

Abstract
Clinical application of recombinant tissue plasminogen activator (rtPA) for stroke is limited by hemorrhagic transformation, which narrows rtPA’s therapeutic window. In addition, mounting evidence indicates that rtPA is potentially neurotoxic if it traverses a compromised blood brain barrier. Here, we demonstrated that pyruvate protects cultured HT22 neuronal and primary microvascular endothelial cells co-cultured with primary astrocytes from oxygen glucose deprivation (OGD)/reoxygenation stress and rtPA cytotoxicity. After 3 or 6 h OGD, cells were reoxygenated with 11 mmol/L glucose±pyruvate (8 mmol/L) and/or rtPA (10 μg/ml). Measured variables included cellular viability (calcein AM and annexin-V/propidium iodide), reactive oxygen species (ROS; mitosox red and 2′,7′-dichlorofluorescein diacetate), NADPH, NADP+ and ATP contents (spectrophotometry), matrix metalloproteinase-2 (MMP2) activities (gelatin zymography), and cellular contents of MMP2, tissue inhibitor of metalloproteinase-2 (TIMP2), and phosphor-activation of anti-apoptotic p70s6 kinase, Akt and Erk (immunoblot). Pyruvate prevented the loss of HT22 cells after 3 h OGD±rtPA. After 6 h OGD, rtPA sharply lowered cell viability; pyruvate dampened this effect. Three hours OGD and 4 h reoxygenation with rtPA increased ROS formation by about 50%. Pyruvate prevented this ROS formation and doubled cellular NADPH/NADP+ ratio and ATP content. In endothelial cell monolayers, 3 h OGD and 24 h reoxygenation increased FITC-dextran leakage, indicating disruption of intercellular junctions. Although rtPA exacerbated this effect, pyruvate prevented it while sharply lowering MMP2/TIMP2 ratio and increasing phosphorylation of p70s6 kinase, Akt and Erk. Pyruvate protects neuronal cells and microvascular endothelium from hypoxia-reoxygenation and cytotoxic action of rtPA while reducing ROS and activating anti-apoptotic signaling. These results support the proposed use of pyruvate as an adjuvant to dampen the side effects of rtPA treatment, thereby extending rtPA’s therapeutic window.
Keywords: Antioxidant, Blood brain barrier, Cell death, Matrix metalloproteinases, Monocarboxylate transporter, Reactive oxygen species
1. Introduction
Ischemic stroke, the third leading cause of death and leading cause of long-term disability in adult Americans (Gillum, 2002), has defied the development of effective treatments. More than 1000 compounds have been tested pre-clinically, but only a select few are approved for clinical application (O’Collins et al., 2006). The only clinical treatment for ischemic stroke approved by the United States Food and Drug Administration, recombinant tissue plasminogen activator (rtPA) saves patients by dissolving thrombi obstructing cerebral blood flow. Nevertheless, only 4–5% of stroke patients benefit from rtPA treatment because its therapeutic window is limited to the first 4.5 h after onset of ischemic stroke (Green, 2008; Hacke et al., 2008). By exacerbating blood brain barrier (BBB) leakage, cerebral edema formation and hemorrhagic transformation (Garcia-Yebenes et al., 2011; Jia et al., 2010), delayed rtPA treatment can worsen outcomes of ischemic stroke. During cerebral ischemia and reperfusion, massive formation of reactive oxygen species (ROS) exacerbates brain damage by activating inflammatory cascades and matrix metalloproteinases (MMPs)(Florczak-Rzepka et al., 2012). Activated MMPs degrade BBB components, allowing inflammatory cells to infiltrate the brain parenchyma (Florczak-Rzepka et al., 2012) and intensify the injury cascade.
