

Summary
Synthesis of dNTPs is required for both DNA replication and DNA repair and is catalyzed by ribonucleotide reductases (RNR), which convert ribonucleotides to their deoxy forms [1, 2]. Maintaining the correct levels of dNTPs for DNA synthesis is important for minimising the mutation rate [3-7], and this is achieved by tight regulation of ribonucleotide reductase [2, 8, 9]. In fission yeast, ribonucleotide reductase is regulated in part by a small protein inhibitor, Spd1, which is degraded in S phase and after DNA damage to allow up-regulation of dNTP supply [10-12]. Spd1 degradation is mediated by the activity of the CRL4Cdt2 ubiquitin ligase complex [5, 13, 14]. This has been reported to be dependent on modulation of Cdt2 levels which are cell cycle regulated, peaking in S phase, and which also increase after DNA damage in a checkpoint-dependent manner [7, 13]. We show here that Cdt2 levels fluctuations are not sufficient to regulate Spd1 proteolysis and that the key step in this event is the interaction of Spd1 with the polymerase processivity factor PCNA, complexed onto DNA. This mechanism thus provides a direct link between DNA synthesis and ribonucleotide reductase regulation.
Keywords: Spd1, Cdt2, PCNA, Cul4, Ddb1, CRL4, degron, PIP box, DNA replication, DNA repair, ribonucleotide reductase, Cdc22, Suc22, fission yeast
Results and discussion
High Cdt2 levels are necessary but not sufficient to induce Spd1 degradation
Spd1 degradation is mediated by the activity of the CRL4Cdt2 ubiquitin ligase complex, which consists of a scaffold protein (Cul4), an adaptor protein (Ddb1) and a substrate-recruiting factor (Cdt2) [5, 13, 14]. As with Sml1 in S. cerevisiae, Spd1 is degraded in S phase and after DNA damage to allow up-regulation of dNTP levels. This has been reported to be dependent on modulation of Cdt2 levels which are cell cycle regulated, peaking in S phase, and which also increase after DNA damage in a checkpoint-dependent manner [7, 13]. Liu et al. [13, 14] reported that Spd1 degradation in S phase is independent of DNA damage checkpoint activation but requires both Rad3 and Chk1 in G2 phase and is driven by increased Cdt2 abundance. Furthermore, Moss et al. [7] reported that Rad3-dependent elevation of Cdt2 levels following double-strand break induction is responsible for Spd1 proteolysis and elevated dNTP levels, facilitating DNA repair.
From previous work it is not clear whether checkpoint activation simply serves to elevate Cdt2 levels or plays an additional role required for Spd1 proteolysis. To address this possibility, we expressed Cdt2 at a high level in a manner that was not dependent on checkpoint activation. Yox1 is an inhibitor of the Mlu1 binding factor (MBF) [15], which is an activator of the transcription of several genes including cdt2. Therefore, deletion of yox1 results in deregulated cdt2 expression (Fig. 1A, ‘log yox1Δ’). In a yox1Δ strain, degradation of Spd1 occurs after DNA damage as in a wild-type strain, but this is no longer Rad3 dependent (Fig. 1 B). We carried out a similar experiment with cells arrested in mitosis using an nda3 block; under these conditions Cdt2 levels are also high (Fig. 1A; ‘mitotic-arrested wt’). Again, degradation of Spd1 following DNA damage is not dependent on Rad3 (Fig. 1 B lower panels). These experiments indicate that the only role of the DNA damage checkpoint in Spd1 proteolysis is to allow cdt2 expression, and that this requirement can be bypassed when cdt2 over-expression is achieved by other pathways.
Figure 1. Increased expression of cdt2 is necessary but not sufficient to induce Spd1 proteolysis.
