatory element binding protein-1c (SREBP-1c) is an essential transcription factor for lipogenic gene expression (4). SREBP-1c is fundamental to the pathogenesis of metabolic diseases, including hepatic steatosis, and has been suggested as a potential therapeutic target (5).
Licorice root from Glycyrrhiza inflata (G. inflata) has been used in traditional and herbal medicines. This species contains unusual phenolic compounds, called retrochalcones, which include licochalcone A to E and echinatin. Licochalcone A (LCA; Fig. 1A upper panel) has been demonstrated to have a variety of pharmacological activities, including anti-bacterial, anti-cancer, and anti-inflammatory activities (6-9). Licochalcone E (LCE; Fig. 1A lower panel) has recently been isolated and characterized from G. inflata (10). The pharmacological efficacy of LCE has been studied extensively. Anti-diabetic (11), anti-inflammatory (12), and cytotoxic effects (13) have been reported for LCE. Moreover, recent report showed the anti-lipogenic effect of LCA in HepG2 cells and mice fed on high fat diet (14). They showed that the basal expression levels of lipogenic gene were decreased by LCA in HepG2 cells. In addition, LCA attenuated high fat diet-induced fat accumulation in the mice. However, the effects of LCA and LCE on LXRα-activated lipogenic gene expression and the role of AMPactivated protein kinase (AMPK) and sirtuin 1 Sirt1) have not yet been investigated.
Fig. 1. The effects of LCA and LCE on cytotoxicity. (A) Chemical structure of licochalcone A (LCA) and licochalcone E (LCE). (B) MTT assays for cell viability. The effect of LCA or LCE (0~10 μM, 12 hr treatment) on cell viability was assessed using MTT assays.

The AMPK/Sirt1 signaling pathway is well established as a key sensor of energy status and an important regulator of glucose and lipid metabolism, therefore making this pathway a promising therapeutic target for both aging (15) and metabolic diseases, including diabetes, obesity, and hepatic steatosis (16). Many natural compounds, like resveratrol and berberine, also activate the AMPK/Sirt1 signaling pathway and have beneficial effects on lipid metabolism and insulin sensitivity (17,18).
Thus, the purpose of this study was to investigate if LCA and LCE could inhibit LXRα-dependent lipogenic gene expression in hepatocytes and, if so, the molecular mechanism. The results of the current study show that LCA and LCE do impair LXRα-mediated SREBP-1c expression and thereby inhibit SREBP-1c target gene expression. Moreover, treatment with LCA and LCE attenuated LXRα- and Retinoid X Receptor-α (RXRα)-activated LXRE transactivation. Our results demonstrate that LC increased AMPK/ Sirt1 activity, which contributes to the inhibition of LXRα-mediated hepatic lipogenesis.
MATERIALS AND METHODS
Materials. Anti-SREBP-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-β-actin GW3965 (Sigma, St. Louis, MO, USA), and anti-Sirt1 (Novus, Littleton, CO, USA) antibodies as well as anti-FAS, anti-ACC, antipAMPK, and anti-AMPK antibodies (all from Cell Signaling, Beverly, MA, USA) were used for experiments. T0901317 (T090) was purchased from Calbiochem (San Diego, CA, USA). LCA and LCE were kindly donated from Dr. SH Cheon (Chonnam National University).
Cell culture and treatment. HepG2 cells were obtained from ATCC (American Type Culture Collection, Manassas, VA, USA). Primary hepatocytes were isolated from male ICR mice. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS, Hyclone, Logan, UT, USA) and 50 units/ml penicillin/streptomycin at 37℃ in a humidified atmosphere with 5% CO2. Cells were plated at 1 × 105 per well in six-well plates, and wells were used when the cells were 70~80% confluent. LCA or LCE (0~10 μM), dissolved in dimethylsulfoxide (DMSO), was added to cells and incubated at 37℃ for the indicated time period. The final concentration of DMSO in culture medium did not exceed 0.1% (v/v) to minimize solvent effects. Control cells were treated with equivalent volumes of DMSO alone. Cells were washed twice with ice-cold phosphate-buffered saline (PBS) before sample preparation.
