

Abstract
Most of the mutations in the presenilin‐1 gene (PS‐1) are associated with familial Alzheimer's disease (AD). However, certain examples can be associated with frontotemporal dementia (FTD). We performed a clinical evaluation of individuals belonging to a family with the FTD phenotype, and additional molecular studies and neuropathological assessment of the proband. The PS‐1 M146V mutation was found in the 50‐year‐old subject (the proband) with family history of early‐onset FTD. Neuropathological examination showed abundant amyloid plaques, widespread neurofibrillary pathology, Pick bodies in the hippocampus and cortex, cortical globose tangles and ubiquitin‐positive nuclear inclusions in white matter oligodendrocytes. We report a kindred with clinical features of FTD, whose proband bore the PS‐1 M146V mutation and showed diffuse Alzheimer's type pathology and Pick bodies on post‐mortem neuropathological examination. As with other mutations within the same codon, this substitution may predispose to both diseases by affecting APP and/or tau processing.
Keywords: Alzheimer's disease, FTD, M146V, oligodendrocytes, Pick bodies, PS‐1 mutations
Introduction
Alzheimer's disease (AD) is the most common dementia worldwide, representing 65–70% of cases in the United States 21 and other countries 14. Originally described in the early 20th century 1, AD represents the end‐stage of a neurodegenerative process that evolves over several decades 10. In general, AD presents as a sporadic disorder; however, in approximately 5% of the cases 2, the disease can have a familial component.
Clinically, patients suffering from the illness tend to present with recent memory disturbance, progressing to visuospatial, attention and language impairment (ie, low verbal fluency and problems with naming). Some patients may present with prominent features of executive dysfunction and language impairment as well as behavioral abnormalities (frontal dysfunction) early in the disease, the so‐called frontal variant AD 15. This variant is often difficult to differentiate from frontotemporal dementia (FTD) 36.
Even though most of the cases of AD are late‐onset and sporadic, early‐onset AD with autosomal dominant inheritance is recognized and is associated with mutations in three different genes: presenilin‐1 (PS‐1) gene located in chromosome 14 32, presenilin 2 (PS‐2) gene located in chromosome 1 27 and β‐Amyloid precursor protein (APP) gene located in chromosome 21 33. Most of the mutations described, approximately 70%, are located in different codons (Arg79Val, Tyr115Cys, Met146Leu, Met146Val, Thr354Ile, etc.) 26, in the PS‐1 gene 17, and in general those patients bearing those genetic defects tend to present as AD, however, certain mutations (ie, Leu113Pro, Gly183Val) in the same gene can be associated with a FTD phenotype 13.
The neuropathological changes of AD are characterized by the presence of β‐Amyloid deposition forming different kind of plaques (ie, diffuse, neuritic, cotton‐wool) and sometimes with deposition in the blood vessels (cerebral amyloid angiopathy; CAA). Additionally, neurofibrillary changes consisting of tangles, dystrophic neurites and neuropil threads are seen; which tend to be more common in the hippocampus and neocortex 4, 18.
The aims of this study are to report an Argentine family with familial dementia, showing an amino acid substitution of methionine (M) to valine (V) at codon 146 of the PS‐1 gene, which clinically behaved as FTD, and secondly, to describe for the first time the neuropathological findings associated with this mutation.
Materials and Methods
Family history and clinical assessment
Informed consent for DNA and neuropathological testing was obtained from the next‐of‐kin. Clinical histories and examination details were obtained from medical notes completed at the time of clinical interview. Family history and complementary clinical information were obtained from alive and healthy members of the kindred (Figure 1).
Figure 1.
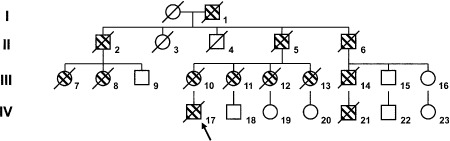
Family pedigree. Dashed symbols reflect individuals affected by dementia, and diagonal lines those who are deceased. The arrow singles out the proband.
A battery of neuropsychological assessments encompassing memory, language, orientation, attention, visuospatial skills and executive functions was administered to the proband. Tests performed included Mini Mental State Examination 9, Trail Making Test form A and form B 23, Boston naming test 16, Signoret memory battery 31 and Digit span of the Wechsler Adult Intelligence Scale 34.