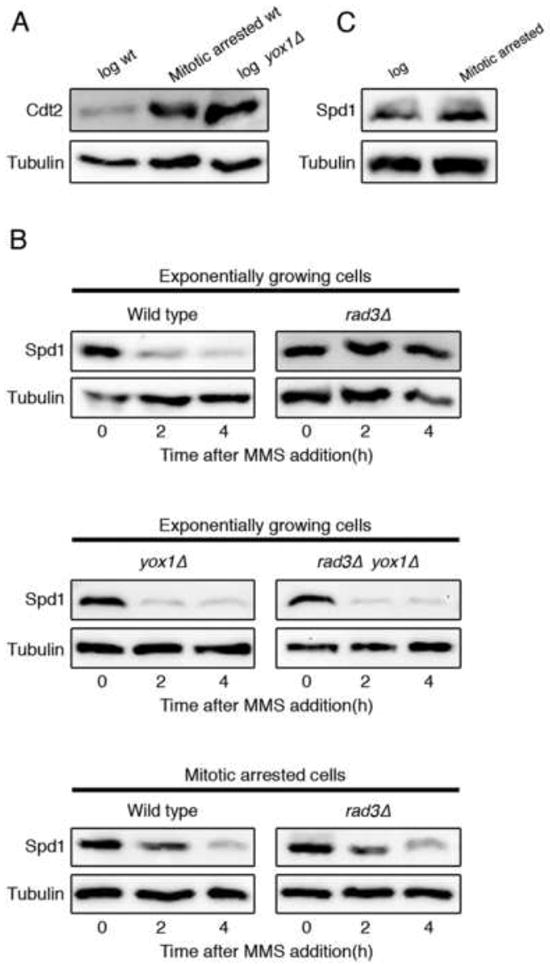
(A) Cdt2-TAP levels in exponentially growing (log) or mitotically-arrested wild type cells (2710) and in exponentially growing yox1Δ mutant cells (2698). (B) Western blot analysis showing Spd1-TAP levels after MMS addition in a wild type strain (1766) and in a rad3Δ (2644) mutant (upper panels); spd1-TAP levels after MMS treatment of yox1Δ (2711) and rad3Δ yox1Δ (2713) cells (middle panels); and Spd1-TAP levels after MMS treatment of mitotically-arrested cells that were either wild-type for checkpoint function (2678) or DNA damage checkpoint defective (rad3Δ) mutant (2677) (lower panels). (C) Spd1-TAP levels in exponentially growing (log) or mitotically-arrested wild type cells (2678). Tubulin is shown as a loading control.
Interestingly, we noted that although Cdt2 levels were high in mitotically-arrested cells, Spd1 levels were not lower than those observed in exponentially growing cells (Fig. 1 C) unless DNA damage was induced (Fig. 1B, lower panels). This observation is at odds with the model where Spd1 regulation is only driven by fluctuations in Cdt2 levels, and suggests that there must be another process induced by DNA damage and S phase that is rate-limiting for Spd1 proteolysis.
Spd1 proteolysis requires chromatin-bound PCNA
Since high Cdt2 levels alone does not seem to be sufficient to induce Spd1 degradation while DNA synthesis is required, it seems likely that an event involved in the replication itself is necessary for proteolysis. Ubiquitylation of several other substrates of CRL4Cdt2, such as Cdt1, p21, E2F, DNA pol η, and Set8 requires interaction of the substrate with the polymerase processivity factor PCNA [16-23]. For Cdt1, Set8, and p21 substrates, it has been shown that ubiquitylation occurs on chromatin and DNA loading of PCNA is required to stimulate substrate ubiquitylation [19, 23]. To determine if Spd1 turnover is regulated by this mechanism, we examined whether inactivation of replication factor C, that blocks loading of PCNA onto DNA, affected Spd1 degradation. Cells arrested in S phase with HU required active Rfc1 for Spd1 degradation (Fig. 2A, left panel). Similarly, Spd1 proteolysis seen after DNA damage is also blocked by Rfc1 inactivation (Fig. 2A, right panel), and thus these observations suggest that Spd1 ubiquitylation and subsequent proteolysis is dependent on DNA-associated PCNA. We also observed that after Rfc1 inactivation Cdt2 levels increased notably but Spd1 was accumulated, confirming that elevated Cdt2 levels are necessary but not sufficient for Spd1 degradation (Fig. S1).
Figure 2. Chromatin-bound PCNA is required for Spd1 proteolysis.
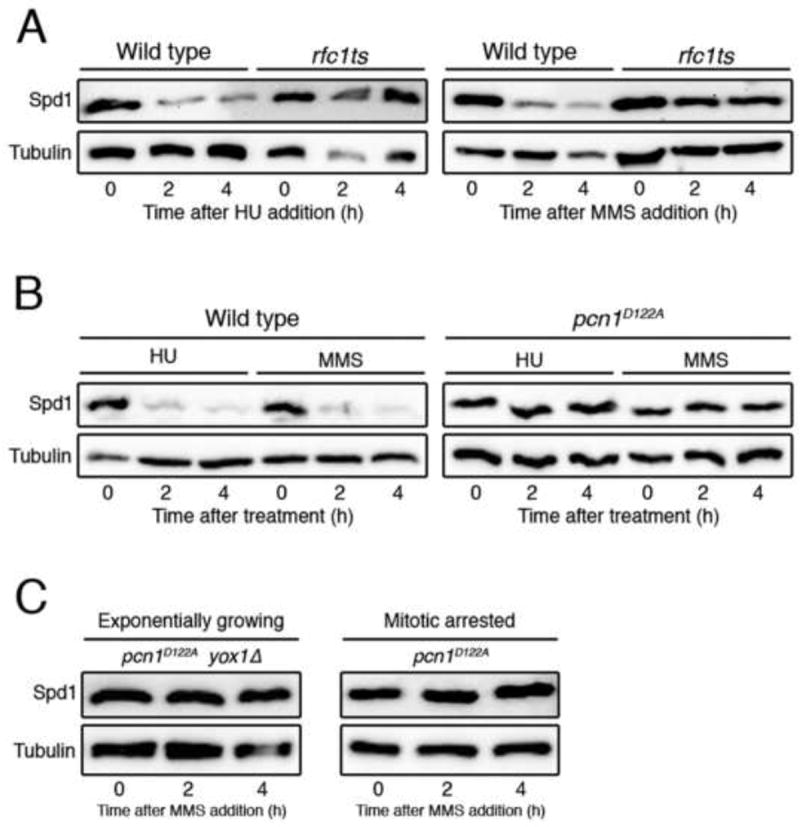
(A) Spd1-TAP levels in a wild type strain (1766) and in an rfc1-44 thermosensitive mutant (2072), treated with HU or MMS at the restrictive temperature (37°C). (B) Western blot analysis showing Spd1-TAP levels after HU or MMS addition in a wild type strain (1766) and a pcn1D122A mutant (2649). (C) Spd1-TAP levels after MMS treatment of pcn1D122A yox1Δ (2755) cells and mitotically-arrested (nda3) pcn1D122A cells (2664). Tubulin is shown as a loading control.