Primary hepatocyte isolation. Primary hepatocytes were isolated and cultured as described previously (19). Briefly, ICR mice were anesthetized with Zoletil (Virbac, France), and the portal vein was cannulated under aseptic conditions. The livers were perfused in situ with Ca2+-free Hank’s balanced saline solution (HBSS) at 37℃ for 5 min. The livers were then perfused for 5 min with HBSS containing 0.05% collagenase and Ca2+ at a perfusion flow rate of 10 ml/min. After perfusion, the livers were minced gently with scissors and resuspended in sterilized PBS. The cell suspensions were then filtered through cell strainers and centrifuged at 50 ×g for 5 min to separate parenchymal and nonparenchymal cells. The viability of isolated hepatocytes estimated by trypan blue staining was usually 80~90%. Isolated hepatocytes were plated on collagen-coated plates and cultured in DMEM containing 50 units/ml penicillin/streptomycin and 10% FBS.
MTT cell viability assay. To measure cytotoxicity, HepG2 cells were plated at a density of 1 × 105 cells/well in 48-well plates and treated with LCA or LCE (0~10 μM, 12 hr). After treatment, viable cells were stained with MTT (0.2 mg/ml, 4 hr). The media were then removed, and the formazan crystals produced in the wells were dissolved in 200 ml of DMSO. The absorbance at 540 nm was measured using an enzyme-linked immunosorbent assay microplate reader (SpectraMAX, Molecular Devices, Sunnyvale, CA, USA). Cell viability was defined relative to the untreated control [i.e., viability (% control) = 100 × (absorbance of treated sample)/(absorbance of control)].
Immunoblot analysis. SDS-polyacrylamide gel electrophoresis and immunoblot analyses were performed according to previously published procedures (20). The protein of interest in cell lysates was isolated using a 7.5% gel electrophoresis and then electophoretically transferred to nitrocellulose paper. After blocking, membranes were incubated with antibodies against SREBP-1 at a dilution of 1 : 1000 at 4℃ overnight, followed by further incubation with a secondary antibody (1 : 5000). Immunoreactive protein was visualized using an ECL chemiluminescence detection kit (Amersham Biosciences, Buckinghamshire, UK). Equal loading of proteins was verified by Coomassie blue staining of gels and β-actin immunoblotting.
RNA isolation and real-time RT-PCR analysis. Total RNA was extracted using Trizol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. To obtain cDNA, total RNA (1 μg) was reverse-transcribed using an oligo(dT)18 primer to obtain cDNA. The cDNA was amplified using a high capacity cDNA synthesis kit (Bioneer, Daejon, Korea) in a thermal cycler (Bio-rad, Hercules, CA, USA). Real-time PCR was performed with StepOneTM (Applied Biosystems, Foster City, CA) using a SYBR® Green premix according to the manufacturer’s instructions (Applied Biosystems). The following primer sequences were used: human SREBP-1c, 5'-CGACATCGAAGACATGCTTCAG-3' (sense) and 5'-GGAAGGCTTCAAGAGAGGAGC-3' (antisense). The relative levels of PCR products were determined using the threshold cycle value. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the reference gene for normalization. Melting curve analysis was performed after amplification to verify the accuracy of the amplicon.
Transient transfection and reporter gene assay. The luciferase reporter plasmid TK-CYP7a-LXRE(X3)-LUC, which contains three tandem copies of the sequence (5'- GCTTTGGTCACTCAAGTTCAAGTTA-3') from the rat Cyp7a1 gene, and the overexpression vector for LXRα were described previously (20). To determine the luciferase activities, we used the dual-luciferase reporter assay system (Promega, Madison, WI, USA). Briefly, HepG2 cells were replated in 12-well plates overnight, serum-starved for 6 hr, and transiently transfected with LXRα, RXRα, TK-CYP7a-LXRE(X3)-LUC, and pRL-TK plasmid, which encodes for Renilla luciferase and is used to normalize transfection efficacy, in the presence of Lipofectamine® Reagent (Invitrogen, San Diego, CA, USA) for 3 hr. Transfected cells were further incubated in DMEM containing 1% FBS for the indicated time periods.
Statistical analysis. One-way analysis of variance (ANOVA) was used to assess the statistical significance of differences among treatment groups. For each statistically significant treatment result, the Newman-Keuls test was used for comparisons between multiple group means. The data were expressed as means ± SEM.
