

Abstract
Cadmium (Cd) exposure contributes to human diseases affecting liver, kidney, lung, and other organ systems, but mechanisms underlying the pleotropic nature of these toxicities are poorly understood. Cd accumulates in humans from dietary, environmental (including cigarette smoke), and occupational sources, and has a twenty-year biologic half-life. Our previous mouse and cell studies showed that environmental low-dose Cd exposure altered protein redox states resulting in stimulation of inflammatory signaling and disruption of the actin cytoskeleton system, suggesting that Cd could impact multiple mechanisms of disease. In the current study, we investigated the effects of acute Cd exposure on the redox proteome and metabolome of mouse liver mitochondria to gain insight into associated toxicological mechanisms and functions. We analyzed redox states of liver mitochondrial proteins by redox proteomics using isotope coded affinity tag (ICAT) combined mass spectrometry. Redox ICAT identified 2687 cysteine-containing peptides (peptidyl Cys) of which 1667 peptidyl Cys (657 proteins) were detected in both control and Cd-exposed samples. Of these, 46% (1247 peptidyl Cys, 547 proteins) were oxidized by Cd more than 1.5-fold relative to controls. Bioinformatics analysis using MetaCore software showed that Cd affected 86 pathways, including 24 Cys in proteins functioning in branched chain amino acid (BCAA) and 14 Cys in proteins functioning in fatty acid (acylcarnitine/carnitine) metabolism. Consistently, high-resolution metabolomics data showed that Cd treatment altered levels of BCAA and carnitine metabolites. Together, these results show that mitochondrial protein redox and metabolites are targets in Cd-induced hepatotoxicity. The results further indicate that redox proteomics and metabolomics can be used in an integrated systems approach to investigate complex disease mechanisms.
Keywords: cysteine proteome, environmental toxicant, metabolome, pathway maps, thiol/disulfide redox state
Abbreviations
- Cys
cysteine
- GSH
glutathione
- GSSG
glutathione disulfide
- ICAT
isotope coded affinity tag
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- MS
mass spectrometry
- MT
metallothionein
- Pr-SH
protein thiol
- Pr-SSG
glutathionylated protein
Cadmium (Cd) is an intensively studied environmental agent with detailed knowledge about a wide range of organ and tissue targets, including hepatotoxicity, nephrotoxicity, pulmonotoxicity, neurotoxicity, bone toxicity, and carcinogenesis. Cd is present in food and tobacco (ATSDR, 2012), and accumulates throughout life due to a long biological half-life (t1/2) of 17–30 year (Goyer, 1997). In addition to accumulation in the kidney (t1/2, 6–38 year), Cd accumulates in human liver (t1/2, 4–19 year) as the main target organ for Cd toxicity according to the Nordberg-Kjellstrom model (ATSDR, 2012). Cd has a high affinity for sulfhydryl (thiol) groups of albumin and metallothionein (MT; Nordberg et al., 1985), resulting in MT functioning as a sequestering molecule for a large fraction of tissue Cd (Shaikh, 1982). Cd-bound MT in the liver can redistribute to the kidney and result in elevated level of Cd after release from MT (Dudley et al., 1985). Moreover, increasing evidence shows that mitochondrial damage is involved in the mechanism of Cd-induced liver cell injury (Belyaeva et al., 2011; Liu et al., 2011; Modi and Katyare, 2009).
Cd exposure interrupts hepatic glycogen storage, increases blood glucose levels (Mamyrbaeva et al., 1998), perturbs lipid composition, enhances lipid peroxidation (Krajcovicova-Kudlackova and Ozdin, 1995; Kudo et al., 1991), and disrupts metabolism of BCAA in rats (Pasternak et al., 2004; Wu et al., 2011). BCAA metabolism and fatty acid metabolism are associated with mitochondria, and related imbalances and disruption may contribute to pathological complications such as obesity, diabetes, abnormality in lipid transport, and cancer. Mitochondria are also sensitive to heavy metals, including Cd, Hg, and Cu, as shown by induction of mitochondrial permeability transition (MPT) pore opening and inhibition of the electron transport chain (Belyaeva et al., 2011; Su et al., 2011). Together, the data show that mitochondrial dysfunction is an important component of hepatotoxicity, but details of the mechanistic basis remain unclear.
Our previous study on the redox dependence of the cysteine (Cys) proteome shows that Cd at a low level stimulated translocation of thioredoxin-1 into cell nuclei and potentiated inflammatory signaling by NF-κB activation, and this event was regulated by actin dynamics (Go et al., 2013b). This study showed that Cd exposure at low levels could disrupt cell signaling by altering central redox processes controlling the Cys proteome. Previous studies with high-dose Cd exposure and our cell study with low-dose Cd treatment show that Cd-induced toxicological cellular responses and elevated oxidative stress mechanisms involve disruption of actin dynamics (Dalle-Donne et al., 2002; Go et al., 2013b; Prozialeck and Niewenhuis, 1991). Cd is not a redox-active molecule but interacts with selenium, thiols of proteins, albumin and MT as discussed above, and small molecules, leading to the condition of redox-disruption-associated oxidative stress, e.g., inactivation and decreased amounts of antioxidants (Gasiewicz and Smith, 1976; Gong and Hart, 1997; Ikediobi et al., 2004; Whanger, 1985). Although multiple studies show that Cd stimulates oxidative stress, the detailed mechanisms for Cd-induced stress signaling associated with changes in protein Cys redox state, and abundance of small molecules and metabolites, are largely unknown. The Cys redox proteome consisting of reversible and irreversible covalent modification links to biological structure and function (Go and Jones, 2013a,b; Go et al., 2013c). Thiol/disulfide redox state and an accumulating redox proteomics base knowledge enable development of integrated redox systems biology to better understand the function and dysfunction of biological systems during oxidative stress. In particular, redox proteomics identifies critical Cys residues associated with protein function and also redox-sensing Cys residues integrating biologic functions (Go and Jones, 2013a). The availability of advanced online bioinformatics software tools enables mapping of relevant toxicologic pathways and networks (Go et al., 2010, 2011, 2013c).
The aim of the present study was to test whether integrated use of advanced mass-spectrometry-based redox proteomics, metabolomics, and pathway mapping tools could reveal potential mechanistic targets of acute Cd exposure on liver mitochondria in mice. Biochemical approaches showed that the glutathione/glutathione disulfide (GSH/GSSG) redox state was significantly oxidized in liver and plasma compared with other organ tissues by acute Cd administration. Under these conditions, redox proteomics and metabolomics identified proteins and small molecules in liver mitochondria affected by acute Cd exposure, and MetaCore (Thomson Reuters, https://portal.genego.com/) analysis showed that Cd-altered molecules were associated with multiple functional pathways. Importantly, out of large numbers of functional pathways, the analyses identified significant associations for both redox proteomics and metabolomics with pathways for BCAA metabolism and fatty acid oxidation. The data show that Cd-induced oxidation of proteins functioning in BCAA and fatty acid metabolism is linked to significant alterations in metabolites associated with these metabolic pathways.
MATERIALS AND METHODS
Mouse exposure to Cd and mitochondria isolation
All animal experiments and husbandry for the studies presented were conducted under the review and approval of the Emory University Institutional Animal Care and Use Committee and were performed in accordance with published National Institutes of Health guidelines. Mice (C57BL/6) purchased from Charles River Laboratories were maintained in the Emory University Division of Animal Resources Facility. Mice (8–10 weeks, 9 males per group) were used to analyze GSH, GSSG, protein thiol (Pr-SH), glutathionylation of protein (Pr-SSG), metallothioneins (MT1/MT2), and MPT in plasma, tissue, and mitochondria. For redox proteomics and metabolomics, mice (8–10 weeks, 3 males per group) were exposed to Cd (CdCl2, Sigma-Aldrich) or saline as control (CR) via intraperitoneal injection (10 mg Cd/kg body weight, 6 h) and euthanized to isolate mitochondria from livers. The dose was selected from concentrations studied by Bartosievicz (Bartosiewicz et al., 2001) following preliminary studies to assure measurable oxidation of Pr-SH, measured by Ellman's reagent, and oxidation of peroxiredoxin-3, measured by redox Western blot (Go et al., 2013a; Roede et al., 2011; data not shown). To address Cd effects on thiol/disulfide redox systems, we examined redox states of Cys-containing peptides (peptidyl Cys), total Pr-SH and the GSH redox system. For mitochondria isolation, the liver excised immediately from euthanized mouse was placed in ice-cold phosphate buffered saline, washed, homogenized, and fractionated by differential centrifugation (Savage et al., 1991).