Pyruvate, a natural glycolytic metabolite, is a powerful antioxidant and energy substrate (Mallet, 2000; Mallet and Sun, 2003). Pyruvate scavenges ROS and increases ATP production in various organs, including brain and heart (Alvarez et al., 2003; Fukushima et al., 2009; Gonzalez-Falcon et al., 2003; Jagtap et al., 2003; Mallet and Sun, 2003; Pan et al., 2012; Yi et al., 2007), so that it could contribute to the recovery of cellular homeostasis by generating ATP and detoxifying ROS under harsh ischemia/reperfusion conditions. Recently we demonstrated in a rat stroke model that intravenous administration of pyruvate during middle cerebral artery occlusion and reperfusion sharply decreased infarct volume while inducing erythropoietin production in the brain and activating erythropoietin’s anti-apoptotic signaling pathway (Ryou et al., 2012). These results led us to propose that pyruvate might protect the neurovascular unit from the detrimental action of rtPA after oxygen-glucose deprivation (OGD). Accordingly, this study tested the hypothesis that post-hypoxic pyruvate treatment protects neuronal cells and microvascular endothelium from ODG/reoxygenation stress and rtPA cytotoxicity by scavenging excessive ROS and stimulating energy production.
2. Results
2.1. Delayed rtPA treatment exacerbates cell death after OGD and reoxygenation
Viability of neuronally derived HT22 cells was assessed by calcein AM labeling after 3 or 6 h OGD followed by 24 h reoxygenation (Fig. 1). While rtPA treatment after 3 h OGD, i.e. within its treatment window, did not worsen cell death vs. OGD control (Fig. 1A), rtPA treatment after 6 h (Fig. 1B) or 10 h (Fig. 1C) OGD was markedly toxic to HT22 neuronal cells. Pyruvate treatment alone after 3, 6, or 10 h OGD appreciably increased cell viability, and pyruvate cotreatment increased viability of rtPA-treated cells after all three OGD-reoxygenation protocols. Prolonged OGD (10 h) decreased the viability about 85% vs. normoxic control, and rtPA treatment after 10 h OGD was lethal to HT22 cells (Fig. 1C). Pyruvate-induced cellular protection after 3 h OGD and 24 h reoxygenation with or without rtPA were confirmed by flow cytometry with annexin-V and propidium iodide staining to detect cell death (Fig. 1D).