RESULTS
The inhibition of LXRα-mediated SREBP-1c expression by LCA or LCE treatment. Colorimetric MTT assays were performed to evaluate the cytotoxicity of LCA or LCE treatment. Because no cytotoxicity was observed with LCA or LCE treatment up to 10 μM for 12 hr in HepG2 cells (Fig. 1B), 10 μM was used as the maximum concentration for in vitro studies. Next, the LXRα synthetic agonist T0901317 T090) and GW3965 compound were used to investigate the effect of LCA and LCE on LXRα-induced lipogenic gene expression. T090 treatment increased SREBP-1 expression. However, LCA or LCE pretreatment markedly inhibited T090-induced SREBP-1c (Fig. 2A). In addition, GW3965, another synthetic LXRα agonist, induced SREBP-1c expression, which was also attenuated by LCA or LCE pretreatment (Fig. 2B). Real-time RT-PCR analyses clearly showed that T090 treatment for 12 hr increased SREBP-1c mRNA levels in HepG2 cells, and that increase was significantly suppressed by LCE treatment (Fig. 2C). A similar pattern of LCE inhibition was observed in the SREBP-1c mRNA levels in primary hepatocytes (Fig. 2D).
Fig. 2. LCA and LCE inhibit T090-induced SREBP-1c protein and mRNA levels. (A) The effects of LCA or LCE on T0901317 (T090)-mediated SREBP-1c expression in HepG2 cells. Immunoblot analyses were performed on lysates of cells treated with LCA (upper) or LCE (lower) for 1 hr with subsequent treatment with 10 μM T090 for 12 hr. (B) The effects of LCA or LCE on GW3965-induced SREBP-1c expression in HepG2 cells. (C) Real-time RT-PCR assays. HepG2 cells were treated with T090 or vehicle in combination with LCE for 12 hr. The transcripts of SREBP-1c genes were analyzed by real-time RT-PCR assays, with GAPDH used as a reference gene for normalization. (D) Primary mouse hepatocytes were treated with LCE in combination with T090 for 12 hr. The levels of SREBP-1c mRNA were analyzed by real-time RT-PCR assays. Data represent the mean ± S.E.M. of 4 separate experiments. The statistical significance of differences between each treatment group and the control (##p < 0.01) or T090 alone (*p<0.05, **p< 0.01) was determined.

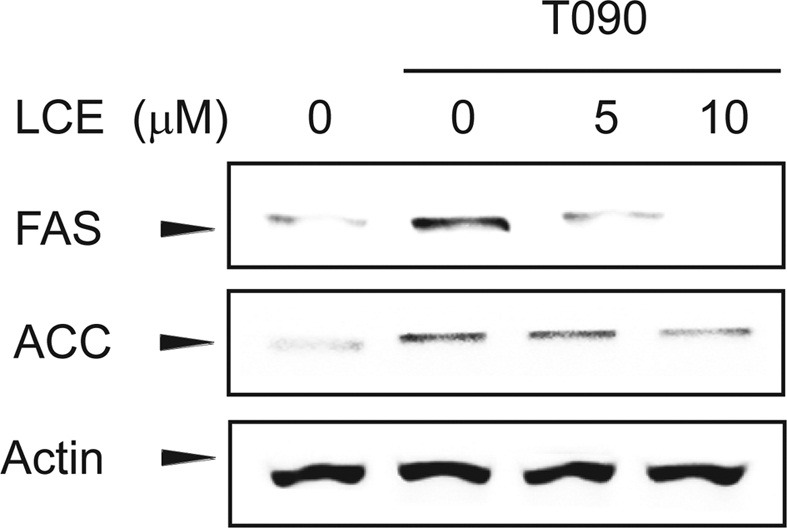
The inhibition of SREBP-1 target gene expression by LCA or LCE treatment. SREBP-1c was initially suggested in nutrition to regulate lipogenic genes, such as those coding for FAS and ACC in the liver and adipose tissue. Because SREBP-1c is a major transcription factor involved in the nutritional regulation of lipogenesis, the effect of LCE pretreatment on SREBP-1c-activated target gene expression was examined. LCE treatment attenuated the ability of T090 to induce the expression of the SREBP-1c target genes encoding FAS and ACC (Fig. 3).
Fig. 3. LCE inhibits T090-induced SREBP-1c target gene expression. Immunoblot analyses were carried out using the lysates of cells treated with LCE for 1 hr with subsequent treatment with 10 μM T090 for 12 hr.