Determination of gene expression levels by quantitative real-time polymerase chain reaction
Total mRNA was isolated from organ tissues (liver, lung, and kidney) harvested from mice challenged with saline or Cd (10 mg/kg body weight, 6 h) using RNeasy mini kit (Qiagen) following the manufacturer's protocol, and reverse transcription was performed to generate cDNA (Clonetech). For quantitative real-time polymerase chain reaction (qRT-PCR), amplification was performed in triplicate on an iCycler IQ Multicolor RT-PCR Detection System (Bio-Rad Laboratories, Hercules, CA) for 30 cycles as follows: 95°C for 30 s, 58°C for 30 s, and 72°C for 1 min. Quantification and melting curves were analyzed with iCycler software. Primers for MT were designed using a program provided by Integrated DNA Technologies (Coralville, Iowa). Details of PCR primer sequences for MT1 and MT2 used in the analyses of extracted RNA for qRT-PCR are as follows: mouse MT1, forward: 5′-ATG GAC CCC AAC TGC TCC TGC TCC ACC-3′, reverse: 5′-GGC ACA GCA CGT GCA CTT GTC CGC-3′; mouse MT2, forward: 5′-ATG GAC CCC AAC TGC TCC TGT GCC-3′, reverse: 5′-GCT GCA CTT GTC GGA AGC CTC TTT-3′.
Assays for Pr-SH level, GSH and GSSG amounts, GSH/GSSG redox potential (EhGSSG), Pr-SSG, and MPT
Liver, lung, and kidney tissues after Cd (or saline) treatment (10 mg/kg body weight) were analyzed for Pr-SH levels using Ellman's reagent (Go et al., 2007; Sedlak and Lindsay, 1968) and GSH and GSSG amounts by high performance liquid chromatography with fluorescence detection (Jones and Liang, 2009). Values were used to calculate the steady-state redox potential for cellular EhGSSG using the measured concentrations, the Nernst equation, and respective EoGSSG (−264 mV, pH 7.4; Jones and Liang, 2009). To measure Pr-SSG, we followed procedures reported previously (Go et al., 2007; Lash and Jones, 1985). Briefly, proteins from the liver mitochondria were precipitated with cold 10% trichloroacetic acid (TCA, Sigma-Aldrich) and precipitated proteins were resuspended, neutralized, and reduced by 5mM DTT. Proteins were reprecipitated, and liberated GSH was analyzed in the supernatant and presented as Pr-SSG (Go et al., 2007). For MPT assay, freshly isolated liver mitochondria from both saline CR and Cd-treated mice were used following procedures described previously (He et al., 2008). MPT was determined spectrometrically at 540 nm with or without presence of 60μM Ca2+ (He et al., 2008; Savage et al., 1991).
Quantification of peptidyl Cys by redox proteomic analysis
Redox isotope coded affinity tag (ICAT) was performed using ICAT-based mass spectrometry (MS; Go et al., 2010, 2011, 2013c). This methodology has been extensively studied and developed to minimize oxidation during sample extraction, processing, and MS (Go et al., 2010, 2011, 2013c). Briefly, freshly isolated liver mitochondria from mice challenged with Cd or saline CR (3 mice for each treatment) were precipitated with ice-cold 10% TCA and 120 μg total protein was used for redox ICAT analysis (Go et al., 2013c). Protein was washed with ice-cold acetone, resuspended in denaturing buffer (50mM Tris, 0.1% SDS, pH 8.5) provided by the manufacturer (AB Sciex, CA), and treated with the biotin-conjugated thiol reagent (Heavy isotopic [H-ICAT]) for 1 h at 37°C. Protein was then reprecipitated by ice-cold 10% TCA for 30 min on ice, pelleted, washed with acetone, and resuspended in 80 μl denaturing buffer. Unlabeled disulfides and sulfenic acids in the proteins were then reduced by TCEP [tris-(2-carboxyethyl phosphine)] and labeled with another biotin-conjugated thiol reagent (Light isotopic [L-ICAT]) at 37°C for 1 h. Samples including both H- and L-ICAT-labeled proteins were digested with trypsin for 18 h, fractionated by cationic exchange followed by avidin purification, and analyzed by MS as described below. ICAT-labeled Cys containing peptides (peptidyl Cys) were identified with an H to L ratio as a measure of the reduced/oxidized state of the protein, expressed as percentage values, and labeled as “% oxidized.” Identified peptidyl Cys were individually processed to eliminate redundancies, matched to proteins based upon amino acid sequences, and verified for consistency between saline CR and Cd treatments. Fold oxidation was calculated by dividing % oxidized (L-ICAT/[L-ICAT + H-ICAT]) of individual peptidyl Cys from Cd treatment by % oxidized of the identical peptidyl Cys from CR.
Mass spectrometry
ICAT-labeled Cys peptides were analyzed by reverse-phase liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS; Go et al., 2013c; Xu et al., 2009). Peptide eluates were monitored in an MS survey scan followed by 10 data-dependent MS/MS scans on an LTQ-Orbitrap ion trap mass spectrometer (Thermo Finnigan, San Jose, CA). The LTQ was used to acquire MS/MS spectra (2 m/z isolation width, 35% collision energy, 5000 AGC target, and 200 ms maximum ion time). The Orbitrap was used to collect MS scans (300–1600 m/z, 1,000,000 AGC target, 500 ms maximum ion time, and resolution 30,000). All data were converted from .raw files to the .dta format using ExtractMS Version 2.0 (Thermo Finnigan). The acquired MS/MS spectra were searched against a concatenated target-decoy human reference database of the National Center for Biotechnology Information using the SEQUEST Sorcerer algorithm (Version 3.11, SAGE-N; Eng et al., 1994); searching parameters included partially tryptic restriction, parent ion mass tolerance (± 20 ppm), dynamic modifications of oxidized Met (+15.9949 Da), differential ICAT-modified Cys (+9.0302 Da), and static ICAT modification of Cys (+227.1270 Da). The peptides were classified by charge state and tryptic state (fully and partial) and first filtered by mass accuracy (10 ppm for high-resolution MS) and then dynamically by increasing XCorr and ΔCn values to reduce protein false discovery rate (FDR) to less than 1%, according to the target-decoy strategy (Elias and Gygi, 2007; Peng et al., 2003). Parameter settings allowed detection of 100% oxidation and 100% reduction as well as H:L ratios for partially oxidized peptidyl Cys. Peptides included for analyses are presented in Supplement 1.
Sample preparation and extraction for metabolomics
Metabolomics using high-resolution MS were performed following the procedures described previously (Roede et al., 2012). Briefly, an aliquot of isolated mitochondria was used to determine protein content using a Bio-Rad DC kit. Equivalent amounts of mitochondria corresponding to 150 μg of mitochondrial protein were used and diluted to a final volume of 50 μl using incubation media that included 220mM mannitol and 2mM hepes (pH 7.4). A mixture of standards consisting of 14 stable isotopic chemicals (see chemicals below) that cover a broad range of chemical properties represented in small molecules were added to 100 μl acetonitrile (ACN; Soltow et al., 2013). An amount of 100 μl of the ACN solution including internal standards was then added to 50 μl of mitochondria samples. Samples were centrifuged at 14,000 × g for 10 min at 4°C to remove protein and supernatants were loaded onto an autosampler maintained at 4°C until injection.
LC-MS metabolic profiling
LC-MS with chromatography as described above was performed using an LTQ Velos Orbitrap (Thermo Fisher, San Diego, CA) mass spectrometer. Optimized operating conditions for the LTQ Orbitrap were used as follows: HESI (heated electrospray ionization) probe with S-lens combination for ESI (electrospray ionization); MS1 mode scanning mass to charge (m/z) range of 85–2000; resolution, 60,000; maximum number of ions collected, 5.00 × 105; the maximum injection time, 5 μl/s; capillary temperature, 275°C; source heater, 45°C; voltage, 4.6 kV; sheath gas, 45; auxillary gas flow, 5; and sweep gas flow, 0. Each sample was run in triplicate with C18 column chromatography and 10 μl injection volume.
Data collection, processing, and analysis
Data were collected continuously over the 10-min chromatographic separation and converted using XCalibur file converter software (Thermo Fisher) for further data processing. The data were processed for peak extraction and quantification of ion intensities using apLCMS (Go et al., 2013c; Yu et al., 2009) with enhanced data extraction using xMSanalyzer (Go et al., 2013c; Uppal et al., 2013). The technical replicates were averaged following the quality assessment, and only the m/z features (ions) with at least 70% signal in one of the test conditions (CR, Cd) were retained. The features were annotated by searching metabolic databases, including Kyoto Encyclopedia of Genes and Genomes (KEGG) and METLIN, for putative matches to known metabolites using a m/z threshold (±10 ppm).
2H3-palmitoylcarnitine uptake
Isolated mitochondria (200 μg) obtained from Cd or CR mouse were incubated with 2H3-pamitoylcarnitine (40μM, Cambridge Isotope Laboratories) for 20 min. Mitochondria were washed with the medium (250mM sucrose, 10mM MOPS, 3mM KH2PO4, 5mM succinate, and 5mM malate) and spun down (8000 × g, 10 min, 4°C), resuspended in the medium (220mM mannitol, 2mM hepes, pH 7.0), and extracted for metabolomics analysis by MS as above. Intensity of 2H3-palmitoylcarnitine (m/z; 403.3617) was calculated by following the method for metabolomics data collection, processing, and analysis as described above.