Fig. 1.
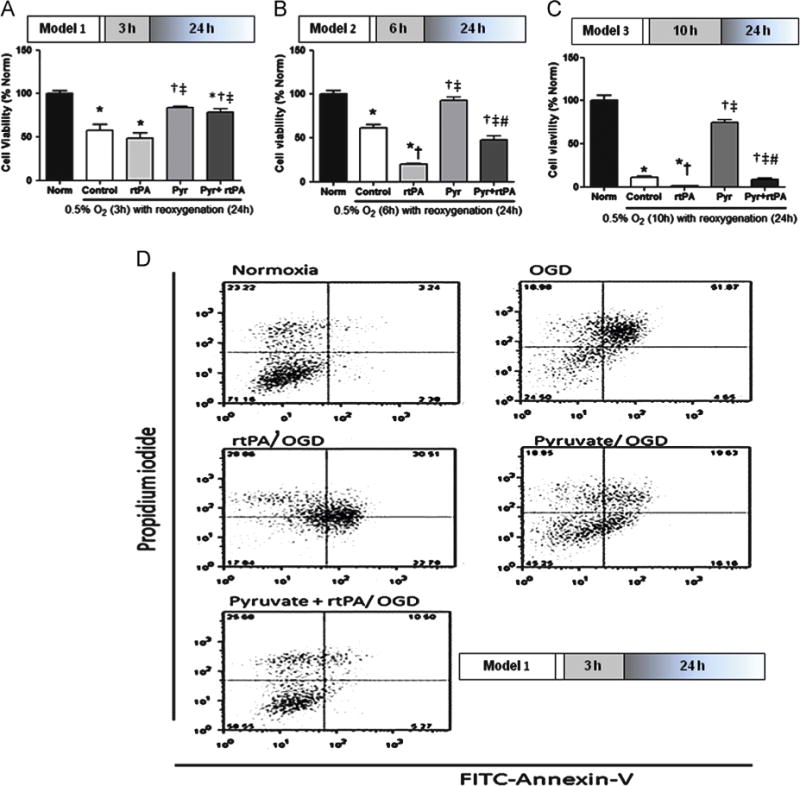
Effects of rtPA treatment after OGD on cell viability. Panels A, B, C: Cell viability assessed by calcein AM assay. Panel A: rtPA (10 μg/ml) treatment beginning at 3 h OGD did not aggravate OGD-induced death of HT22 neuronal cells. Panel B: Delayed rtPA treatment after 6 h OGD exacerbated cell death vs. control. Pyruvate (8 mmol/L) protected the cells from OGD and rtPA treatment in both conditions. Panel C: 10 h OGD decreased viability about 85% vs. normoxic control, and killed about 100% of cells with rtPA treatment. Pyruvate salvaged cells from rtPA toxicity, but viability was not better than OGD control. *p < 0.05 vs. Normoxia, †p <0.05 vs. Control, ‡p<0.05 vs. rtPA, #p<0.05 vs. Pyruvate. Panel D: Flow cytometry analysis after annexin-V/propidium iodide stain confirmed rtPA toxicity and pyruvate’s cytoprotective function.
2.2. Pyruvate enhances monocarboxylate transporter (MCT) expression
Three hours of OGD and 24 h reoxygenation increased content of MCT, the major plasma membrane mechanism for pyruvate uptake, vs. normoxic HT22 cells (Fig. 2A). rtPA blunted the post-OGD enhancement of MCT expression. Pyruvate alone or in combination with rtPA maintained normal HT22 cell morphology and augmented MCT2 content. Under normoxia, MCT2 protein content fell with rtPA treatment, but was restored with pyruvate (8 mmol/L)±rtPA treatment (Fig. 2B).
Fig. 2.
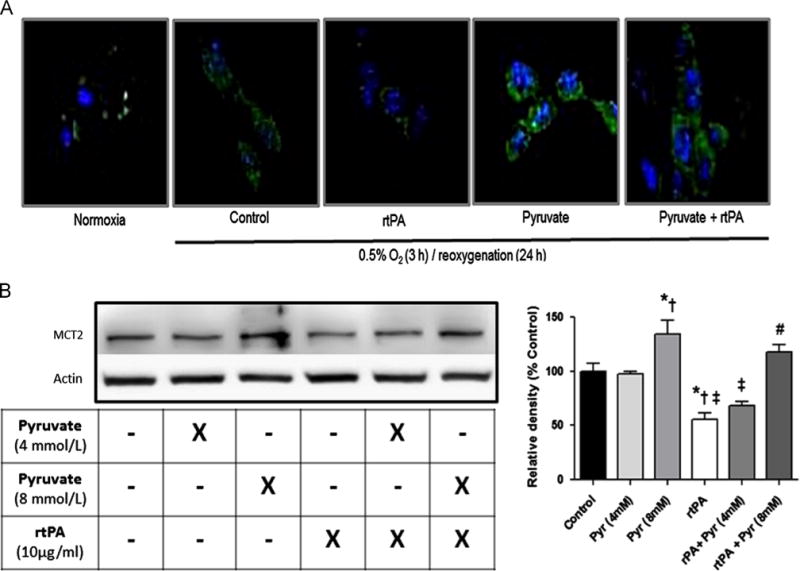
Monocarboxylate transporter-2 content in HT22 cells. Panel A: MCT2 increased after 3 h OGD, but rtPA dampened OGD-associated increased MCT2 expression. Pyruvate increased MCT-2 expression in both absence and presence of rtPA. Green: MCT2 immunolabeling; blue: DAPI labeling of nuclei. Panel B: In normoxic cells, pyruvate increased MCT2 protein content in a concentration dependent manner, and rtPA significantly reduced MCT-2 contents. Pyruvate dampened negative effect of rtPA on MCT2 protein content. Pyruvate weakens rtPA’s negative effects on MCT-2 expression. *p<0.05 vs. Control, †p<0.05 vs. pyruvate 4mM, ‡ p<0.05 vs. pyruvate 8mM, # p<0.05 vs. rtPA+pyruvate (4mM).