Neuroimaging studies (proband) included brain magnetic resonance imaging (MRI), computed tomography (CT) scan and brain single‐photon emission computed tomography (SPECT) with the intravenous administration of 30 mCi of 99mTc‐ECD.
Neuropathological assessment
A brain autopsy was performed on the proband (IV‐13, Figure 1). The brain weighed 1150 g; the left hemibrain was fixed in 10% formalin and the right hemibrain frozen at −80°C. The left hemisphere was sectioned 2 weeks after fixation. Samples of fixed tissue were obtained from the superior‐middle frontal gyri, superior‐middle temporal gyri, inferior parietal gyrus, calcarine cortex, middle hippocampal formation at the level of the lateral geniculate body, basal ganglia (putamen, globus pallidum, and caudate), middle thalamus, cerebellum, mesencephalon, pons and medulla. The samples were processed for paraffin embedding, then 10‐μm thick sections were stained with hematoxylin and eosin, and the modified Bielschowsky silver impregnation. In addition, sections were immunostained for ubiquitin (NCL‐Ubiq; 1:500; Novocastra Reagents, Newcastle, UK), β‐Amyloid (NCL‐β‐Amyloid; 1:150; Novocastra Reagents), phospho‐tau (NCL‐Tau‐2; 1:1000; Novocastra Reagents), 3R tau (RD‐3; 1:500; Millipore, Billerica, MA, USA), 4R tau (RD‐4; 1:200; Millipore), TAR DNA‐binding protein 43 (TDP‐43; 1:100; ProteinTech Group, Inc., Chicago, IL, USA) and for alpha‐synuclein (NCL‐Asyn; 1:50; Novocastra Reagents). The neuritic plaque density was assigned a semi‐quantitative and age‐adjusted score (0, A, B, or C) according to the guidelines of Consortium to Establish a Registry for Alzheimer's Disease (CERAD) 18. The staging of neurofibrillary tangles was assigned a score (0 to 6) according to the Braak system 4.
Molecular analysis
DNA was isolated from blood sample using a QIAGEN kit. The entire open reading frame of PSEN1 (including exon/intron boundaries) of the patient was analyzed as previously described 25. Mutations were searched by direct inspection of the fluorescent chromatographs using the SeqScape software (version 1.0) (Applied Biosystems, Foster City, CA, USA) Also, exons 9–13 from the microtubule associated protein tau (MAPT) gene were screened for mutations.
Results
Family history and clinical features
The family (Figure 1) was originally from Lisbon, Portugal, then moved to Argentina (around Buenos Aires) in the early 20th century.
The proband (IV‐17, Figure 1) was a 51‐year‐old male with secondary school education who at age 39 years sought medical consultation due to “progressive social disorder” characterized by gradual loss of initiative (apathy), disinhibition and loss of concern at work. A brain SPECT showed mild hypoperfusion in the left temporal, bilateral fronto‐orbital, and right parietal regions (Figure 2D). MRI displayed ventricular dilatation with decreased volume of both medial temporal lobes. Neuropsychological assessment showed executive dysfunction, anomia and memory loss. Based on the available clinical data (Lund and Manchester), a diagnosis of the behavioral variant of frontotemporal dementia was established (Table 1). The electroencephalogram (EEG) preformed in 1999 showed signs of diffuse disorganization. The progressed with extrapyramidal manifestations including rigidity, akinesia and movement disorders, but without tremor. An EEG performed in 2003 (age 44) displayed signs of disorganization and bilateral sharp waves. CT scan performed during the same year displayed bilateral ventricular dilatation. An MRI in 2004 (age 45) showed diffuse hyperintense images (T2 MRI) predominantly located in the occipital white matter, and progressive atrophy of the frontal and temporal regions (Figure 2A–C). During the ensuing years, his clinical state worsened and progressed with myoclonus, seizures and mutism. The patient died in early 2008 and a brain autopsy was performed in our unit.
Figure 2.