Biological network analysis by MetaCore
Functional redox proteomic and metabolomic networks affected by protein redox state and abundance of metabolites, respectively, were analyzed using MetaCore (Thomson Reuters, https://portal.genego.com/), METLIN (Scripps Center For metabolomics, http://metlin.scripps.edu/metabo_search_alt2.php), and KEGG (http://www.genome.jp/kegg/). For pathway analysis associated with Cd-oxidized proteins, peptidyl Cys with fold oxidation value ≥1.5 were analyzed by loading information of protein ID and fold oxidation value of oxidized peptidyl Cys into MetaCore (Version 6.13, build 43450). For pathway analysis associated with Cd-altered abundance of metabolites, 542 significantly increased and decreased features were loaded to MetaCore. Ranking of relevant integrated pathways was based on hypergeometric p-values (Levine et al., 2006). Pathway map analysis was specified with a significance level for the FDR filter by setting FDR threshold level at 0.05.
RESULTS
The Effect of Cd on Oxidative Stress
To evaluate oxidative stress induced by acute Cd exposure, plasma (Figs. 1A and B) and different organ tissues including liver (C), lung (D), and kidney (E) were examined for redox states (redox potential, Eh) of thiol/disulfide couples (cysteine [Cys]/cystine [CySS, oxidized form of cysteine], GSH/GSSG). The results showed that Cd significantly decreased GSH in plasma (CR, 18.8 ± 2.0μM; Cd, 8.0 ± 1.1μM) and liver (CR, 9.3 ± 1.7mM; Cd, 4.6 ± 1.1mM), and oxidized GSH redox potential (A, Plasma EhGSSG [CR, −141.3 ± 2.1 mV; Cd, −129.8 ± 3.7 mV], C, liver EhGSSG [CR, −249.1 ± 3.0 mV; Cd, −237.8 ± 4.8 mV]). Consistent with these results, total Pr-SH measured by Ellman's reagent showed that liver Pr-SH were significantly decreased by Cd (Fig. 2A). On the other hand, no significant effect was observed in plasma concentration of Cys and CySS, or EhCySS (B). Similarly, no significant Cd effect was observed on GSH, GSSG, or EhGSSG levels for lung and kidney tissues (D, E). The lack of significant effects in kidney and lung may have been due to the short time frame for these studies. In contrast, qRT-PCR analysis on MT expression by Cd show that mRNA levels of MT-1 (Fig. 2B) and MT-2 (Fig. 2C) were significantly elevated by Cd in all tissues compared with CR. These data suggest that liver is more susceptible to oxidative stress induced by Cd under the conditions used, although MT expression examined for cellular responses to metal was substantially elevated in all organ tissues.
FIG. 1.

GSH, GSSG, and GSH/GSSG redox potential in plasma and liver by acute Cd exposure. Plasma (A, B) and organ tissues (C–E) collected from mice (9 mice per group) exposed to saline control (0 Cd) or Cd (10 mg/kg body weight, 10 Cd) for 6 h were analyzed for redox states of thiol/disulfide couples, GSH, GSSG (A, top) and Cys and CySS amounts (B–E, top), and respective GSH/GSSG (A, C–E; bottom) and Cys/CySS (B, bottom) redox potentials (EhGSSG and EhCySS). *p < 0.05 versus CR group.
FIG. 2.

Cd decreased total protein-bound thiols and increased mRNA expression of metallothionein (MT). Tissues including liver, lung, and kidney isolated from mice treated with Cd as described above were measured for total protein-bound thiol (Pr-SH) by Ellman's reagent (A). MT-1 (B) and MT-2 (C) expression levels (mRNA level) were measured by quantitative real-time PCR in different tissues as a measure of cellular response to the metal stress, Cd. *p < 0.05 versus CR group.
Mitochondrial Dysfunction in Liver by Cd
Because the Cd effects on liver appeared to be significant and more susceptible to oxidation than other tissues, we focused on examining liver mitochondria to determine acute Cd-exposure-induced toxicological mechanisms associated with liver metabolism. Considerable evidence using in vitro cells and isolated mitochondria show that Cd-induced mitochondrial dysfunction occurs following activation of the MPT (Belyaeva et al., 2011; Lee et al., 2004; Zhang et al., 2008). The MPT, a high-amplitude swelling of mitochondria (Hunter et al., 1976) triggered by the opening of a pore, allows rapid influx of ions. The MPT pore is activated by Ca2+ in the presence of diverse molecules such as heavy metal ions (Gunter and Pfeiffer, 1990). Therefore, to examine acute Cd effect on liver mitochondrial function, liver mitochondria isolated from mice administered with Cd or saline were analyzed for MPT assay (Figs. 3A and B). Both in the presence and absence of 60μM Ca2+, mitochondria from Cd-treated mice showed that MPT is more activated than that of control mice (CR, Fig. 3A; Cd, Fig. 3B) suggesting that Cd exposure impaired mitochondrial function. Cd-induced mitochondrial oxidative damage was assessed by measuring the GSH/GSSG redox system. Results showed that mitochondria isolated from Cd-treated mice had significantly decreased GSH (Fig. 3C), oxidized EhGSSG (Fig. 3D), decreased Pr-SH (Fig. 3E), and elevated glutathionylation of proteins (Pr-SSG; Fig. 3F). These data support previous findings that acute exposure to Cd is associated with mitochondrial oxidative stress and damage in liver.
FIG. 3.
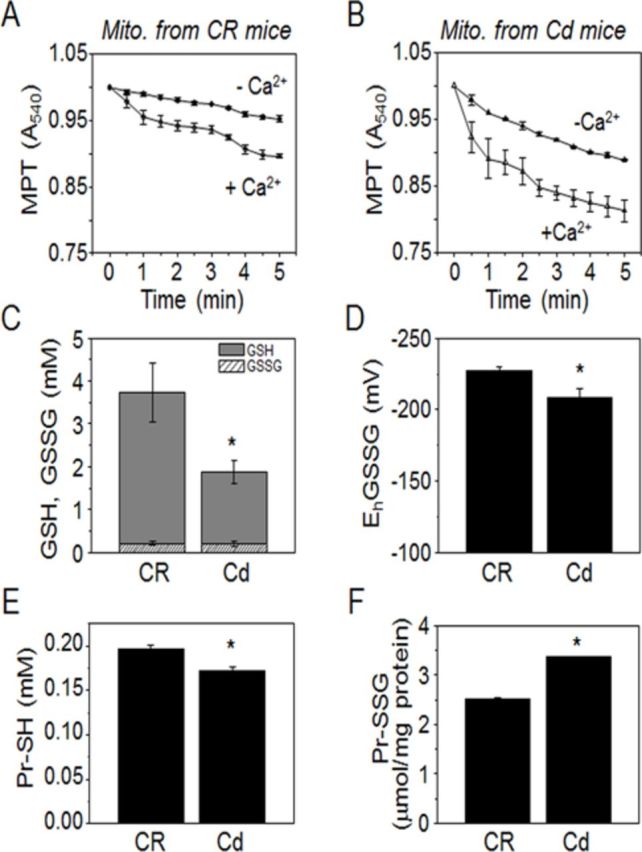
Liver mitochondrial GSH, protein thiol, and functional impairment by Cd exposure. Isolated mitochondria from mice exposed to saline CR and Cd (10 mg/kg body weight) for 6 h were used to measure mitochondrial permeability transition (MPT; A, B) and determine GSH/GSSG related oxidative stress (C–F). Mitochondria were suspended in media containing 3mM inorganic phosphate and 5mM sodium succinate with or without 60μM Ca2+ at 20°C, and activation of MPT was monitored at 540 nm. The MPT from Cd-exposed mice (B) were more activated than CR (A) in both presence and absence of Ca2+. Redox states of isolated mitochondria from CR and Cd were examined by measuring amounts of GSH/GSSG (C), GSH/GSSG redox potential (EhGSSG, D), and concentration of protein-bound thiol (Pr-SH, E) and glutathionylated protein (Pr-SSG, F). *p < 0.05 versus CR group. Data are representative of experiments from 9 mice for each CR and Cd treatment.