To test more directly whether PCNA is required for Spd1 degradation we examined a mutant of PCNA that is defective for CRL4Cdt2-mediated ubiquitylation. Havens et al. [24] have recently found that mutating the surface of PCNA that surrounds the PCNA-interacting protein (PIP) binding site prevents CRL4Cdt2-mediated proteolysis. This mutation (D122A) has no major effect on binding of the PIP degron to PCNA, but rather prevents recruitment of CRL4Cdt2 to PCNA. Strikingly, we find that PCNAD122A blocks Spd1 proteolysis after arresting cells in S phase with HU or exposure to DNA-damaging MMS (Fig. 2B). Furthermore, this mutation prevented Spd1 degradation after MMS treatment even in a yox1Δ background or in mitotically-arrested cells (Fig. 2C), where Cdt2 is highly abundant, implying that PCNA has a critical role in this process.
From previous work it is expected that stabilization of Spd1 would impair genome stability and S phase execution owing to RNR inhibition [5, 13, 14]. This is most clearly apparent in the effect of PCNAD122A on pre-meiotic S phase, which is very sensitive to down regulation of RNR activity [5]. PCNAD122A slows pre-meiotic S phase, but this is suppressed by deletion of the spd1 gene (Fig. 3A), arguing that Spd1 is an important target for S phase execution. We also observed that in the vegetative cell cycle, pcn1D122A cells are elongated but this is again suppressed by spd1 deletion (Fig. 3B). A plausible explanation is that failure to degrade Spd1 leads to a reduced dNTP supply for S phase and consequent impaired replication or DNA damage which causes a checkpoint delay to mitotic entry. Consistent with this interpretation, we were unable to construct a pcn1D122A strain where the repair and checkpoint pathways are inactivated by deletion of the rad3 gene, unless the spd1 gene was deleted as well (Fig. 3B). To confirm the synthetic lethality of pcn1D122A and rad3Δ, we used a temperature-sensitive rad3 allele to construct the double mutant. This pcn1D122A rad3ts mutant is inviable at the restrictive temperature, and this is partially suppressed by deletion of spd1 (Fig. 3C). Finally, we also observe elevated spontaneous minichromosome loss rate in the pcn1D122A strain, which is largely suppressed by deletion of the spd1 gene, again implying failure to degrade Spd1 promotes genome instability (Fig. 3D). Taken together, these observations indicate that in unperturbed cells, defects in Spd1 proteolysis caused by the pcn1D122A mutation, and consequent effects on dNTP supply, lead to defects in DNA replication or DNA damage that are only tolerated in checkpoint-proficient cells. This highlights the importance of the PCNA function in coordinating RNR activity for DNA replication and genome stability.
Figure 3. PCNAD122A-induced defects are suppressed by spd1 deletion.