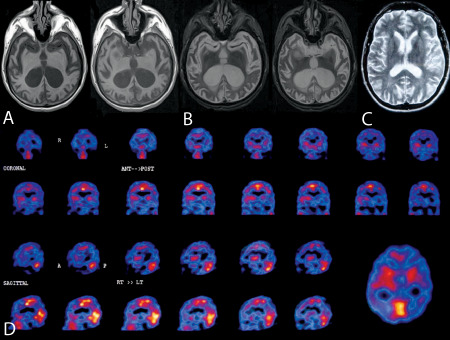
Neuroimaging from the proband. A–C. Brain magnetic resonance imaging at age 45 showing severe bilateral cortical and subcortical atrophy. D. Brain single‐photon emission computed tomography: Left temporal region, bilateral fronto‐orbital and parietal right hypoperfusion.
Table 1.
Neuropsychological Battery (2 years after onset of symptoms). Tests are expressed in total score
| Test | Patient | Mean | SD |
|---|---|---|---|
| Mini mental state exam (Folstein) | 20/30 | 28.1 | 1.3 |
| Clock drawing test | 4 | 6.1 | 1.1 |
| Episodic memory | |||
| Immediate logical memory | 1 | 8.3 | 0.9 |
| Delayed logical memory | 0 | 7.8 | 1.2 |
| Verbal learning | 4 | 7.7 | 0.7 |
| Delayed recall | 1 | 7.4 | 0.9 |
| Recognition | 6 | 11.5 | 0.5 |
| Attention | |||
| Digit span | 5 | 5.2 | 0.9 |
| Trail making test A | 120 | 47.2 | 17.9 |
| Language | |||
| Boston naming test | 44 | 50.7 | 5.1 |
| Semantic fluency (COWA) | 13 | 16.5 | 2.3 |
| Phonologic fluency (COWA) | 9 | 9.8 | 3.7 |
| Executive function | |||
| Trail making test B | 500 | 119.4 | 50.2 |
Scant clinical information was available from the other members of the family. However, according to reports from relatives, subjects III‐10 (mother), III‐11, III‐12, III‐13, III‐14 and IV‐21 died before the age of 50 with clinical diagnosis of FTD (Figure 1). Clinical information available from the rest of affected subjects showed only progressive dementia.
Neuropathology
Gross examination of the fixed hemisphere showed atrophy mainly in the frontal lobe and superior and middle temporal gyri; an atrophic hippocampal formation, and severe enlargement of the ventricular system. The substantia nigra and locus coeruleus were depigmented (Figure 3).
Figure 3.
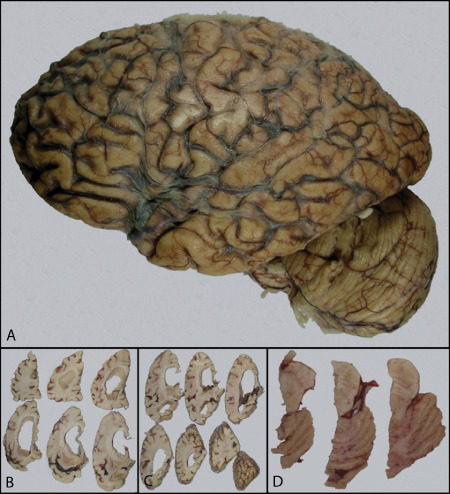
Left hemisphere. Displays atrophy of frontal and temporal cortices (A) Coronal sections showing prominent hydrocephalus (B), hippocampal atrophy (C) and pallor of the substantia nigra and locus coeruleus.
Microscopic examination revealed moderate spongiform changes in the superficial cortical layers, and diffuse neuronal loss throughout the cortex. The substantia nigra showed loss of pigmented neurons, pigment incontinence, gliosis and occasional Lewy bodies. Microscopic characteristics are summarized in Table 2.
Table 2.