2.3. Generation of reactive oxygen species (ROS) in reoxygenated HT22 cells
Formation of ROS in HT22 cells was detected from mitosox red and 2′,7′-dichlorofluorescein diacetate (H2DCFDA) fluorescence (Fig. 3). Three hours of OGD and 4 h reoxygenation slightly increased mitosox red fluorescence and increased H2DCFDA fluorescence by approximately 30%. Although rtPA did not provoke ROS formation in normoxic cells (Fig. 3A), after OGD and reoxygenation it intensified mitosox reddetectable ROS formation and tended to increase H2DCFDA fluorescence. These fluorescence measurements confirmed pyruvate’s antioxidant capabilities: pyruvate decreased ROS formation to the respective normoxia values, whether given alone or in combination with rtPA.
Fig. 3.
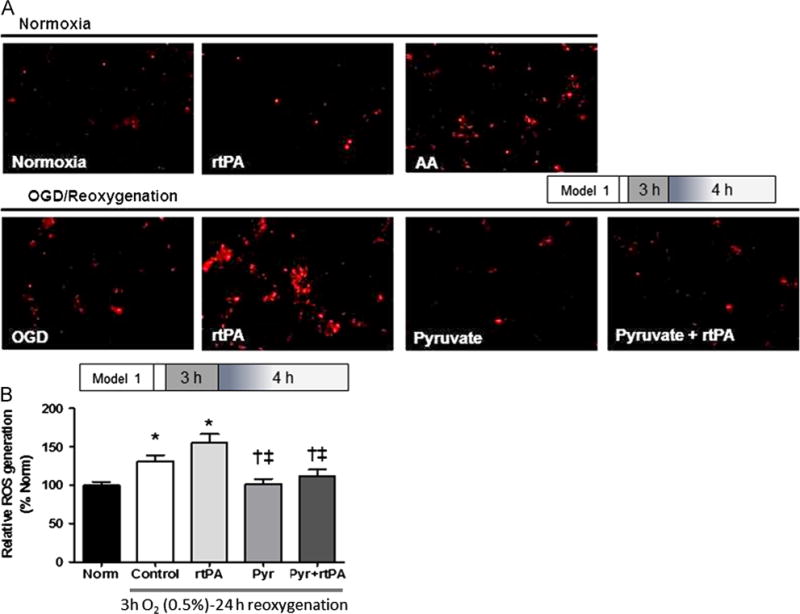
Pyruvate prevented ROS generated by OGD/reoxygenation and rtPA. (A) Mitosox Red stain of HT22 neuronal cells shows increased ROS with rtPA treatment after OGD/reoxygenation, but no effect of rtPA in the absence of OGD. Pyruvate suppresses post-OGD ROS formation±rtPA. Exposure of cells to ROS-generating antimycin A (AA) provided an OGD-independent positive control. (B) H2DCFDA stain confirms increased ROS in cells subjected to OGD±rtPA treatment, and prevention of these ROS by pyruvate (Pyr). Norm: normoxia. *p<0.05 vs. Normoxia, †p<0.05 vs. Control, ‡p<0.05 vs. rtPA.
2.4. ATP content and NADPH/NADP+ redox state
ATP concentration of normoxic control was 5 μmol/L/mg protein. Cellular ATP content fell by 60% after 3 h OGD and 24 h reoxygenation (Fig. 4A). rtPA unexpectedly prevented this ATP depletion. Pyruvate increased ATP content approximately six-fold in the absence and 2.5-fold in the presence of rtPA. NADPH/NADP+ ratio, a measure of the poise of this pivotal antioxidant system, was not appreciably altered by OGD-reoxygenation per se or by rtPA treatment during reoxygenation (Fig. 4B), but was sharply increased by pyruvate alone or in combination with rtPA, in accordance with pyruvate’s enhancement of cellular antioxidant redox states.