The inhibition of LXRα-dependent CYP7A1 gene induction by LCA or LCE treatment. To determine if LXRα activity was altered by LCA or LCE treatment, reporter gene assays employing the LXR response element were used. LCA or LCE treatment diminished the transcriptional activity of LXRα (Fig. 4). LXRα and RXRα transfection increased the transactivation of LXRE-luciferase activity as previously reported (20), and treatment with either LCA or LCE attenuated that LXRE-luciferase activity (Fig. 4). These results demonstrate that both LCA and LCE inhibit LXRα-mediated lipogenic gene expression via LXRα inactivation.
Fig. 4. LCA and LCE inhibit LXRα-/RXRα-dependent LXREluciferase activity. LXRE-luciferase transactivation was determined using lysates of HepG2 cells treated with LCA or LCE for 12 hr after transfection of plasmids encoding for LXRα and RXRα. Data represent the mean ± S.E. of 4 separate experiments. The statistical significance of differences between each treatment group and the control (##p< 0.01) or T090 without LCA or LCE (**p< 0.01) was determined.

The activation of the AMPK/Sirt1 signaling pathway by LCA or LCE treatment. AMPK has been reported to inhibit cleavage and transcriptional activation of SREBP-1c via direct phosphorylation (20,21). Sirt1 expression and activity increased during fasting and resulted in deacetylation and inhibition of SREBP-1c activity and target gene expression (22). Thus AMPK/Sirt1-mediated inactivation of SREBP-1c may offer therapeutic strategies to combat metabolic disorders, such as steatosis, insulin resistance, and ath-erosclerosis. Therefore, the effects of LCE treatment on AMPK and Sirt1 were assessed. The time course of AMPK phosphorylation in response to 10 μM LCE (Fig. 5A) was determined. Phosphorylation of AMPK increased 0.5~6 hr after LCE treatment. In contrast, by 12 hr of LCE treatment, AMPK phosphorylation returned to basal levels. Next, the time course of Sirt1 expression in response to LCE was examined. Consistent with AMPK phosphorylation, Sirt1 expression increased after LCE treatment and peaked at 3~6 hr after LCE treatment. These results suggest that LCE-mediated repression of T090-induced LXRα target gene expression might be due to AMPK/Sirt1 activation (Fig. 5C).
Fig. 5. The LCE-induced activation of AMPK and Sirt1. (A) Immunoblot analysis for LCE-induced AMPK phosphorylation. Levels of AMPK phosphorylation were determined in cell lysates incubated with 10 μM LCE for 30 min to 12 hr. (B) Immunoblot analysis for LCE-induced Sirt1 expression. Results were confirmed in 3 replicate experiments. (C) Schematic diagram illustrating the mechanism by which LCA and LCE inhibit LXRα-mediated SREBP-1c expression and lipogenic gene expression via the AMPK/Sirt1 pathway.

DISCUSSION
Nonalcoholic fatty liver disease (NAFLD), an epidemic global health problem, is a term used to describe a wide range of conditions from simple steatosis to steatohepatitis and cirrhosis. NAFLD is associated with metabolic disorders including obesity, insulin resistance, hyperdislipidemia, and high blood pressure (23). The main cause of NAFLD is the accumulation of fat within hepatocytes, and steatosis is defined as fat accumulation in more than 5% of hepatocytes (24). Hepatic steatosis partially manifests due to increased lipogenesis and reduced fatty acid b-oxidation in the liver. To prevent or reverse NAFLD, it is essential to understand the precise mechanism of lipogenesis in hepatocytes.
LXR serves as a sterol sensor and plays a pivotal role in regulating the expression of genes involved in hepatic lipogenesis. LXR can directly promote the expression of SREBP-1c via two LXREs in the SREBP-1c promoter (25), which result in the upregulation of lipogenic genes. The LXR agonist, T090, showed lipogenic effects through the transcriptional induction of genes associated with fatty acid synthesis, including SREBP1c, ACC, and FAS under certain physiological conditions (26). In the case of β-oxidation, the LXR agonist (T090) increased peroxisomal β-oxidation as a counterregulatory response to the hypertriglyceridemia in vivo. Throughout this study, the representative retrochalcone compounds LCA and LCE significantly inhibited SREBP-1c expression via LXRα inactivation in hepatocytes, as further demonstrated by the suppression of SREBP-1c target gene expression (e.g., FAS and ACC).