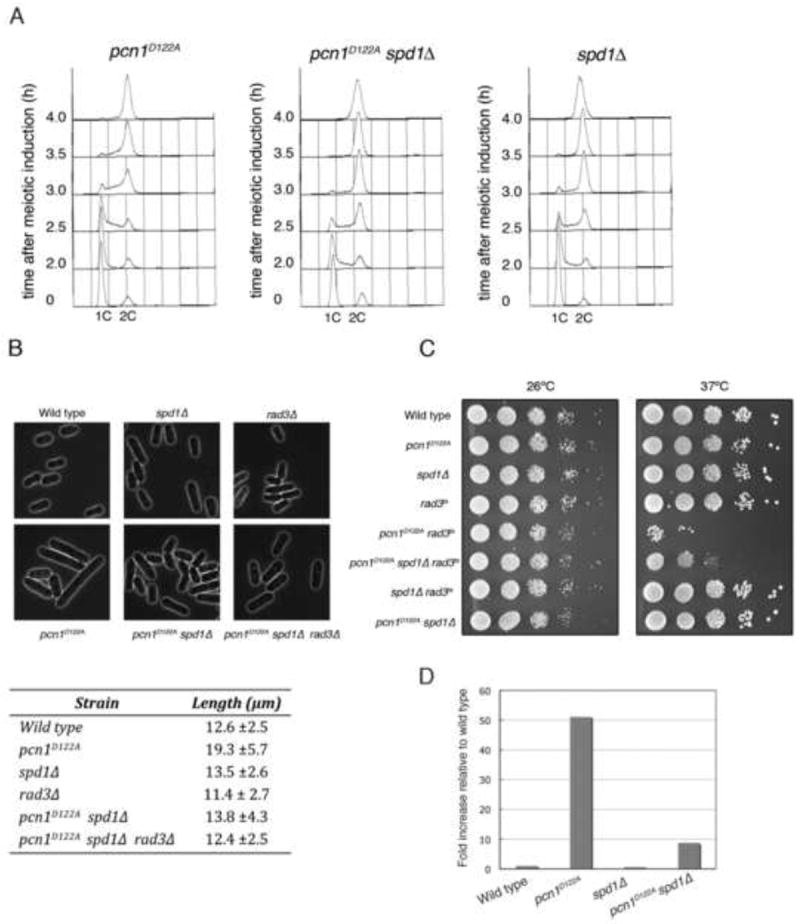
(A) pat1 strains also containing pcn1D122A (2912), pcn1D122A spd1Δ (2915), or spd1Δ (2930) mutations were arrested in G1 by nitrogen starvation, and then released from the block at 34°C to inactivate Pat1 and induce meiosis. The progress of pre-meiotic S phase was followed by flow cytometry. (B) Upper panels show images of exponentially growing cells from wild-type (137), spd1Δ (2671), rad3Δ (1811), pcn1D122A (2738), pcn1D122A spd1Δ (2747) and pcn1D122A spd1Δ rad3Δ (2842) cultures; average cell lengths (± standard deviation) are shown in the lower panel (150 cells were measured for each strain). (C) Viability of wild-type (137), pcn1D122A (2738), spd1Δ (2671), rad3ts (2839), pcn1D122A rad3ts (2887), pcn1D122A spd1Δ rad3ts (2888), spd1Δ rad3ts (2889) and pcn1D122A spd1Δ (2747) strains on YE3S at 26°C and 37°C analyzed by spot test. (D) Rate of minichromosome loss in wild-type (2836), pcn1D122A (2898), spd1Δ (2885) and pcn1D122A spd1Δ (2900) strains.
It has been previously reported that the DNA damage sensitivity of cdt2Δ mutants is not reversed by spd1 deletion [13]. In concordance with this observation, pcn1D122A cells are sensitive to DNA damaging agents, but this sensitivity is not suppressed by deletion of spd1 (Fig. S2A). We also find that cdt2Δ phenotypes are not enhanced in a double pcn1D122A cdt2Δ mutant (Fig. S2B and C), which argues that both mutations act by just blocking the same pathway, i.e. proteolysis of CRL4Cdt2 targets, and that the pcn1D122A mutation does not cause other defects in PCNA function. The simplest explanation for these findings is that while Spd1 is a key target of CRL4Cdt2 proteolysis in unperturbed cells, stabilization of other targets, via cdt2Δ or pcn1D122A, impairs the ability of cells to survive DNA damaging agents. This interpretation is also broadly consistent with the findings of Holmberg et al. [5], who concluded that insufficient RNR activity contributes to 50% of observed mutations in strains defective in CRL4 function.
Spd1 contains a PIP degron that is important for its proteolysis
Most targets of CRL4Cdt2-mediated ubiquitylation contain a PIP degron consisting of a ‘classical’ PIP consensus sequence with TD before the aromatic residues and a positively-charged amino acid downstream [19]. Spd1 does not contain a clear match to this consensus and no Spd1-PCNA interaction has been detected so far. In addition, extensive mutagenesis of Spd1 did not identify a degron sequence [11]. Nevertheless, a weak match to this consensus is in fact found in Spd1 at position 30 that is conserved in the Schizosaccharomyces genus (Fig. 4A). A C-terminally truncated Spd1 mutant (Spd11-43) retaining this PIP degron, is degraded similar to wild-type Spd1 following HU treatment (Fig.4B). In contrast, when the N-terminus of Spd1 including this PIP box, is deleted, proteolysis after HU is significantly reduced (Fig. 4B, Spd144-124). Mutating conserved amino acids in the PIP box (Fig. 4A, Spd1MPIP) also reduced Spd1 proteolysis after HU treatment (Fig. 4B). All of these Spd1 derivatives included a nuclear localization sequence (NLS) as, unlike wild type Spd1, they showed a pancellular distribution (data not shown), and we wanted to ensure that effects on proteolysis were not due to a secondary effect of inefficient nuclear accumulation, which would preclude interaction with PCNA.