Microscopic characteristics
| Technique | Microscopic features |
|---|---|
| Bielschowsky | CERAD Score C. |
| Braak SCORE VI. | |
| Pick bodies, also in the hippocampus. | |
| β‐Amyloid immunohistochemistry | Frequent deposits with occasional cotton‐wool appearance, more abundant in layer IV in temporal cortex, in layer III‐IV in frontal cortex, in layer III in parietal cortex and in layer IV in occipital cortex. |
| Positive blood vessels and surrounding neuropil. | |
| Deposits also detected in mesencephalon, pons, medulla and cerebellum. | |
| Phospho‐tau immunohistochemistry | Presence of both regular and globose tangles. |
| Occasional ballooned neurons in the cortex, substantia nigra, pons (locus coeruleus) and medulla. | |
| Pick bodies in the cortex and hippocampus. | |
| Neuropil threads throughout the tissue. | |
| 3R‐tau immunohistochemistry | Cortical pre‐tangles, tangles, ghost tangles, Pick bodies, neuropil threads, and plaques more frequent in layers III and IV. |
| Hippocampal pre‐tangles, tangles, ghost tangles, dystrophic neurites and Pick bodies in the dentate. | |
| Thalamic globose tangles, occasional ballooned neurons and dystrophic neurites. | |
| Basal ganglia with occasional pre‐tangles and tangles. | |
| Cerebellum, mesencephalon, pons, and medulla with occasional intraneuronal deposits and dystrophic neurites. | |
| 4R‐tau immunohistochemistry | Occasional cortical pre‐tangles, tangles, ghost tangles, dystrophic neurites, plaques and ballooned neurons. |
| The hippocampus showed dystrophic neurites, ghost tangles, and occasional pre‐tangles. | |
| Occasional thalamic globose tangles, dystrophic neurites and intraneuronal inclusions. | |
| Basal ganglia with occasional pre‐tangles, tangles and isolated granular deposits in the neuropil. | |
| Cerebellum with occasional intraneuronal deposits and dystrophic neurites. | |
| Mesencephalon, medulla and pons with dystrophic neurites, occasional tangles and intraneuronal deposits. | |
| Ubiquitin immunohistochemistry | Positivity in plaques, tangles, threads and Pick bodies. |
| Presence of minute nuclear inclusions in oligodendroglial cells. | |
| TDP‐43 | No pathological inclusions detected. |
Lewy bodies present in the substantia nigra were positive for alpha‐synuclein. Interestingly, minute ubiquitin and alpha‐synuclein positive nuclear inclusions were seen in oligodendroglial cells (Figure 4). Also, occasional neurons from basal ganglia displayed cytoplasmic granular inclusions positive with immunohistochemistry for alpha‐synuclein (Figure 4). TDP‐43 immunostain was negative. According to CERAD criteria 18, the clinical history of dementia and plaque frequency score of C, warranted the diagnosis of definitive Alzheimer's disease. Using the Braak classification for tangles, a score of VI was assigned 4. The presence of Pick bodies in the hippocampus and cortex rendered a diagnosis of Pick's disease (PiD).
Figure 4.
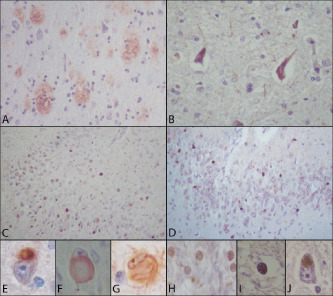
Microscopic examination. Cortical diffuse plaques (A). Frontal cortex displaying neurofibrillary changes and tangles with immunohistochemistry for hyperphosphorylated tau (B). Hyperphosphorylated tau (C) and RD3 hyperphosphrylated tau (D) positive Pick bodies in the dentate gyrus. Cortical tau‐positive Pick body (E). Cortical ballooned neuron appreciated with tau immunohistochemistry positive at the cytoplasmic border (F). Occasional cortical pyramidal neurons displayed globose tangles with hematoxylin and eosin, and tau (G). White matter showed many oligodendrocytes with nuclear granular inclusions positive with immunohistochemistry for ubiquitin (H) and alpha‐synuclein (I). Occasional neurons from basal ganglia displayed cytoplasmic granular inclusions positive with immunohistochemistry for alpha‐synuclein (I). (β‐Amyloid, NCL‐β‐Amyloid, Novocastra Reagents; phospho‐tau, NCL‐Tau‐2, Novocastra Reagents; 3R tau, RD‐3, Millipore; ubiquitin, NCL‐Ubiq, Novocastra Reagents; alpha‐synuclein, NCL‐Asyn, Novocastra Reagents).