In a recent study, AMPK was shown to directly phosphorylate and suppress LXRα activity (20). Other groups have also shown that AMPK inhibits activation and expression of SREBP-1c via direct phosphorylation (21). AMPK activators, including metformin and resveratrol, have been shown to inhibit SREPB-1c expression and prevent the progression of hepatic steatosis (19,27). Here, we showed that AMPK phosphorylation in response to LCE increased at 0.5~6 hr (Fig. 5A). These results suggest that suppression of SREBP-1c and its target genes in response to LCA and LCE treatment could be due to AMPK-mediated LXR/ SREBP-1c inhibition.
Sirt1, the mammalian ortholog of yeast Sir2, is an evolutionarily conserved NAD+-dependent protein deacetylase. It has been implicated in mediating the anti-aging effects of calorie restriction by regulating energy metabolism (28). Genetic or pharmacological activation of Sirt1 has beneficial effects on aging-associated medical complications, including both metabolic and neurodegenerative disorders (29). Our results showed that LCE treatment increased Sirt1 expression, which peaked 3~6 hr after LCE treatment. A recent report showed that SIRT1 enhances LXRα activity and target gene expression, including that of ABCA1, and this is accompanied by LXRα deacetylation (30). However, deacetylation of SREBP-1c by Sirt1 decreases SREBP-1c activity (22). In addition, pharmacological activators of Sirt1 inhibit SREBP-1c and its target gene expression both in vitro and in vivo, accompanied by decreased hepatic lipid and cholesterol levels (31). This discrepancy needs to be further studied to fully evaluate the role of Sirt1 on the LXR-SREBP1 axis.
Taken together, the data indicate that AMPK/SIRT1/SREBP axis is an attractive therapeutic target for the treatment of NAFLD. Activated AMPK increases the cellular NAD+/NADH ratio through transcriptional regulation of nicotinamide phosphoribosyltransferase (NAMPT), an essential co-factor for SIRT1 activity (32). Reciprocally, the activation of Sirt1 leads to the activation of AMPK. SIRT1 deacetylates liver kinase B1 (LKB1), a representative upstream kinase of AMPK, facilitating the ability of LKB1 to phosphorylate AMPK (17). However, further study is needed to fully elucidate the relationship between AMPK and SIRT1 and their roles in the regulation of hepatic metabolic process.
In conclusion, our study demonstrated for the first time that both LCA and LCE attenuated LXRα-mediated lipogenic gene expression by affecting LXRα-induced SREBP- 1c expression. The LC-mediated inhibition of lipogenesis may be due to increased AMPK phosphorylation and Sirt1 expression. Therefore, our study provides crucial information that would support the use of LCA and LCE as novel therapeutic candidates for the prevention or treatment of NAFLD and NASH.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
Acknowledgments
Financial Support: This study was supported by the Korea Science and Engineering Foundation (KOSEF) funded by the Korean government (MOST) (Grant No. 2011-0009835).