Figure 4. Spd1 and PCNA interact in vivo and a PIP box in Spd1 is important for this interaction.
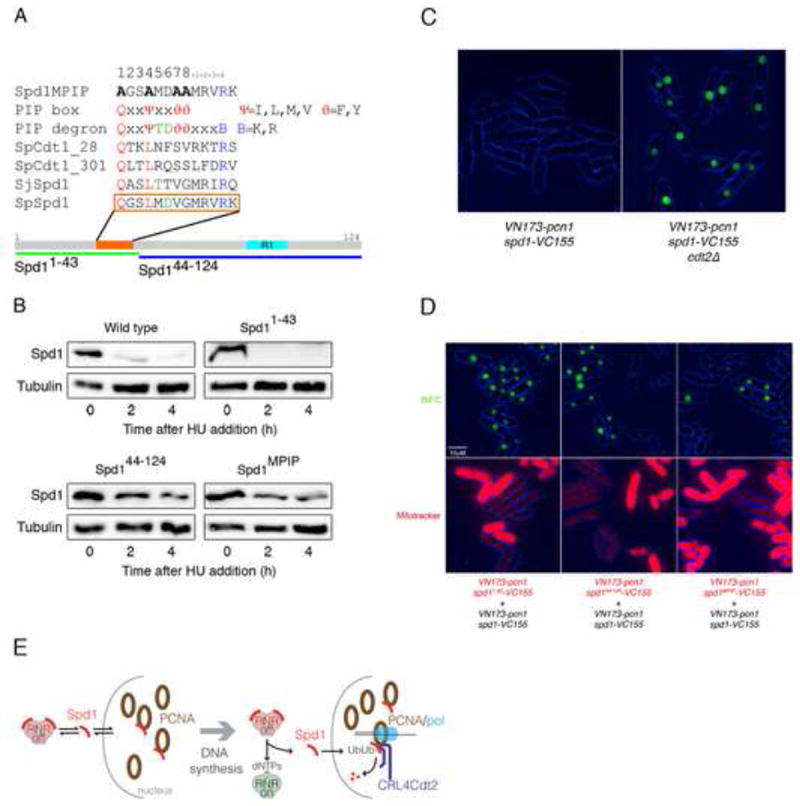
(A) Identification of a sequence (orange bar and box) in Spd1 (grey bar) that partially matches the PIP degron consensus sequence. Other sequences shown are (bottom to top): corresponding region of S. japonicus Spd1 (SjSpd1); PIP degrons in S. pombe Cdt1 (SpCdt1_301, SpCdt1_28 [18]): PIP degron and PIP consensus sequences [30]; and the mutated sequence of Spd1MPIP. Lower green and blue bars show the Spd11-43 and Spd144-124 deletion mutants. The cyan bar labelled “R1’ represents the location of sequence similarity with a region of Sm11 known to interact with the R1 subunit of RNR [11]. (B) Western blot analysis of Spd1-YFP-NLS levels after HU addition in a wild-type strain (2672), the Spd11-43 mutant (2612), the Spd144-124 mutant (2744) and the Spd1MPIP mutant (2673). Tubulin is shown as a loading control. (C) BiFC of exponentially growing live cells expressing VN173-pcn1 and spd1-VC155 in a wild type (2536) and in a cdt2Δ background (2546). (D) Comparison of the intensity of BiFC in exponentially growing live cells; a wild type (2752) and an spd11-43 (2750) strain (left panels), a wild type (2752) and an spd144-124 (2751) strain (middle panels), and a wild type (2752) and an spd1MPIP (2748) (right panels) strain were compared. Phase (blue) and BiFC signal (green) channels are merged (upper panels). In all the combinations, cells carrying the mutation in Spd1 were stained with MitoTracker® Red to allow direct comparison between the two strains (lower panels). All the strains were in a cdt2Δ background to stabilize Spd1 and comparison of Spd1 levels in these strains is shown in Fig. S3B. (E) Model for RNR activity control by PCNA: left panel represents an unperturbed cell in G1, G2 or M phase that is not subject to DNA damage. Spd1 shuttles between nucleus and cytoplasm, interacting and inhibiting RNR in the cytoplasm and possibly interacting with free PCNA in the nucleus. CRL4Cdt2 does not ubiquitylate nuclear Spd1 as PCNA is not DNA bound. Right panel represents the situation when S phase starts or DNA synthesis associated with DNA damage repair is occurring. Spd1 shuttles from cytoplasm to nucleus but in the nucleus, following interaction with chromatin-bound PCNA, it is ubiquitylated and proteolyzed. The net reduction in Spd1 levels leads to RNR activation.