Molecular analysis
Sequence analysis revealed a heterozygous A>G transition at position 436 of the cDNA (ATG to GTG) leading to an amino acid substitution of methionine (M) to valine (V) at codon 146 in exon 6 of the PS‐1 gene. No mutations were found in the exons analyzed from the MAPT gene.
Discussion
This case of familial dementia associated (or caused by) with a PS‐1 M146V mutation 26 presented with typical clinical features of FTD, and showed mainly frontal and temporal atrophy on both neuroimaging and macroscopic evaluation at autopsy. Histopathologically, the coexistence of both PiD and AD pathology were noted throughout the brain. Whereas AD is the most common dementing disorder worldwide, the prevalence of familial AD is much lower than the FTD group (5%) 14, and its genetic causes are mainly located in the APP gene, PS‐1 and the PS‐2. Taken together, the mutations present in those genes are responsible for approximately 30–50% of autosomal dominant AD cases. In FTD, the spectrum of genetic factors is different. More specifically, MAPT gene, the granulin gene and a gene encoding the chromatin modifying protein 2B are recognized. Although rare, PS‐1 mutations may be associated with isolated FTD phenotype in a minority of cases 6, 13.
Over the last years, cases presenting with clinical features of AD, displaying both AD and FTD pathology (Pick's disease in some cases), but with a mutation present in the PS‐1—a gene typically linked to AD—have been described 5, 12. Nevertheless, those cases presented clinically as AD, and in contrast to our case, the patient was diagnosed with FTD and a PS‐1 mutation (M146V) was detected, an uncommon association 24. Another case was described with the same mutation and similar presentation, but with solely AD clinicopathological features 28.
The neuropathological features of both disorders have in rare instances been identified by other authors 12. In most reports, the clinical presentation resembles AD 12, which further highlights the uniqueness of the present case. To the best of our knowledge, the neuropathological features associated with this mutation have been reported in an isolated kindred 28. Although histopathologic similarities to the present case are noted, tau pathology typical of PiD was seen, which was also present in a PS‐1 Met146Leu kindred previously described 12. PS‐1 mutations are known to influence the γ secretase activity by either enhancing its activity, and therefore increasing the amount of β‐Amyloid‐42 over total β‐Amyloid 29, or by inhibiting it 3. It is also proposed that its absence or mutation can also influence the amount of tau hyperphosphorylation in knockout mice 8, or in autopsied brains 30. Our case shows other microscopic characteristics such as the presence of globose tangles, the amount of β‐Amyloid that was present even in the brain stem, and the ubiquitin‐positive inclusions in the oligodendroglial cells, glial elements that were previously described either to be affected in transgenic mouse models of AD with the PS‐1 M146V mutation 7, or could show also those type of intranuclear inclusions in occasional cases of frontotemporal 20 or multiple system atrophy 19, and in experimental models of stress 11. Although the presence of alpha‐synuclein deposits found in the brain could be related to the enhancement by β‐Amyloid previously described 22, the location (granular intraneuronal in basal ganglia, and intranuclear in oligodendrocytes) could be related to a certain phenomena of alpha‐synuclein mislocalization described in animal models of PS‐1 deficiency 35 and multiple system atrophy 24.
Whether this or any other mutation in the PS‐1 gene is responsible for the FTD phenotype alone or accompanied by AD remains unclear. In fact, the clinical and neuropathological variability demonstrated among cases with different or even with the same PS‐1 mutation lends credence to that notion, and emphasizes the idea that there should be another factor, either polymorphic or mutational, that would contribute to that variation.
In conclusion, we report an Argentine patient with a family history of dementia, who presented with clinical features of FTD, associated with the M146V mutation of the PS‐1 gene and histopathological changes of both PiD and AD.
Acknowledgments
This work was supported by the Department of Research, Institute for Neurological Research (FLENI).
Conflict of interest: All of the authors declare no conflict of interest.