Fig. 4.
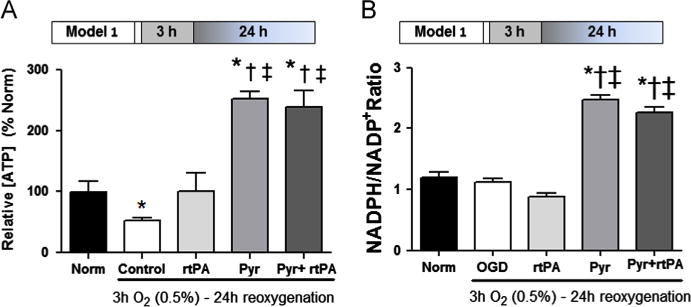
Pyruvate increases ATP production and NADPH/NADP+ ratio. Panel A: OGD lowered ATP content of HT22 cells by 60%. Pyruvate (Pyr) sharply increased ATP content in the absence and presence of rtPA. Panel B: Pyruvate sharply increased NADPH/NADP+ ratio in post-OGD cells in the absence and presence of rtPA. *p < 0.05 vs. Normoxia (Norm), †p < 0.05 vs. Control, ‡p < 0.05 vs. rtPA.
2.5. Matrix metalloproteinase-2 (MMP2) activity and endothelial integrity
Gel zymography (Fig. 5A) revealed an intense MMP2 activation by rtPA, but pyruvate suppressed MMP2 activity even in the presence of rtPA. Pyruvate powerfully increased the cellular content of the endogenous MMP inhibitor TIMP2, and thereby sharply lowered MMP2/TIMP2 content ratio (Fig. 5B) in parallel with the suppression of MMP2 activity.
Fig. 5.
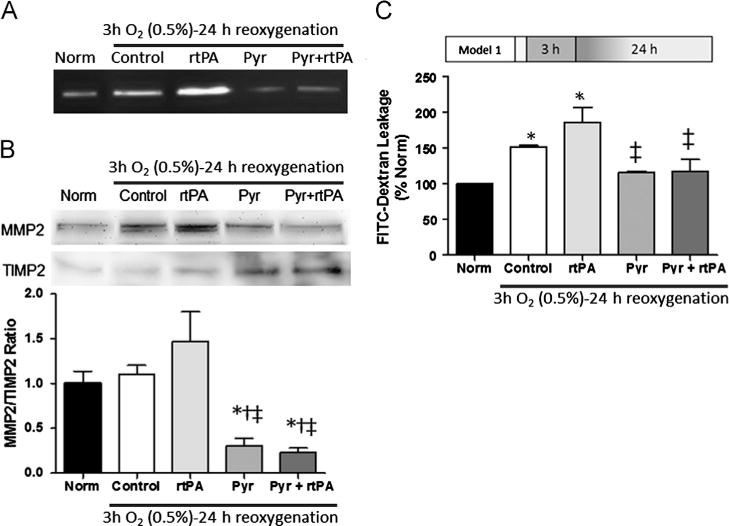
Pyruvate inhibits MMP 2 and preserves integrity of microvascular endothelium. Panel A: Gelatin-zymography revealed MMP2 activation by rtPA and inhibition by pyruvate in confluent vascular endothelial cell cultures. Panel B: Immunoblots showed pyruvate decreased MMP2 content and increased TIMP2 content. Panel C: Increased MMP2 activity coincides with increased leakage of FITC-dextran in endothelial cells treated with rtPA. Pyruvate cotreatment limited dextran leakage to the normoxic level. *p<0.05 vs. Normoxia (Norm), †p<0.05 vs. Control, ‡p<0.05 vs. rtPA.