Spd1 interaction with PCNA is reduced by mutation of the PIP box
We used bimolecular fluorescence complementation (BiFC) [25, 26] to determine if interaction between PCNA and Spd1 can be detected in live cells. PCNA and Spd1 were tagged with the N and C-terminal domains of Venus-YFP respectively and expressed from their native promoters. No fluorescence was observed when the tagged proteins were in a cdt2+ background (Fig. 4C). However, when co-expressed in a cdt2Δ background to stabilize Spd1, nuclear YFP fluorescence was seen in all cells, irrespective of cell-cycle stage (Fig. 4C). This indicates that Spd1-PCNA interaction occurs in vivo and can be detected provided that the turnover of Spd1 is blocked by lack of Cdt2. An interaction between Spd1 and PCNA was also detectable by immunoprecipitation, using purified PCNA and in vitro expressed Spd1 (Fig. S3A).
We tested the Spd1 mutants to see how removal of the PIP box affected interaction with PCNA. As with the proteolysis experiments, these all included a C-terminal NLS and western blotting was used to show that the mutations do not have a significant effect on Spd1 levels in a cdt2Δ mutant background (Fig. S3B). Spd11-43, retaining the PIP box, showed a BiFC signal similar to that seen with wild-type Spd1 (Fig. 4D; note that in these experiments we marked one of the strains with a mitochondrial stain to allow direct comparison of two strains in the same image). However, Spd144-124 showed a reduced BiFC signal as did the Spd1MPIP mutant (Fig. 4D). We note that deleting or mutating the PIP box only reduces and does not eliminate the BiFC signal and Spd1 proteolysis, possibly indicating that there are other PCNA-interacting regions in Spd1.
Taken together, the results described here indicate that the role of the DNA damage checkpoint activation in Spd1 degradation is simply to provide an adequate level of Cdt2 at times of the cell cycle when levels are low, and high Cdt2 levels are not sufficient to promote Spd1 degradation. Significantly, we demonstrate that the critical step in Spd1 proteolysis, and therefore in RNR up-regulation, is the interaction of Spd1 with DNA-bound PCNA. Since PCNA serves as an essential polymerase processivity factor in S phase, as well promoting repair synthesis by pol δ and other repair polymerases, this provides a direct mechanism to synchronize DNA synthesis with stimulation of RNR to up-regulate dNTP production. This mechanism is distinct from proteolysis of Sml1 in S. cerevisiae. In this organism, which lacks the CRL4Cdt2 pathway, checkpoint activation in S phase and after DNA damage leads to Sml1 phosphorylation and this modified version of the protein is ubiquitylated by the Rad6-Ubr2-Mub1 E2/E3 ubiquitin ligase complex [27, 28].
The type 1a class of RNRs, found in eukaryotes and eubacteria, consists of a heterotetramer composed of two large catalytic R1 subunits, and two small R2 subunits which generate the tyrosyl free radical required for catalysis (for reviews see [1, 2]). Spd1 inhibits RNR activity most probably by binding to the R1/Cdc22 subunit [10], although it has also been implicated in promoting the nuclear import of the R2/Suc22 subunit, facilitating R1-R2 interactions and binding to the R2 subunit [11]. One implication of our findings is that Spd1 is degraded via interaction with a nuclear protein, while its mode of inhibition appears to require interaction with R1/Cdc22, which is predominantly cytoplasmic. RNR is thought to be active in the cytoplasm, with the R2/Suc22 subunit translocating from the nucleus to the cytoplasm to increase RNR activity. In a model to reconcile these observations, Spd1 may shuttle between nucleus and cytoplasm and, in the absence of DNA synthesis, Spd1 is stable during nuclear transit (Fig. 4E, left panel). During S phase or DNA synthesis associated with repair, nuclear Spd1 interacts with chromatin-associated PCNA, leading to its ubiquitylation and proteolysis, and the net reduction of Spd1 levels causes RNR activation (Fig. 4E, right panel). Spd1 proteolysis may also contribute to RNR activation by reducing nuclear import of the R2/Suc22 subunit, thus increasing cytoplasmic R2/Suc22 levels, although the mechanism of this is unclear [11]. Although we note that the active form of the enzyme is thought to be cytoplasmic, the PCNA-Spd1 interaction potentially can target RNR to sites of DNA synthesis. In mammalian cells, Tip60 has been reported to localize RNR to sites of DNA damage, which may be important for providing adequate dNTP at repair sites at times of the cell cycle when dNTP pools are low [29].