References
- 1.Jakobsson T., Treuter E., Gustafsson J.A., Steffensen K.R. Liver X receptor biology and pharmacology: new pathways, challenges and opportunities. Trends Pharmacol. Sci. 012;33:394–404. doi: 10.1016/j.tips.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 2.Akiyama T.E., Sakai S., Lambert G., Nicol C.J., Matsusue K., Pimprale S., Lee Y.H., Ricote M., Glass C.K., Brewer H.B. Jr., Gonzalez F.J. Conditional disruption of the peroxisome proliferator-activated receptor gamma gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Mol. Cell Biol. (2002);22:2607–2619. doi: 10.1128/MCB.22.8.2607-2619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schultz J.R., Tu H., Luk A., Repa J.J., Medina J.C., Li L., Schwendner S., Wang S., Thoolen M., Mangelsdorf D.J., Lustig K.D., Shan B. Role of LXRs in control of lipogenesis. Genes Dev. (2000);14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Repa J.J., Liang G., Ou J., Bashmakov Y., Lobaccaro J.M., Shimomura I., Shan B., Brown M.S., Goldstein J.L., Mangelsdorf D.J. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. (2000);14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferre P., Foufelle F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes. Metab. (2010);12(Suppl 2):83–92. doi: 10.1111/j.1463-1326.2010.01275.x. [DOI] [PubMed] [Google Scholar]
- 6.Kim J.K., Shin E.K., Park J.H., Kim Y.H., Park J.H. Antitumor and antimetastatic effects of licochalcone A in mouse models. J. Mol. Med. (Berlin) (2010);88:829–838. doi: 10.1007/s00109-010-0625-2. [DOI] [PubMed] [Google Scholar]
- 7.Chu X., Ci X., Wei M., Yang X., Cao Q., Guan M., Li H., Deng Y., Feng H., Deng X. Licochalcone a inhibits lipopolysaccharide-induced inflammatory response in vitro and in vivo. J. Agric. Food Chem. (2012);60:3947–3954. doi: 10.1021/jf2051587. [DOI] [PubMed] [Google Scholar]
- 8.Friis-Møller A., Chen M., Fuursted K., Christensen S.B., Kharazmi A. In vitro antimycobacterial and antilegionella activity of licochalcone A from Chinese licorice roots. Planta Med. (2002);68:416–419. doi: 10.1055/s-2002-32087. [DOI] [PubMed] [Google Scholar]
- 9.Haraguchi H., Ishikawa H., Mizutani K., Tamura Y., Kinoshita T. Antioxidative and superoxide scavenging activities of retrochalcones in Glycyrrhiza inflata. Bioorg. Med. Chem. (1998);6:339–347. doi: 10.1016/S0968-0896(97)10034-7. [DOI] [PubMed] [Google Scholar]
- 10.Yoon G., Jung Y.D., Cheon S.H. Cytotoxic allyl retrochalcone from the roots of Glycyrrhiza inflata. Chem. Pharm. Bull. (Tokyo) (2005);53:694–695. doi: 10.1248/cpb.53.694. [DOI] [PubMed] [Google Scholar]
- 11.Park H.G., Bak E.J., Woo G.H., Kim J.M., Quan Z., Yoon H.K., Cheon S.H., Yoon G., Yoo Y.J., Na Y., Cha J.H. Licochalcone E has an antidiabetic effect. J. Nutr. Biochem. (2012);23:759–767. doi: 10.1016/j.jnutbio.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 12.Lee H.N., Cho H.J., Lim do Y., Kang Y.H., Lee K.W., Park J.H. Mechanisms by which licochalcone e exhibits potent anti-inflammatory properties: studies with phorbol ester-treated mouse skin and lipopolysaccharide-stimulated murine macrophages. Int. J. Mol. Sci. (2013);14:10926–10943. doi: 10.3390/ijms140610926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoon G., Kang B.Y., Cheon S.H. Topoisomerase I inhibition and cytotoxicity of licochalcones A and E from Glycyrrhiza inflata. Arch. Pharmacal Res. (2007);30:313–316. doi: 10.1007/BF02977611. [DOI] [PubMed] [Google Scholar]
- 14.Quan H.Y., Kim S.J., Kim do Y., Jo H.K., Kim G.W., Chung S.H. Licochalcone A regulates hepatic lipid metabolism through activation of AMP-activated protein kinase. Fitoterapia. (2013);86:208–216. doi: 10.1016/j.fitote.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y., Liang Y., Vanhoutte P.M. SIRT1 and AMPK in regulating mammalian senescence: a critical review and a working model. FEBS Lett. (2011);585:986–994. doi: 10.1016/j.febslet.2010.11.047. [DOI] [PubMed] [Google Scholar]