Cd-Induced Oxidation of Peptidyl Cys Measured in Liver Mitochondria by Redox ICAT Mass Spectrometry
Cd is not redox-active; however, many studies show that cells and mice respond to Cd similarly as to oxidative stress (Dailianis et al., 2009; Go et al., 2013b; Gong and Hart, 1997; Ikediobi et al., 2004). To determine whether acute Cd-induced mitochondrial oxidative stress is associated with alteration in redox states of liver mitochondrial proteins, we used redox ICAT/MS-based proteomics to measure percent oxidation of peptidyl Cys of proteins. To quantify redox states of peptidyl Cys in mitochondria, mouse liver in three biological replicates of each saline CR and Cd (10 mg/kg body weight) were used. Redox ICAT analysis requires that each peptide be detected in both heavy isotope and light isotope form to allow determination of the percentage oxidation (Go et al., 2013c). Consequently, some data were missing for individual experiments (Supplement 1). We filtered peptidyl Cys for fold oxidation (Cd/CR) by Cd treatment compared with control, keeping those with values higher than 1.5 fold change (50% more oxidized in Cd than CR) in mouse liver. The distribution of % oxidation of identical peptidyl Cys detected from CR and Cd treatment is shown in histograms, with a mean % oxidation indicated by arrows (CR, mean = 8.9%, n = 1667; Cd, mean = 15.7%, n = 1667; Fig. 4A). Cd-induced fold-change oxidation of these peptides was characterized and can be visualized in Figure 4B. These data show that 75% of these peptidyl Cys were oxidized more than 50% (≥ 1.5-fold) by acute Cd exposure (4B). A complete list of these peptidyl Cys, along with proteins containing these sequences, is given in Supplement 1. Total numbers of peptidyl Cys with ≥1.5-fold oxidation were 1247 corresponding to 547 proteins (Supplement 1). These redox proteomics data show that acute Cd exposure causes extensive oxidation of the mitochondrial proteome.
FIG. 4.

Cd-induced oxidation in liver mitochondrial redox proteome. Freshly isolated liver mitochondria (120 μg protein) obtained from saline CR (3 mice) and Cd (3 mice) were analyzed for redox proteomics using redox ICAT-based mass spectrometry (MS). (A) Distribution of the oxidized states (% oxidized) of the Cys-including peptide (peptidyl Cys) relevant to 655 proteins from CR (blue; n = 1667) and Cd (red; n = 1667). The mean value of % oxidation from 1667 peptidyl Cys from CR and Cd were 8.9% and 15.7%, respectively. (B) Pie chart showing the distribution of peptidyl Cys according to the measured fold oxidation. Of 1667 peptidyl Cys, 77% were oxidized more than 50% (1.5-fold) by Cd treatment. (C) MetaCore Bioinformatics software identified significant pathway maps that are regulated by Cd-oxidized peptidyl Cys (fold oxidation ≥ 1.5, n = 1247). Of the 86 statistically significant pathways, the top 10 pathways are shown (C).
Pathways for Branched Amino Acid Metabolism are Significantly Affected by Cd
To identify functional pathways and networks affected by Cd exposure, we performed pathway enrichment analysis using MetaCore software (https://portal.genego.com/). This approach was used to overcome the limitation of the large number of comparisons of individual peptidyl Cys, which result in few significant differences for individual peptidyl Cys after FDR correction (Supplement 1). MetaCore uses the Fisher's exact test to test for over-representation of peptidyl Cys relative to those expected to occur randomly, adjusting for multiple sample testing using FDR. Peptidyl Cys were selected that were oxidized ≥ 1.5-fold by Cd. These included 1247 peptidyl Cys from 547 proteins and were analyzed using MetaCore with an FDR threshold level at 0.05. The results showed that Cd-oxidized proteins were significantly associated with 86 pathways (Supplement 2, A). Of these, the top 10 pathways ranked according to p-value are shown in Figure 4C. These included BCAA metabolism, oxidative phosphorylation, tryptophan, butanoate and pyruvate metabolism, branched chain fatty acid oxidation, and lysine metabolism. The most significant pathway was for BCAA (leucine, isoleucine, and valine) metabolism (p = 1.5 × 10−17; Supplement 2A and Fig. 5) matching with 24 objects (rectangular box; https://portal.genego.com/cgi/imagemap.cgi?id=2315&channel=Other§ion=Workflow). The detailed information about these Cd-oxidized peptides involved in BCAA metabolism is shown in Table 1.
FIG. 5.

Cd-affected significant mitochondrial pathways for branched chain amino acid metabolism. Among significant pathways associated with Cd-oxidized proteins/peptidyl Cys and Cd-increased metabolites, the pathways for branched chain amino acids (leucine, isoleucine, and valine) metabolism are shown. Redox ICAT/MS-identified 24 peptidyl Cys (rectangular box; p = 1.5 × 10−17) oxidized by Cd and high-resolution metabolomics-identified 3 metabolites (oval) increased by Cd are shown in these pathways including total 80 proteins and metabolites.
TABLE 1.
Cd-Oxidized Peptidyl Cys Involved in Leucine, Isoleucine, and Valine Metabolism Pathways as Measured by Redox ICAT. Cd-oxidized Cys residue of peptidyl Cys is indicated as bold and italic letter.
| Protein | Accession | Peptide | Fold ox: Cd/CR (p-value) |
|---|---|---|---|
| Isobutyryl-CoA dehydrogenase | NP_080138.2 | ITFC32IDPSLGLNEEQK | 3.5 (0.01) |
| INVASC281SLGAAHASVILTQEHLK | 2.6 (0.50) | ||
| Short/branched chain specific acyl-CoA dehydrogenase | NP_080102.1 | GITC234FLVDR | 3.5 (0.08) |
| Enoyl-CoA hydratase | NP_444349.1 | LVEEAIQC225AEK | 4.7 (0.03) |
| Carnitine O-acetyltransferase | NP_031786.2 | SIFTVC287LDK | 3.3 (ND) |
| 3-hydroxyisobutyryl-CoA hydrolase | NP_666220.1 | AFC94AGGDIK | 3.1 (ND) |
| Hydroxyacyl-coenzyme A dehydrogenase | NP_032238.2 | TFESLVDFC201K | 4.6 (0.12) |
| 3-ketoacyl-CoA thiolase | NP_803421.1 | VVGYFVSGC287DPTIMGIGPVPAINGALK | 7.6 (0.15) |
| Methylmalonate-semialdehyde dehydrogenase | NP_598803.1 | VC368NLIDSGTK | 3.0 (0.06) |
| 3-ketoacyl-CoA thiolase B | NP_666342.1 | VNPLGGAIALGHPLGC381TGAR | 380.0 (0.02) |
| DC177LIPMGITSENVAER | |||
| 4-aminobutyrate aminotransferase | NP_766549.2 | GRGTFC449SFDTPDEAIR | 3.5 (0.06) |
| Aldehyde dehydrogenase | NP_033786.1 | KTFPTVNPSTGEVIC68QVAEGNKEDVDK | 2.8 (0.03) |
| Propionyl-CoA carboxylase beta chain | NP_080111.1 | EC293HDPSDR | 4.4 (ND) |
| Methylmalonyl-CoA mutase | NP_032676.2 | IEEC469AAR | 3.7 (0.05) |
| Hydroxymethylglutaryl-CoA lyase | NP_032280.2 | LLEAGDFIC307QALNR | 4.0 (0.13) |
| Hydroxymethylglutaryl-CoA synthase | NP_032282.2 | LSTNNGNMYTSSLYGC389LASLLSHHSAQELAGSR | 2.9 (0.32) |
| Methylcrotonoyl-CoA carboxylase beta chain | NP_084302.1 | MVAAVAC431AK | 4.6 (0.06) |
| VSGVEC131MIVANDATVK | 2.3 (0.34) | ||
| 3-methylcrotonyl-CoA carboxylase alpha chain | NP_076133.3 | DQSDQC186LR | 3.4 (0.004) |
| ESVC505QAALGLILK | 2.9 (0.33) | ||
| Isovaleryl-CoA dehydrogenase | NP_062800.1 | AC348DEGHIIPK | 2.8 (0.09) |
ND, not determined.
Cd-Induced Alterations in Abundance of Small Molecules (Metabolites) Measured in Liver Mitochondria by High-Resolution Mass Spectrometry
To compare with redox proteomics results and provide information regarding Cd effects on liver mitochondrial metabolome, we performed metabolomics analysis of the same mitochondria using high-resolution MS (Roede et al., 2012). The MS data show that a total 4745 m/z features (ions) were detected. Of these, 542 m/z features (increased by Cd, 250 m/z; decreased by Cd, 292 m/z) were significantly different comparing between Cd and CR mice after FDR correction. Cd-altered 542 features were searched for accurate mass matches using METLIN (http://metlin.scripps.edu/metabo_search_alt2.php). This analysis provides information including m/z, METLIN ID, and KEGG ID if the data are available. The KEGG IDs of significantly altered m/z features were loaded into MetaCore. MetaCore identified 29 significant pathways associated with increased or decreased features in the mitochondria (Supplement 2, B). Of these, 10 significant pathways associated with Cd-altered 542 features are shown (Fig. 6). This result supports the redox proteomics data that BCAA metabolism is significantly affected by Cd. Cd-increased three metabolites including succinyl Coenzyme A (M + 2Na + H, 912.1024), isobutyryl CoA (M + 2H, 419.5858), and acetyl CoA (M + H, 810.1329) as shown in the pathway (oval shape box, Fig. 5, Table 2).
FIG. 6.

Cd-altered mitochondrial metabolome. Isolated mitochondria prepared for redox proteomics were used for metabolomics by high-resolution mass spectrometry. Top 10 pathways associated with 542 small molecules (m/z features) that are significantly affected by Cd were ranked by MetaCore Bioinformatics software according to p-value.
TABLE 2.