So far, no Spd1 orthologue has been identified in Metazoa, but the small size of this protein and its low sequence conservation through evolution makes detection of any related proteins difficult. In addition, the intrinsically disordered nature of Spd1 [11] could mean that proteins without any sequence similarity could perform similar roles in higher organisms. However, given the conservation of the CRL4Cdt2 pathway, identification of mammalian protein inhibitors of RNR might be facilitated by inactivation of this mechanism.
Proteolysis of CRL4Cdt2 targets, triggered by PCNA chromatin binding is emerging as an important mechanism in DNA replication control (reviewed in [30]). Degradation of Cdt1 blocks Mcm chromatin binding directly, while p21 proteolysis leads to nuclear export of the Cdc6 licensing factor [21], and down-regulation of Set8 in S phase also seems to be important for blocking re-replication [31, 32]. Our results here, showing that an inhibitor of the elongation step of replication is degraded simultaneously with destruction of licensing activators, emphasizes how PCNA activation of CRL4Cdt2 is a master switch in the transition from G1 to S phase.
Methods
Methods, strains and oligos used are given in the Supplementary Information.
Supplementary Material
Highlights.
High Cdt2 levels are necessary but not sufficient to induce Spd1 degradation.
Spd1 interacts with PCNA in vivo.
Chromatin binding of PCNA synchronizes Spd1 proteolysis with DNA synthesis.
Acknowledgments
We are grateful to Jürg Bähler, Tony Carr, and Tim Humphrey for strains and plasmids. We thank Beata Grallert, Tim Humphrey, Olaf Nielsen for comments and Sue Cotterill for advice on protein expression. This work was supported by CRUK grants C814/A7985 and C814/A9035, the EP Abraham Cephalosporin Trust and a fellowship to IS from the Junta de Extremadura, Spain (Consejería de Economía, Comercio e Innovación).
References
- 1.Jordan A, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 1998;67:71–98. doi: 10.1146/annurev.biochem.67.1.71. [DOI] [PubMed] [Google Scholar]
- 2.Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 3.Bebenek K, Roberts JD, Kunkel TA. The effects of dNTP pool imbalances on frameshift fidelity during DNA replication. J Biol Chem. 1992;267:3589–3596. [PubMed] [Google Scholar]
- 4.Chabes A, Georgieva B, Domkin V, Zhao X, Rothstein R, Thelander L. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell. 2003;112:391–401. doi: 10.1016/s0092-8674(03)00075-8. [DOI] [PubMed] [Google Scholar]
- 5.Holmberg C, Fleck O, Hansen HA, Liu C, Slaaby R, Carr AM, Nielsen O. Ddb1 controls genome stability and meiosis in fission yeast. Genes Dev. 2005;19:853–862. doi: 10.1101/gad.329905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathews CK. DNA precursor metabolism and genomic stability. FASEB J. 2006;20:1300–1314. doi: 10.1096/fj.06-5730rev. [DOI] [PubMed] [Google Scholar]
- 7.Moss J, Tinline-Purvis H, Walker CA, Folkes LK, Stratford MR, Hayles J, Hoe KL, Kim DU, Park HO, Kearsey SE, et al. Break-induced ATR and Ddb1-Cul4(Cdt)(2) ubiquitin ligase-dependent nucleotide synthesis promotes homologous recombination repair in fission yeast. Genes Dev. 2010;24:2705–2716. doi: 10.1101/gad.1970810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elledge SJ, Zhou Z, Allen JB, Navas TA. DNA damage and cell cycle regulation of ribonucleotide reductase. Bioessays. 1993;15:333–339. doi: 10.1002/bies.950150507. [DOI] [PubMed] [Google Scholar]
- 9.Reichard P. Ribonucleotide reductases: substrate specificity by allostery. Biochem Biophys Res Commun. 2010;396:19–23. doi: 10.1016/j.bbrc.2010.02.108. [DOI] [PubMed] [Google Scholar]
- 10.Hakansson P, Dahl L, Chilkova O, Domkin V, Thelander L. The Schizosaccharomyces pombe replication inhibitor Spd1 regulates ribonucleotide reductase activity and dNTPs by binding to the large Cdc22 subunit. J Biol Chem. 2006;281:1778–1783. doi: 10.1074/jbc.M511716200. [DOI] [PubMed] [Google Scholar]
- 11.Nestoras K, Mohammed AH, Schreurs AS, Fleck O, Watson AT, Poitelea M, O’Shea C, Chahwan C, Holmberg C, Kragelund BB, et al. Regulation of ribonucleotide reductase by Spd1 involves multiple mechanisms. Genes Dev. 2010;24:1145–1159. doi: 10.1101/gad.561910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woollard A, Basi G, Nurse P. A novel S phase inhibitor in fission yeast. Embo J. 1996;15:4603–4612. [PMC free article] [PubMed] [Google Scholar]