The inter-endothelial cell tight junctions which effect the blood–brain barrier are targets of MMP2-catalyzed proteolysis. Tight junction integrity was assessed by measuring leakage of FITC-dextran across endothelial cell monolayers. Subjecting these monolayers to 3 h OGD and 24 h reoxygenation increased FITC-dextran leakage by 50% in the absence and 80% in the presence of rtPA (Fig. 5C). Pyruvate, either alone or coadministered with rtPA, prevented the increased leakage of FITC-dextran induced by OGD-reoxygenation. Thus, pyruvate maintained the barrier function of the confluent endothelial cells, even in the presence of rtPA.
2.6. Impact of OGD-reoxygenation, rtPA and pyruvate on anti-apoptotic signaling kinases
After 3 h OGD, rtPA activated mTOR and its target protein kinase, p70S6 kinase, but activation of mTOR signaling pathway was not detected in rtPA treatment group after 6 h OGD (Fig. 6A). Pyruvate activated mTOR and p70s6 kinase, and was able to activate mTOR pathway despite the negative influence of rtPA. Pyruvate activated the protective protein kinases Erk and Akt after 3 h OGD and 24 h reoxygenation, but rtPA alone did not activate Akt and Erk (Fig. 6B).
Fig. 6.
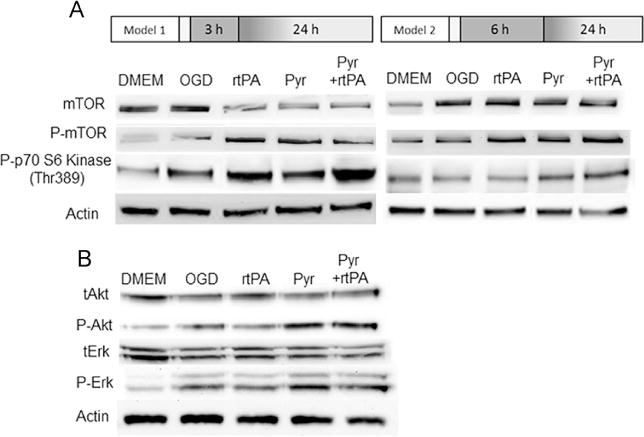
Pyruvate activation of signaling kinases in neuronal cells. Panel A: Phosphorylation of mTOR and p70S6 kinase was assessed by immunoblot at 4 h reoxygenation after 3 and 6 h of OGD. p70S6 kinase was activated by rtPA administered after 3 h OGD, but inactivated by delayed rtPA treatment. Pyruvate±rtPA activates p70S6 kinase in OGD both protocols. Panel B: Pyruvate but not rtPA phosphor-activates Akt and Erk.
3. Discussion
Despite extensive preclinical research and large-scale clinical trials, rtPA remains the only treatment approved by the Food and Drug Administration for ischemic stroke. Recombinant tPA cleaves plasminogen into plasmin, an enzyme that degrades intravascular thrombi thereby recommencing nutritive blood flow to the ischemic brain. Unfortunately, rtPA’s therapeutic window is limited to the first 4.5 h after onset of ischemic stroke (Hacke et al., 2008), which is generally before many stroke victims arrive at the hospital or complete diagnostic procedures. Consequently, only 4–5% of stroke victims can benefit from treatment. Moreover, rtPA treatment initiated after the therapeutic window may even worsen ischemic brain injury. rtPA-induced brain damage has been found to be associated with increased ROS (Takamiya et al., 2012), so coadministration of rtPA with agents that dampen ROS production might afford robust cerebroprotection. However, conventional antioxidants have been found generally ineffective at protecting ischemic brain or preserving neurocognitive function in stroke victims (Sutherland et al., 2012).