References
- 1. Alzheimer A (1906) Uber einen eigenartigen schweren Krankheitsprozess der Hirnrinde. Neurol Centralbl 25:1134. [Google Scholar]
- 2. Alzheimer's‐Association (2009) Alzheimer's disease facts and figures. Alzheimers Dement 5:1–80. [DOI] [PubMed] [Google Scholar]
- 3. Amtul Z, Lewis PA, Piper S, Crook R, Baker M, Findlay K et al (2002) A presenilin 1 mutation associated with familial frontotemporal dementia inhibits g‐secretase cleavage of APP and notch. Neurobiol Dis 9:269–273. [DOI] [PubMed] [Google Scholar]
- 4. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 5. Bruni AC, Bernardi L, Colao R, Rubino E, Smirne N, Frangipane F et al (2010) Worldwide distribution of PSEN1 Met146Leu mutation: a large variability for a founder mutation. Neurology 74:798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dermaut B, Kumar‐Singh S, Engelborghs S, Theuns J, Rademakers R, Saerens J et al (2004) A novel presenilin 1 mutation associated with Pick's disease but not B‐amyloid plaques. Ann Neurol 55:617–626. [DOI] [PubMed] [Google Scholar]
- 7. Desai MK, Guercio BJ, Narrow WC, Bowers WJ (2011) An Alzheimer's disease‐relevant presenilin‐1 mutation augments amyloid‐beta‐induced oligodendrocyte dysfunction. Glia 59:627–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feng R, Wang H, Wang J, Shrom D, Zeng X, Tsien JZ (2004) Forebrain degeneration and ventricle enlargement caused by double knockout of Alzheimer's presenilin‐1 and presenilin‐2. Proc Natl Acad Sci USA 101:8162–8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Folstein MF, Folstein SE, McHugh PR (1975) “Mini‐Mental State”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12:189–198. [DOI] [PubMed] [Google Scholar]
- 10. Godbolt A, Cipolotti L, Watt H, Fox NC, Janssen JC, Rossor MN (2004) The natural history of Alzheimer disease: a longitudinal presymptomatic and symptomatic study of a familial cohort. Arch Neurol 61:1743–1748. [DOI] [PubMed] [Google Scholar]
- 11. Goldbaum O, Richter‐Landsberg C (2004) Proteolytic stress causes heat shock protein induction, tau ubiquitination, and the recruitment of ubiquitin to tau‐positive aggregates in oligodendrocytes in culture. J Neurosci 24:5748–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Halliday GM, Song Y, Lepar G, Brooks W, Kwok J, Kersaitis C et al (2005) Pick bodies in a family with presenilin‐1 Alzheimer's disease. Ann Neurol 57:139–143. [DOI] [PubMed] [Google Scholar]
- 13. Hutton M (2004) Presenilin mutations associated with fronto‐temporal dementia. Ann Neurol 55:604–606. [DOI] [PubMed] [Google Scholar]
- 14. Ikeda M, Ishikawa T, Tanabe H (2004) Epidemiology of frontotemporal lobar degeneration. Dement Geriatr Cogn Disord 17:265–268. [DOI] [PubMed] [Google Scholar]
- 15. Johnson J, Head E, Kim R, Starr A, Cotman C (1999) Clinical and pathological evidence for a frontal variant of Alzheimer disease. Arch Neurol 56:1233–1239. [DOI] [PubMed] [Google Scholar]
- 16. Kaplan E, Goodglass H, Weintraub S (1983) Boston Naming Test. Lea & Febiger: Philadelphia. [Google Scholar]
- 17. Lowe J, Mirra S, Hyman BT, Dickson D (2008) Greenfield's Neuropathology. Hodder Arnold: London. [Google Scholar]
- 18. Mirra S, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41:479–486. [DOI] [PubMed] [Google Scholar]
- 19. Murayama S, Arima K, Nakazato Y, Satoh J, Oda M, Inose T (1992) Immunocytochemical and ultrastructural studies of neuronal and oligodendroglial cytoplasmic inclusions in multiple system atrophy. Oligodendroglial cytoplasmic inclusions. Acta Neuropathol 84:32–38. [DOI] [PubMed] [Google Scholar]