Cd-Elevated Mitochondrial Levels of Metabolites Associated with Branched Chain Amino Acid Metabolism
| Name (METLIN/KEGG ID) | Adduct form (m/z) | Feature intensity (average) | Fold change in abundance | |
|---|---|---|---|---|
| CR | Cd | Cd/CR | ||
| Succinyl CoA (444/C00091) | M +2Na + H (912.1024) | 65268.38 | 76314.64 | 1.2 |
| Isobutyryl CoA (445/C00630) | M + 2H (419.5858) | 16278.03 | 38970.67 | 2.4 |
| Acetyl CoA (36661/C00024) | M + H (810.1329) | 15948.28 | 19159.82 | 1.2 |
Cd-Affected Pathways for Fatty Acid Oxidation
Previous studies showed that deficiency and mutation of enzymes involved in long-chain fatty acid oxidation resulted in abnormal clinical phenotypes (Andresen et al., 1999; Gregersen et al., 2001). The current study of redox proteomics identified Cd-affected significant pathways for long-chain fatty acid oxidation (p = 1.93 × 10−6; Supplement 2, Fig. 7) involving oxidation of 46 peptidyl Cys corresponding to 14 different proteins (Table 3). Metabolomics analyses also identified several metabolic features associated with carnitine/acylcarnitine and acyl-CoA metabolism (Table 4) supporting pathway analysis of redox proteomics data. These had different patterns of increase and decrease suggesting that there are multiple protein targets disrupted by Cd, illustrated by a pathway map of long-chain fatty acid oxidation metabolism (Fig. 7), also provided in the online link https://portal.genego.com/cgi/imagemap.cgi?id=832&channel=Other§ion=Workflow. The Cd-induced effects on specific proteins and metabolites of long-chain fatty acid oxidation and CoA-linked carnitine metabolism including carnitine acetyltransferase (CAT), carnitine palmitoyl transferase (CPTII), carnitine/acylcarnitine carrier (CAC), with oxidized Cys residues, and associated metabolites (acetylcarnitine, palmitoylcarnitine, and stearylcarnitine) are presented in a schematic diagram of the carnitine/acylcarnitine transport system (Fig. 8A). Cd-induced disruption of carnitine metabolism was confirmed by a 2H3-palmitoylcarnitine uptake experiment. Results show that 2H3-palmitoylcarnitine measured by LC-high-resolution MS was substantially lower in mitochondria obtained from Cd-treated mouse compared with CR mouse (saline, see Fig. 8B). This data confirms the Cd-induced disruption of mitochondrial CAC function through oxidation of critical Cys residues (Table 3).
FIG. 7.

Cd-affected significant mitochondrial pathways for long-chain fatty acid oxidation metabolism. Redox ICAT/MS-identified 13 peptidyl Cys (rectangular box; p = 1.9 × 10−6) oxidized by Cd and high-resolution metabolomics-identified 12 metabolites (oval) are shown in these pathways including total 83 proteins and metabolites.
TABLE 3.
Cd-Induced Oxidation of Peptidyl Cys Associated with Long-Chain Fatty Acid Oxidation/Carnitine Metabolism. Cd-oxidized Cys residue of peptidyl Cys is indicated as bold and italic letter.
| Protein | Accession | Peptide | Fold ox: Cd/CR (p-value) |
|---|---|---|---|
| Mitochondrial carnitine/acylcarnitine carrier protein | NP_065266.1 | LQTQPPSLSGQPPMYSGTLDC58FRK | 4.1 (0.11) |
| C136LLQIQASSGENK | 4.0 (0.14) | ||
| YSGTLDC155AKK | 3.1 (0.03) | ||
| Peroxisomal carnitine O-octanoyltransferase | NP_076222.1 | SC458TVEAVR | 2.8 (0.14) |
| AFVFDVLHEGC210LITPPELLR | 2.3 (ND) | ||
| C228SNEPVGPSIAALTSEER | 1.9 (0.29) | ||
| MLFSTC166K | 1.8 (ND) | ||
| SC584PETDAEK | 1.6 (ND) | ||
| Carnitine O-palmitoyltransferase 2 | NP_034079.2 | KTEVLC84K | 1.9 (0.12) |
| C512SEAFVR | 1.7 (ND) | ||
| Carnitine O-acetyltransferase | NP_031786.2 | SIFTVC287LDK | 3.3 (ND) |
| Very long chain specific acyl-CoA dehydrogenase | NP_059062.1 | VASGQALAAFC216LTEPSSGSDVASIR | 3.2 (0.09) |
| VADEC434IQIMGGMGFMKEPGVER | 2.8 (ND) | ||
| LFVALQGC478MDK | 2.7 (0.21) | ||
| C216LTEPSSGSDVASIR | 2.4 (0.07) | ||
| SSAIPSPC238GK | 1.6 (ND) | ||
| Enoyl-CoA hydratase | NP_444349.1 | LVEEAIQC225AEK | 4.7 (0.03) |
| TFQDC111YSSK | 1.8 (0.18) | ||
| Acetyl-CoA acyltransferase | NP_663533.1 | DGGQYALVAAC459AAGGQGHAMIVEAYPK | 2.4 (0.18) |
| GGSLSLGHPFGATGC436R | 1.9 (0.32) | ||
| 3-ketoacyl-CoA thiolase | NP-803421.1 | VVGYFVSGC287DPTIMG | 7.6 (0.15) |
| EDC179DRYALQSQQR | 2.2 (0.13) | ||
| Hydroxymethylglutaryl-CoA synthase | NP_032282.2 | LLEAGDFIC307QALNR | 4.0 (0.13) |
| GYVSC170ALGC#PYEGK | 2.4 (0.05) | ||
| NANC141SIEESFQR | 2.0 (0.07) | ||
| VAQATC323KL | 1.5 (0.38) | ||
| Hydroxyacyl-coenzyme A dehydrogenase | NP_032238.2 | TFESLVDFC201K | 4.6 (0.12) |
| HPVSC211K | 2.0 (0.16) | ||
| Long-chain specific acyl-CoA dehydrogenase | NP_031407.2 | AFVDSC351LQLHETK | 4.2 (0.15) |
| THIC342VTR | 2.7 (0.46) | ||
| C166IGAIAMTEPGAGSDLQGVR | 1.7 (0.35) | ||
| Long-chain fatty acid-CoA ligase 1 | NP_032007.2 | VKPKPPEPEDLAIIC275FTSGTTGNPK | 6.3 (0.29) |
| RGLQGSFEELC626R | 4.6 (0.26) | ||
| EVAELAEC133IGSGLIQK | 4.1 (0.07) | ||
| ALKPPC55DLSMQSVEIAGTTDGIR | 3.5 (0.03) | ||
| LLLEGVENKLTPC221LK | 4.0 (0.13) | ||
| KC242GVEIISLK | 2.4 (0.33) | ||
| GEGEVC510VK | 2.1 (0.08) | ||
| GIQVSNNGPC109LGSR | 1.7 (0.17) | ||
| Medium-chain specific acyl-CoA dehydrogenase | NP_031408.1 | C159VTEPSAGSDVAAIK | 1.7 (0.02) |
| Long-chain fatty acid-CoA ligase 5 | NP_082252.1 | C226GVEMLSLHDAENIGKENFK | 11.8 (ND) |
| AEYLGSC121LLHK | 2.4 (0.09) | ||
| VGTPVAC471NFVK | 1.8 (0.18) | ||
| GLAVSDNGPC93LGYR | 1.7 (0.14) | ||
| LVQGILFSC322GGK | 1.7 (0.37) | ||
| ADIPVVIC185DTPQK | 1.6 (0.16) |
ND, not determined.
TABLE 4.
High-Resolution Metabolomics-Identified Metabolites/Small Molecules/mz Features Associated with Fatty Acid Oxidation (Carnitine/Acylcarnitine) Metabolism
| Name (METLIN/KEGG ID) | Adduct form (m/z) | Feature intensity (average) | Fold change in abundance | |
|---|---|---|---|---|
| CR | Cd | Cd/CR | ||
| Palmitoyl-CoA (65501/C00154) | M + 2Na – H (1050.3160) | 5518 | 33275 | 6.0 |
| (E)-but-2-enoyl-CoA (96242) | M + H + Na (429.5690) | 5006 | 23521 | 4.7 |
| Propionyl-L-carnitine (965/C03017) | M + H (218.1387) | 16397 | 54162 | 3.3 |
| Malonylcarnitine (6484) | M + H (248.1129) | 10587 | 19678 | 1.8 |
| Acetylcarnitine (956/C02571) | M + H (204.1216) | 116795 | 143306 | 1.2 |
| Acetyl-CoA (36661/C00024) | M + H (810.1329) | 15948 | 19160 | 1.2 |
| 3-oxotetradecanoyl-CoA (58188/C05261) | M + 2Na – H (1036.2640) | 135927 | 129959 | 0.95 |
| Carnitine (52/C00318) | M + H-H2O (144.1009) | 157503 | 147972 | 0.9 |
| Palmitoyl-L-carnitine (961/C02990) | M+ 2Na – H (444.3060) | 151892 | 146795 | 0.9 |
| Tetradecanoyl-CoA (3707/C02593) | M + Na (1000.3028) | 63773 | 55352 | 0.87 |
| (S)-3-hydroxyhexadecanoyl-CoA (58185/C05258) | M + H + Na (522.6681) | 9303 | 6309 | 0.7 |
| (S)-3-Hydroxytetradecanoyl-CoA (58187/05260) | M + H (994.3158) | 101590 | 61571 | 0.6 |
| 3-oxohexadecanoyl-CoA (58405/C05259) | M + Na (1042.3134) | 924767 | 493014 | 0.5 |
| Decanoyl CoA (36654/C05274) | M + 3H (308.0909) | 394196 | 97524 | 0.3 |
FIG. 8.