- 13.Liu C, Poitelea M, Watson A, Yoshida SH, Shimoda C, Holmberg C, Nielsen O, Carr AM. Transactivation of Schizosaccharomyces pombe cdt2+ stimulates a Pcu4-Ddb1-CSN ubiquitin ligase. Embo J. 2005;24:3940–3951. doi: 10.1038/sj.emboj.7600854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu C, Powell KA, Mundt K, Wu L, Carr AM, Caspari T. Cop9/signalosome subunits and Pcu4 regulate ribonucleotide reductase by both checkpoint-dependent and -independent mechanisms. Genes Dev. 2003;17:1130–1140. doi: 10.1101/gad.1090803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez-Escoda B, Ivanova T, Calvo IA, Alves-Rodrigues I, Hidalgo E, Ayte J. Yox1 links MBF-dependent transcription to completion of DNA synthesis. EMBO Rep. 2011;12:84–89. doi: 10.1038/embor.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 2008;22:2496–2506. doi: 10.1101/gad.1676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chuang LC, Yew PR. Proliferating cell nuclear antigen recruits cyclin-dependent kinase inhibitor Xic1 to DNA and couples its proteolysis to DNA polymerase switching. J Biol Chem. 2005;280:35299–35309. doi: 10.1074/jbc.M506429200. [DOI] [PubMed] [Google Scholar]
- 18.Guarino E, Shepherd ME, Salguero I, Hua H, Deegan RS, Kearsey SE. Cdt1 proteolysis is promoted by dual PIP degrons and is modulated by PCNA ubiquitylation. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Havens CG, Walter JC. Docking of a specialized PIP Box onto chromatin-bound PCNA creates a degron for the ubiquitin ligase CRL4Cdt2. Mol Cell. 2009;35:93–104. doi: 10.1016/j.molcel.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim SH, Michael WM. Regulated proteolysis of DNA polymerase eta during the DNA-damage response in C. elegans. Mol Cell. 2008;32:757–766. doi: 10.1016/j.molcel.2008.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim Y, Starostina NG, Kipreos ET. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 2008;22:2507–2519. doi: 10.1101/gad.1703708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shibutani ST, de la Cruz AF, Tran V, Turbyfill WJ, 3rd, Reis T, Edgar BA, Duronio RJ. Intrinsic negative cell cycle regulation provided by PIP box- and Cul4Cdt2-mediated destruction of E2f1 during S phase. Dev Cell. 2008;15:890–900. doi: 10.1016/j.devcel.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Centore RC, Havens CG, Manning AL, Li JM, Flynn RL, Tse A, Jin J, Dyson NJ, Walter JC, Zou L. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol Cell. 2010;40:22–33. doi: 10.1016/j.molcel.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Havens C, Shobnam N, Guarino E, Centore R, Zou L, Kearsey S, Walter J. Direct Role for Proliferating Cell Nuclear Antigen (PCNA) in Substrate Recognition by the E3 Ubiquitin Ligase CRL4Cdt2. J Biol Chem. 2012 doi: 10.1074/jbc.M111.337683. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akman G, MacNeill SA. MCM-GINS and MCM-MCM interactions in vivo visualised by bimolecular fluorescence complementation in fission yeast. BMC Cell Biol. 2009;10:12. doi: 10.1186/1471-2121-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu CD, Kerppola TK. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat Biotechnol. 2003;21:539–545. doi: 10.1038/nbt816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andreson BL, Gupta A, Georgieva BP, Rothstein R. The ribonucleotide reductase inhibitor, Sml1, is sequentially phosphorylated, ubiquitylated and degraded in response to DNA damage. Nucleic Acids Res. 2010;38:6490–6501. doi: 10.1093/nar/gkq552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao X, Chabes A, Domkin V, Thelander L, Rothstein R. The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. Embo J. 2001;20:3544–3553. doi: 10.1093/emboj/20.13.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niida H, Katsuno Y, Sengoku M, Shimada M, Yukawa M, Ikura M, Ikura T, Kohno K, Shima H, Suzuki H, et al. Essential role of Tip60-dependent recruitment of ribonucleotide reductase at DNA damage sites in DNA repair during G1 phase. Genes Dev. 2010;24:333–338. doi: 10.1101/gad.1863810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Havens CG, Walter JC. Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 2011;25:1568–1582. doi: 10.1101/gad.2068611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol Cell. 2010;40:9–21. doi: 10.1016/j.molcel.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, Julien E. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol. 2010;12:1086–1093. doi: 10.1038/ncb2113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.