- 16.Canto C., Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. (2009);20:98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hou X., Xu S., Maitland-Toolan K.A., Sato K., Jiang B., Ido Y., Lan F., Walsh K., Wierzbicki M., Verbeuren T.J., Cohen R.A., Zang M. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. (2008);283:20015–20026. doi: 10.1074/jbc.M802187200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomes A.P., Duarte F.V., Nunes P., Hubbard B.P., Teodoro J.S., Varela A.T., Jones J.G., Sinclair D.A., Palmeira C.M., Rolo A.P. Berberine protects against high fat dietinduced dysfunction in muscle mitochondria by inducing SIRT1-dependent mitochondrial biogenesis. Biochim. Biophys. Acta. (2012);1822:185–195. doi: 10.1016/j.bbadis.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin S.H., Yang J.H., Shin B.Y., Seo K., Shin S.M., Cho I.J., Ki S.H. Resveratrol inhibits LXRalpha-dependent hepatic lipogenesis through novel antioxidant Sestrin2 gene induction. Toxicol. Appl. Pharmacol. (2013);271:95–105. doi: 10.1016/j.taap.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 20.Hwahng S.H., Ki S.H., Bae E.J., Kim H.E., Kim S.G. Role of adenosine monophosphate-activated protein kinase-p70 ribosomal S6 kinase-1 pathway in repression of liver X receptor-alpha-dependent lipogenic gene induction and hepatic steatosis by a novel class of dithiolethiones. Hepatology. (2009);49:1913–1925. doi: 10.1002/hep.22887. [DOI] [PubMed] [Google Scholar]
- 21.Li Y., Xu S., Mihaylova M.M., Zheng B., Hou X., Jiang B., Park O., Luo Z., Lefai E., Shyy J.Y., Gao B., Wierzbicki M., Verbeuren T.J., Shaw R.J., Cohen R.A., Zang M. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. (2011);13:376–388. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ponugoti B., Kim D.H., Xiao Z., Smith Z., Miao J., Zang M., Wu S.Y., Chiang C.M., Veenstra T.D., Kemper J.K. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. (2010);285:33959–33970. doi: 10.1074/jbc.M110.122978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaggini M., Morelli M., Buzzigoli E., DeFronzo R.A., Bugianesi E., Gastaldelli A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients. (2013);5:1544–1560. doi: 10.3390/nu5051544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ludwig J., Viggiano T.R., McGill D.B., Oh B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. (1980);55:434–438. [PubMed] [Google Scholar]
- 25.Yoshikawa T., Shimano H., Amemiya-Kudo M., Yahagi N., Hasty A.H., Matsuzaka T., Okazaki H., Tamura Y., Iizuka Y., Ohashi K., Osuga J., Harada K., Gotoda T., Kimura S., Ishibashi S., Yamada N. Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol. Cell Biol. (2001);21:2991–3000. doi: 10.1128/MCB.21.9.2991-3000.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chisholm J.W., Hong J., Mills S.A., Lawn R.M. The LXR ligand T0901317 induces severe lipogenesis in the db/db diabetic mouse. J. Lipid Res. (2003);44:2039–2048. doi: 10.1194/jlr.M300135-JLR200. [DOI] [PubMed] [Google Scholar]
- 27.Yap F., Craddock L., Yang J. Mechanism of AMPK suppression of LXR-dependent Srebp-1c transcription. Int. J. Biol. Sci. (2011);7:645–650. doi: 10.7150/ijbs.7.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schug T.T., Li X. Sirtuin 1 in lipid metabolism and obesity. Ann. Med. (2011);43:198–211. doi: 10.3109/07853890.2010.547211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hubbard B.P., Sinclair D.A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci. (2014);35:146–154. doi: 10.1016/j.tips.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X., Zhang S., Blander G., Tse J.G., Krieger M., Guarente L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol. Cell. (2007);28:91–106. doi: 10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 31.Walker A.K., Yang F., Jiang K., Ji J.Y., Watts J.L., Purushotham A., Boss O., Hirsch M.L., Ribich S., Smith J.J., Israelian K., Westphal C.H., Rodgers J.T., Shioda T., Elson S.L., Mulligan P., Najafi-Shoushtari H., Black J.C., Thakur J.K., Kadyk L.C., Whetstine J.R., Mostoslavsky R., Puigserver P., Li X., Dyson N.J., Hart A.C., Naar A.M. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. (2010);24:1403–1417. doi: 10.1101/gad.1901210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brandauer J., Vienberg S.G., Andersen M.A., Ringholm S., Risis S., Larsen P.S., Kristensen J.M., Frøsig C., Leick L., Fentz J., Jørgensen S., Kiens B., Wojtaszewski J.F., Richter E.A., Zierath J.R., Goodyear L.J., Pilegaard H., Treebak J.T. AMP-activated protein kinase regulates nicotinamide phosphoribosyl transferase expression in skeletal muscle. J. Physiol. (2013);591:5207–5220. doi: 10.1113/jphysiol.2013.259515. [DOI] [PMC free article] [PubMed] [Google Scholar]