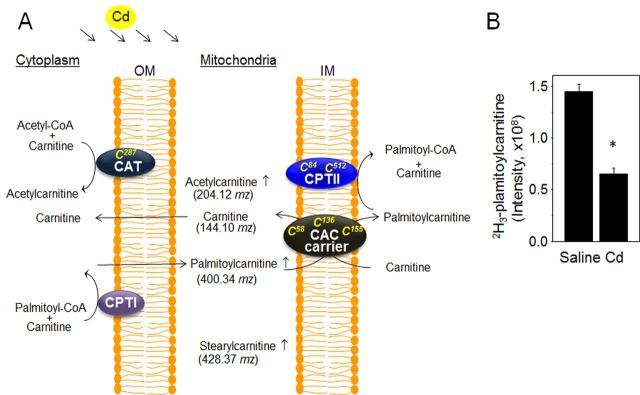
Cd-affected carnitine/acylcarnitine metabolism. A schematic diagram for Cd-affected carnitine/acylcarnitine transport from cytoplasm to mitochondria is shown (A). Several critical proteins (CAT, CPTI, CPTII, and CAC carrier) regulating this transport system are indicated with oxidation in specific Cys residues by Cd exposure. Metabolites identified by metabolomics, including carnitine, acetylcarnitine, palmitoylcarnitine, and stearylcarnitine, are shown with m/z and altered abundance in the mitochondria by Cd treatment (A). (B) Mitochondrial palmitoylcarnitine level (2H3-palmitoylcarnitine) was measured by mass spectrometer (m/z, 403.3617) after treating a mouse with Cd or saline for 6 h. *p = 0.008, n = 3.
DISCUSSION
The present study demonstrates that metabolomics data can be combined with redox proteomics data to enhance detection of mechanisms involved in toxicity. The approach can be used in a targeted way to test specific hypotheses, while gaining information on other potentially relevant mechanisms. In addition, the approach can be used with unbiased bioinformatics tools to identify target pathways representative of the metabolites and proteins detected. By providing information on dozens of biochemical pathways along with multiple functional protein networks, the results can yield more in depth understanding of complex toxic mechanisms. Extension to combinations of chemicals can also potentially clarify synergistic mechanisms of toxicity.
In addition to skeletal muscle and heart, the liver is a target organ of long-chain fatty acid oxidation defects. An increasing number of studies show a close relationship between liver disease and disruption in mitochondrial metabolism; e.g., deficiency and mutation of succinyl CoA ligase and medium/long-chain acyl-CoA dehydrogenase resulted in liver diseases, mitochondrial hepatoencephalomyopathy, and liver failure (Mashek et al., 2007; Van Hove et al., 2010). Mitochondria regulate many metabolic pathways in cells and different tissues by integrating signaling networks and pathways, and play a pivotal role in intracellular energy-generating processes, cell survival, and death. Accordingly, mitochondrial dysfunction due to metabolic pathway interruption has been implicated in various metabolic diseases, such as aging and diabetes (Newsholme et al., 2012). Therefore, our findings of Cd-mediated interruption of fatty acid oxidation suggest potential effects of Cd exposure as a sensitizing agent that may exacerbate diseases already involving mitochondrial dysfunction.
Evaluation of functional relevance in association with redox changes of Cys residues in proteins appears to be an important task to fulfill. The three peptidyl Cys of CAC protein (Cys58, Cys136, Cys155, NP_065266.1; Fig. 8) were found to be oxidized fourfold more by Cd treatment compared with CR (see Table 3). These Cys residues have been previously reported to function importantly in carnitine transport between intermembrane space and matrix (Giangregorio et al., 2007). Additionally, carnitine O-acetyltransferase (NP_031786.2), also known as carnitine palmitoyltransferase I (CPT1), was oxidized by Cd (3.8 fold oxidation of Cys287; Table 3, Fig. 8). This enzyme catalyzes the chemical reaction “acyl-CoA + carnitine ↔ CoA + O-acylcarnitine” (Bieber, 1988). Although the exact mechanism of CPT1 is not known, our data support the previous finding by Shoaf (Shoaf et al., 1986) that Cd inhibited carnitine acylation by inhibiting CPT1 activity via Cd binding to a thiol group of Cys on this enzyme. CPT1 disorder is known to be a risk factor for hepatic disease and other health problems (Collins et al., 2010); therefore, changes in the redox state of CPT1 by Cd could be associated with potentiation of liver dysfunction. Finally, we showed that medium, long, and very long chain acyl-CoA dehydrogenases are oxidized by Cd treatment. These enzymes possess redox-sensitive Cys residues (Okamura-Ikeda et al., 1985), and oxidation of these residues may account for treatment-related increases in palmitoyl-CoA and but-2-enoyl-CoA.
In addition to the effect of protein Cys redox change, consequences from altered abundance of metabolites will also influence cellular metabolism and function. Carnitine is a quaternary ammonium compound synthesized from Lys and Met in the liver and kidney (Steiber et al., 2004). Carnitine is required for transporting fatty acid from the cytoplasm to the mitochondria during lipid metabolism via β-oxidation to generate metabolic energy. Cd altered levels of several metabolites associated with carnitine and CoA in the mitochondria, such as palmitoyl-L-carnitine, propionyl-L-carnitine, malonylcarnitine, palmitoyl-CoA, and several acyl-CoA (see Table 4). Because carnitine and acylcarnitine play an important role in the protection against oxidative stress (Selim et al., 2012; Yari et al., 2010), altered mitochondrial levels of acylcarnitines by Cd could influence mitochondrial protection from oxidative stress.
Recent studies have addressed an important role of BCAA in energy metabolism associated with longevity of a broad range of species including mammals (Valerio et al., 2011). The results suggested that alteration in BCAA metabolism by malnutrition resulted in decreased lifespan accompanied by increased oxidative stress and disrupted mitochondrial biogenesis (Valerio et al., 2011). With regard to Cd effect on BCAA metabolism, a study using nuclear magnetic resonance (NMR) spectroscopy in Silene cucubalus cell showed that BCAA and glutamine amounts were decreased as a consequence of Cd exposure (Bailey et al., 2003). Another study using Wistar rats showed that Cd (50 mg/kg/day) significantly decreased levels of all BCAA including valine, leucine, and isoleucine in blood serum and the kidney, and valine in liver compared with those in control rats (Pasternak et al., 2004). More recently, an NMR-based metabolomic study in green mussels exposed to Cd for 1 week showed significant increase in BCAA (Wu and Wang, 2010). Additionally, alterations in BCAA metabolism can be an indicator of enhanced amino acid degradation in response to altered energy production via fatty acid oxidation. In line with this hypothesis, our data show Cd-mediated increases in succinyl, isobutyryl, and acetyl-CoA as well as propionyl-L-carnitine, all of which are markers of BCAA breakdown and protein catabolism. Accordingly, our metabolomics data with altered levels of BCAA by Cd supports previous findings and also provides information on additional target molecules affected by Cd exposure.
An application of omics technology together with available bioinformatics software enabled identification of functionally significant pathways and a broad range of proteins and small molecules affected by Cd. However, biological functions of Cys residues in proteins and metabolic changes resulted from altered abundance of small molecules are largely unidentified. Therefore, further studies are needed to improve our knowledge in this under-studied area, i.e., association of small molecules and redox-sensitive Cys with mitochondrial functional responses.
In conclusion, exposure to the environmental toxicant Cd significantly increased oxidative stress by diminishing GSH and total Pr-SH levels, and oxidizing GSH/GSSG redox potential in plasma and liver. Especially in the liver, Cd appeared to elevate oxidative stress in association with mitochondrial impairment, significant oxidation in the mitochondrial GSH/GSSG redox system, and increased sensitivity to MPT. The results from redox proteomics and metabolomics with bioinformatics supported disruption in mitochondrial metabolic pathways. This integrated omics approach identified Cd-affected significant pathways and networks for BCAA regulation and long-chain fatty acid oxidation, suggesting that acute Cd exposure substantially impacts mitochondrial metabolism and liver physiology. Moreover, our study establishes that integrated redox proteomics and metabolomics can be used as a powerful approach to determine environmental-toxicant-induced effects in complex systems.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institutes of Health (ES009047, HL113451, AG038746, and ES019116).