- 20. Pirici D, Vandenberghe R, Rademakers R, Dermaut B, Cruts M, Vennekens K et al (2006) Characterization of ubiquitinated intraneuronal inclusions in a novel Belgian frontotemporal lobar degeneration family. J Neuropathol Exp Neurol 65:289–301. [DOI] [PubMed] [Google Scholar]
- 21. Plassman B, Langa K, Fisher G, Heeringa S, Weir D, Mea O (2007) Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology 29:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pletnikova O, West N, Lee M, Rudow G, Skolasky R, Dawson T et al (2005) Abeta deposition is associated with enhanced cortical alpha‐synuclein lesions in Lewy body diseases. Neurobiol Aging 26:1183–1192. [DOI] [PubMed] [Google Scholar]
- 23. Reitan R (1958) Validity of the trail making test as an indication of organic brain damage. Percept Mot Skills 8:271–276. [Google Scholar]
- 24. Rockenstein E, Ubhi K, Inglis C, Mante M, Patrick C, Adame A, Masliah E (2012) Neuronal to oligodendroglial α‐synuclein redistribution in a double transgenic model of multiple system atrophy. Neuroreport 23:259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rogaeva E, Bergeron C, Sato C, Moliaka I, Kawarai T, Toulina A et al (2003) PS1 Alzheimer's disease family with spastic paraplegia: the search for a gene modifier. Neurology 61:1005–1007. [DOI] [PubMed] [Google Scholar]
- 26. Rogaeva EA, Fafel KC, Song YQ, Medeiros H, Sato C, Liang Y et al (2001) Screening for PS1 mutations in a referral‐based series of AD cases. 21 Novel mutations. Neurology 57:621–625. [DOI] [PubMed] [Google Scholar]
- 27. Schellenberg GD, Bird TD, Wijsman EM, Orr HT, Anderson L, Nemens E et al (1992) Genetic linkage evidence for a familial Alzheimer's disease locus on chromosome 14. Science 258:668–671. [DOI] [PubMed] [Google Scholar]
- 28. Scholl M, Almkvist O, Bogdanovic N, Wall A, Langstrom B, Viitanen M, Nordberg A (2011) Time course of glucose metabolism in relation to cognitive performance and postmortem neuropathology in Met146Val PSEN1 mutation carriers. J Alzheimers Dis 24:495–506. [DOI] [PubMed] [Google Scholar]
- 29. Shepherd C, McCann H, Halliday GM (2009) Variations in the neuropathology of familial Alzheimer's disease. Acta Neuropathol 118:37–52. [DOI] [PubMed] [Google Scholar]
- 30. Shepherd CE, Gregory GC, Vickers JC, Brooks WS, Kwok JB, Schofield PR et al (2004) Positional effects of presenilin‐1 mutations on tau phosphorylation in cortical plaques. Neurobiol Dis 15:115–119. [DOI] [PubMed] [Google Scholar]
- 31. Signoret J, Whiteley A (1979) A memory battery scale. Int Neuropsychol Soc Bull 9:2–26. [Google Scholar]
- 32. St George‐Hyslop P, Haines J, Rogaev E, Mortilla M, Vaula G, Pericak‐Vance M et al (1992) Genetic evidence for a novel familial Alzheimer's disease locus on chromosome 14. Nat Genet 2:330–334. [DOI] [PubMed] [Google Scholar]
- 33. St George‐Hyslop PH, Tanzi RE, Polinsky RJ, Haines JL, Nee L, Watkins PC et al (1987) The genetic defect causing familial Alzheimer's disease maps on chromosome 21. Science 235:885–890. [DOI] [PubMed] [Google Scholar]
- 34. Wechsler D (1981) WAIS‐R Manual: Wechsler Adult Intelligence Scale‐Revised. Psychological Corporation: New : York. [Google Scholar]
- 35. Wilson C, Murphy D, Giasson B, Zhang B, Trojanowski J, Lee V (2004) Degradative organelles containing mislocalized alpha‐and beta‐synuclein proliferate in presenilin‐1 null neurons. J Cell Biol 165:335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Woodward M, Jacova C, Black S, Kertesz A, Mackenzie I, Feldman H (2009) Differentiating the frontal variant of Alzheimer's disease. Int J Geriatr Psychiatry 25:732–738. [DOI] [PubMed] [Google Scholar]