Supplementary Material
Acknowledgments
We thank Dr Nick Seyfried, Duc Duong (Biochemistry, Proteomics Core, Emory University), ViLinh Tran, and Karan Uppal for mass spectrometry analysis and data processing.
REFERENCES
- Andresen B. S., Olpin S., Poorthuis B. J., Scholte H. R., Vianey-Saban C., Wanders R., Ijlst L., Morris A., Pourfarzam M., Bartlett K., et al. Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am. J. Hum. Genet. 1999;64:479–494. doi: 10.1086/302261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ATSDR. Toxicological profile for cadmium. 2012. Available at: http://www.atsdr.cdc.gov/toxprofiles/tp5.pdf. Accessed February 6, 2006. [PubMed]
- Bailey N. J., Oven M., Holmes E., Nicholson J. K., Zenk M. H. Metabolomic analysis of the consequences of cadmium exposure in Silene cucubalus cell cultures via 1H NMR spectroscopy and chemometrics. Phytochemistry. 2003;62:851–858. doi: 10.1016/s0031-9422(02)00719-7. [DOI] [PubMed] [Google Scholar]
- Bartosiewicz M., Penn S., Buckpitt A. Applications of gene arrays in environmental toxicology: Fingerprints of gene regulation associated with cadmium chloride, benzo(a)pyrene, and trichloroethylene. Environ. Health Perspect. 2001;109:71–74. doi: 10.1289/ehp.0110971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyaeva E. A., Korotkov S. M., Saris N. E. In vitro modulation of heavy metal-induced rat liver mitochondria dysfunction: A comparison of copper and mercury with cadmium. J. Trace Elem. Med. Biol. 2011;25(Suppl. 1):S63–S73. doi: 10.1016/j.jtemb.2010.10.007. [DOI] [PubMed] [Google Scholar]
- Bieber L. L. Carnitine. Annu. Rev. Biochem. 1988;57:261–283. doi: 10.1146/annurev.bi.57.070188.001401. [DOI] [PubMed] [Google Scholar]
- Collins S. A., Sinclair G., McIntosh S., Bamforth F., Thompson R., Sobol I., Osborne G., Corriveau A., Santos M., Hanley B., et al. Carnitine palmitoyltransferase 1A (CPT1A) P479L prevalence in live newborns in Yukon, Northwest Territories, and Nunavut. Mol. Genet. Metab. 2010;101:200–204. doi: 10.1016/j.ymgme.2010.07.013. [DOI] [PubMed] [Google Scholar]
- Dailianis S., Patetsini E., Kaloyianni M. The role of signalling molecules on actin glutathionylation and protein carbonylation induced by cadmium in haemocytes of mussel Mytilus galloprovincialis (Lmk) J. Exp. Biol. 2009;212(Pt 22):3612–3620. doi: 10.1242/jeb.030817. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I., Rossi R., Giustarini D., Gagliano N., Di Simplicio P., Colombo R., Milzani A. Methionine oxidation as a major cause of the functional impairment of oxidized actin. Free Radic. Biol. Med. 2002;32:927–937. doi: 10.1016/s0891-5849(02)00799-2. [DOI] [PubMed] [Google Scholar]
- Dudley R. E., Gammal L. M., Klaassen C. D. Cadmium-induced hepatic and renal injury in chronically exposed rats: Likely role of hepatic cadmium-metallothionein in nephrotoxicity. Toxicol. Appl. Pharmacol. 1985;77:414–426. doi: 10.1016/0041-008x(85)90181-4. [DOI] [PubMed] [Google Scholar]
- Elias J. E., Gygi S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Eng J., McCormack A., Yates J. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Gasiewicz T. A., Smith J. C. Interactions of cadmium and selenium in rat plasma in vivo and in vitro. Biochim. Biophys. Acta. 1976;428:113–121. doi: 10.1016/0304-4165(76)90113-6. [DOI] [PubMed] [Google Scholar]
- Giangregorio N., Tonazzi A., Indiveri C., Palmieri F. Conformation-dependent accessibility of Cys-136 and Cys-155 of the mitochondrial rat carnitine/acylcarnitine carrier to membrane-impermeable SH reagents. Biochim. Biophys. Acta. 2007;1767:1331–1339. doi: 10.1016/j.bbabio.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Go Y. M., Duong D. M., Peng J., Jones D. P. Protein cysteines map to functional networks according to steady-state level of oxidation. J. Proteomics Bioinform. 2011;4:196–209. doi: 10.4172/jpb.1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go Y. M., Jones D. P. Thiol/disulfide redox states in signaling and sensing. Crit. Rev. Biochem. Mol. Biol. 2013a;48:173–181. doi: 10.3109/10409238.2013.764840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go Y. M., Jones D. P. The redox proteome. J. Biol. Chem. 2013b;288:26512–26520. doi: 10.1074/jbc.R113.464131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go Y. M., Orr M., Jones D. P. Actin cytoskeleton redox proteome oxidation by cadmium. Am. J. Physiol. 2013a;305:L831–L843. doi: 10.1152/ajplung.00203.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go Y. M., Orr M., Jones D. P. Increased nuclear thioredoxin-1 potentiates cadmium-induced cytotoxicity. Toxicol. Sci. 2013b;131:84–94. doi: 10.1093/toxsci/kfs271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go Y. M., Park H., Koval M., Orr M., Reed M., Liang Y., Smith D., Pohl J., Jones D. P. A key role for mitochondria in endothelial signaling by plasma cysteine/cystine redox potential. Free Radic. Biol. Med. 2010;48:275–283. doi: 10.1016/j.freeradbiomed.2009.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go Y. M., Roede J. R., Walkers D., Duong D. M., Seyfried N. T., Orr M., Liang Y., Pennell K. D., Jones D. P. Selective targeting of the cysteine proteome by thioredoxin and glutathione redox systems. Mol. Cell. Proteomics. 2013c;12:3285–3296. doi: 10.1074/mcp.M113.030437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go Y. M., Ziegler T. R., Johnson J. M., Gu L., Hansen J. M., Jones D. P. Selective protection of nuclear thioredoxin-1 and glutathione redox systems against oxidation during glucose and glutamine deficiency in human colonic epithelial cells. Free Radic. Biol. Med. 2007;42:363–370. doi: 10.1016/j.freeradbiomed.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Q., Hart B. A. Effect of thiols on cadmium-induced expression of metallothionein and other oxidant stress genes in rat lung epithelial cells. Toxicology. 1997;119:179–191. doi: 10.1016/s0300-483x(96)03608-6. [DOI] [PubMed] [Google Scholar]
- Goyer R. A. Toxic and essential metal interactions. Annu. Rev. Nutr. 1997;17:37–50. doi: 10.1146/annurev.nutr.17.1.37. [DOI] [PubMed] [Google Scholar]
- Gregersen N., Andresen B. S., Corydon M. J., Corydon T. J., Olsen R. K., Bolund L., Bross P. Mutation analysis in mitochondrial fatty acid oxidation defects: Exemplified by acyl-CoA dehydrogenase deficiencies, with special focus on genotype-phenotype relationship. Hum. Mutat. 2001;18:169–189. doi: 10.1002/humu.1174. [DOI] [PubMed] [Google Scholar]
- Gunter T. E., Pfeiffer D. R. Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 1990;258(5 Pt 1):C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- He M., Cai J., Go Y. M., Johnson J. M., Martin W. D., Hansen J. M., Jones D. P. Identification of thioredoxin-2 as a regulator of the mitochondrial permeability transition. Toxicol. Sci. 2008;105:44–50. doi: 10.1093/toxsci/kfn116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter D. R., Haworth R. A., Southard J. H. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J. Biol. Chem. 1976;251:5069–5077. [PubMed] [Google Scholar]
- Ikediobi C. O., Badisa V. L., Ayuk-Takem L. T., Latinwo L. M., West J. Response of antioxidant enzymes and redox metabolites to cadmium-induced oxidative stress in CRL-1439 normal rat liver cells. Int. J. Mol. Med. 2004;14:87–92. [PubMed] [Google Scholar]
- Jones D. P., Liang Y. Measuring the poise of thiol/disulfide couples in vivo. Free Radic. Biol. Med. 2009;47:1329–1338. doi: 10.1016/j.freeradbiomed.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajcovicova-Kudlackova M., Ozdin L. Effect of fatty acid composition, cadmium and vitamin E intake on prooxidative-antioxidative state of rat liver. Vet. Med. (Praha) 1995;40:293–298. [PubMed] [Google Scholar]
- Kudo N., Nakagawa Y., Waku K., Kawashima Y., Kozuka H. Prevention by zinc of cadmium inhibition of stearoyl-CoA desaturase in rat liver. Toxicology. 1991;68:133–142. doi: 10.1016/0300-483x(91)90016-t. [DOI] [PubMed] [Google Scholar]
- Lash L. H., Jones D. P. Distribution of oxidized and reduced forms of glutathione and cysteine in rat plasma. Arch. Biochem. Biophys. 1985;240:583–592. doi: 10.1016/0003-9861(85)90065-7. [DOI] [PubMed] [Google Scholar]
- Lee W. K., Bork U., Thevenod F. Mitochondria as a target of cadmium nephrotoxicity: Induction of swelling and cytochrome C release. Toxicol. Mech. Methods. 2004;14:67–71. doi: 10.1080/15376520490257509. [DOI] [PubMed] [Google Scholar]
- Levine D. M., Haynor D. R., Castle J. C., Stepaniants S. B., Pellegrini M., Mao M., Johnson J. M. Pathway and gene-set activation measurement from mRNA expression data: The tissue distribution of human pathways. Genome Biol. 2006;7:R93. doi: 10.1186/gb-2006-7-10-r93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., He W., Yan C., Qi Y., Zhang Y. Roles of reactive oxygen species and mitochondria in cadmium-induced injury of liver cells. Toxicol. Ind. Health. 2011;27:249–256. doi: 10.1177/0748233710386408. [DOI] [PubMed] [Google Scholar]
- Mamyrbaeva Z., Shalakhmetova T. M., Kudriavtseva M. V., Kudriavtsev B. N. [Effect of cadmium sulfate and strontium chloride on the glycogen content in hepatocytes of rats of various ages] Tsitologiia. 1998;40:432–444. [PubMed] [Google Scholar]
- Mashek D. G., Li L. O., Coleman R. A. Long-chain acyl-CoA synthetases and fatty acid channeling. Future Lipidol. 2007;2:465–476. doi: 10.2217/17460875.2.4.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi H. R., Katyare S. S. Effect of treatment with cadmium on structure-function relationships in rat liver mitochondria: Studies on oxidative energy metabolism and lipid/phospholipids profiles. J. Membr. Biol. 2009;232:47–57. doi: 10.1007/s00232-009-9217-x. [DOI] [PubMed] [Google Scholar]
- Newsholme P., Gaudel C., Krause M. Mitochondria and diabetes. An intriguing pathogenetic role. Adv. Exp. Med. Biol. 2012;942:235–247. doi: 10.1007/978-94-007-2869-1_10. [DOI] [PubMed] [Google Scholar]
- Nordberg G. F., Kjellström T., Nordberg M. Kinetics and metabolism. Boca Raton, FL: CRC Press; 1985. [Google Scholar]
- Okamura-Ikeda K., Ikeda Y., Tanaka K. An essential cysteine residue located in the vicinity of the FAD-binding site in short-chain, medium-chain, and long-chain acyl-CoA dehydrogenases from rat liver mitochondria. J. Biol. Chem. 1985;260:1338–1345. [PubMed] [Google Scholar]
- Pasternak K., Szpetnar M., Boguszewska A. Magnesium and branched chain amino acids in rats intoxicated with Pb2+ or Cd2+ and receiving certain bioflavonoids or bioflavonoids with glutamine. Magnes. Res. 2004;17:72–78. [PubMed] [Google Scholar]
- Peng J., Elias J. E., Thoreen C. C., Licklider L. J., Gygi S. P. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: The yeast proteome. J. Proteome Res. 2003;2:43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- Prozialeck W. C., Niewenhuis R. J. Cadmium (Cd2+) disrupts intercellular junctions and actin filaments in LLC-PK1 cells. Toxicol. Appl. Pharmacol. 1991;107:81–97. doi: 10.1016/0041-008x(91)90333-a. [DOI] [PubMed] [Google Scholar]
- Roede J. R., Hansen J. M., Go Y. M., Jones D. P. Maneb and paraquat-mediated neurotoxicity: Involvement of peroxiredoxin/thioredoxin system. Toxicol. Sci. 2011;121:368–375. doi: 10.1093/toxsci/kfr058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roede J. R., Park Y., Li S., Strobel F. H., Jones D. P. Detailed mitochondrial phenotyping by high resolution metabolomics. PLoS One. 2012;7:e33020. doi: 10.1371/journal.pone.0033020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage M. K., Jones D. P., Reed D. J. Calcium- and phosphate-dependent release and loading of glutathione by liver mitochondria. Arch. Biochem. Biophys. 1991;290:51–56. doi: 10.1016/0003-9861(91)90590-f. [DOI] [PubMed] [Google Scholar]
- Sedlak J., Lindsay R. H. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman's reagent. Anal. Biochem. 1968;25:192–205. doi: 10.1016/0003-2697(68)90092-4. [DOI] [PubMed] [Google Scholar]
- Selim M. E., Rashed el H. A., Aleisa N. A., Daghestani M. H. The protection role of heat shock protein 70 (HSP-70) in the testes of cadmium-exposed rats. Bioinformation. 2012;8:58–64. doi: 10.6026/97320630008058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh Z. A. Metallothionein as a storage protein for cadmium: Its toxicological implications. Dev. Toxicol. Environ. Sci. 1982;9:69–76. [PubMed] [Google Scholar]
- Shoaf A. R., Jarmer S., Harbison R. D. Heavy metal inhibition of carnitine acetyltransferase activity in human placental syncytiotrophoblast: Possible site of action of HgCl2, CH3HgCl, and CdCl2. Teratog. Carcinog. Mutagen. 1986;6:351–360. doi: 10.1002/tcm.1770060502. [DOI] [PubMed] [Google Scholar]
- Soltow Q. A., Strobel F. H., Mansfield K. G., Wachtman L., Park Y., Jones D. P. High-performance metabolic profiling with dual chromatography-Fourier-transform mass spectrometry (DC-FTMS) for study of the exposome. Metabolomics. 2013;9:132–143. doi: 10.1007/s11306-011-0332-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiber A., Kerner J., Hoppel C. L. Carnitine: A nutritional, biosynthetic, and functional perspective. Mol. Aspects Med. 2004;25:455–473. doi: 10.1016/j.mam.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Su R., Wang R., Guo S., Cao H., Pan J., Li C., Shi D., Tang Z. In vitro effect of copper chloride exposure on reactive oxygen species generation and respiratory chain complex activities of mitochondria isolated from broiler liver. Biol. Trace Elem. Res. 2011;144:668–677. doi: 10.1007/s12011-011-9039-4. [DOI] [PubMed] [Google Scholar]
- Uppal K., Soltow Q. A., Strobel F. H., Pittard W. S., Gernert K. M., Yu T., Jones D. P. xMSanalyzer: Automated pipeline for improved feature detection and downstream analysis of large-scale, non-targeted metabolomics data. BMC Bioinformatics. 2013;14:15. doi: 10.1186/1471-2105-14-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerio A., D'Antona G., Nisoli E. Branched-chain amino acids, mitochondrial biogenesis, and healthspan: An evolutionary perspective. Aging (Albany NY) 2011;3:464–478. doi: 10.18632/aging.100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hove J. L., Saenz M. S., Thomas J. A., Gallagher R. C., Lovell M. A., Fenton L. Z., Shanske S., Myers S. M., Wanders R. J., Ruiter J., et al. Succinyl-CoA ligase deficiency: A mitochondrial hepatoencephalomyopathy. Pediatr. Res. 2010;68:159–164. doi: 10.1203/PDR.0b013e3181e5c3a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whanger P. D. Metabolic interactions of selenium with cadmium, mercury, and silver. Adv. Nutr. Res. 1985;7:221–250. doi: 10.1007/978-1-4613-2529-1_9. [DOI] [PubMed] [Google Scholar]
- Wu H., Liu X., Zhao J., Yu J. NMR-based metabolomic investigations on the differential responses in adductor muscles from two pedigrees of Manila clam Ruditapes philippinarum to Cadmium and Zinc. Mar. Drugs. 2011;9:1566–1579. doi: 10.3390/md9091566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Wang W. X. NMR-based metabolomic studies on the toxicological effects of cadmium and copper on green mussels Perna viridis. Aquat. Toxicol. 2010;100:339–345. doi: 10.1016/j.aquatox.2010.08.005. [DOI] [PubMed] [Google Scholar]
- Xu P., Duong D. M., Peng J. Systematical optimization of reverse-phase chromatography for shotgun proteomics. J. Proteome Res. 2009;8:3944–3950. doi: 10.1021/pr900251d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yari A., Asadi M. H., Bahadoran H., Dashtnavard H., Imani H., Naghii M. R. Cadmium toxicity in spermatogenesis and protective effects of L-carnitine in adult male rats. Biol. Trace Elem. Res. 2010;137:216–225. doi: 10.1007/s12011-009-8577-5. [DOI] [PubMed] [Google Scholar]
- Yu T., Park Y., Johnson J. M., Jones D. P. apLCMS—adaptive processing of high-resolution LC/MS data. Bioinformatics. 2009;25:1930–1936. doi: 10.1093/bioinformatics/btp291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N. N., Mao W. P., Wei C. J., Zhou L., Liu H. Y., Feng J. [Influence of cadmium on structure and functions of mitochondria in hepatic cells] Zhonghua Yi Xue Za Zhi. 2008;88:1350–1353. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.