Unlike many antioxidants, pyruvate readily traverses the blood brain barrier; a high-capacity monocarboxylate transport mechanism (Halestrap and Wilson, 2012) delivers pyruvate to the brain parenchyma, providing energy-yielding fuel as well as ROS scavenging. In our rat middle cerebral artery occlusion-reperfusion stroke model, intravenous pyruvate treatment afforded robust, 85% reduction in infarct volume, suppressed neuronal apoptosis and activated anti-apoptotic signaling cascades (Ryou et al., 2012). The current study demonstrated that pyruvate protects neuronal-lineage cultured cells from OGD/reoxygenation injury and from rtPA toxicity, and that decreased ROS formation, increased monocarboxylate transporter content, ATP content and antioxidant NADPH/NADP+ redox state, suppression of matrix metalloproteinsase activity, preservation of microvascular endothelial integrity, and activation of anti-apoptotic signaling cascades accompanied the neuroprotection. Collectively, these effects of pyruvate could not only exert direct protection of neurons and preservation of tight junctions between MVEC in ischemic brain, but also potentially prolong rtPA’s therapeutic efficacy.
Increased hemorrhagic transformation is the most extensively documented consequence of delayed-rtPA treatment in ischemic stroke patients (Garcia-Yebenes et al., 2011). Hemorrhagic transformation is closely associated with the activation of MMPs. The role of ROS in the activation of MMPs (Gurjar et al., 2001; Lin et al., 2012) and the impact of activated MMPs on BBB injury are well documented (Florczak-Rzepka et al., 2012; Gurjar et al., 2001; Jin et al., 2010; Lin et al., 2012; Morancho et al., 2010). Aside from degrading the extracellular matrix, MMPs invading the cytoplasm via endocytosis can damage proteins and organelles, and impede DNA transcription and repair (Florczak-Rzepka et al., 2012). In cultured astrocyte and brain tissues, ROS up-regulate expression of c-Fos and c-Jun, the subunits of the transcriptional factor, AP-1 (Florczak-Rzepka et al., 2012), which activates AP-1-driven MMP-9 transcription by binding to the MMP-9 promoter. Furthermore, ROS enhance MMP-2′s gelatinolytic activity in a rodent cardiac hypertrophy model (Rizzi et al., 2012). Thus, neutralization of ROS might be a potential target for minimizing rtPA’s harmful effects on the BBB after ischemia-reperfusion. Pyruvate’s antioxidant capabilities have been documented in various experimental models (Jagtap et al., 2003; Wang et al., 2007). Pyruvate’s α-ketocarboxylate structure permits the direct neutralization of peroxides and peroxynitrite, and its enhancement of NADPH/NADP+ sustains the redox state of glutathione, a pivotal endogenous antioxidant (Mallet and Sun, 2003). Furthermore, pyruvate can indirectly contribute to ROS detoxification by inducing synthesis of erythropoietin, a cytokine with antioxidant properties (Toba et al., 2012). We reported that pyruvate increases HIF-1-driven mRNA expression and protein content of erythropoietin by stabilizing the O2-regulated HIF-1α (Ryou et al., 2009, 2012). In accordance with pyruvate’s antioxidant properties, the current study demonstrated that pyruvate enhanced NADPH/NADP+ redox state, suppressed ROS generation after OGD and reoxygenation, and maintained the integrity of vascular endothelial tight junctions. Furthermore, rtPA-increased ROS was associated with MMP-2 activation and disruption of vascular endothelial monolayers.
Pyruvate’s energy-yielding capabilities as an oxidizable fuel complement its antioxidant properties. Here, pyruvate sharply increased ATP content of post-hypoxic HT22 neuronal cells with and without rtPA cotreatment. Increased production of energy for ion transport and other essential cellular functions may contribute importantly to pyruvate protection of neuronal-lineage HT22 cells. However, it should be noted that the immediate hydrolysis products of ATP hydrolysis, cytosolic free ADP and inorganic phosphate, could not be measured reliably due to their limited contents in the cell extracts, which precluded computation of Gibbs free energy of ATP hydrolysis, the definitive measure of thermodynamic energy state in neurons. Nevertheless, restoration of ATP synthesis may be a critical contributor to pyruvate-induced cytoprotection against OGD stress.
To exert neuroprotection, therapeutic agents must surmount the BBB. Although erythropoietin is a promising candidate for ischemic stroke treatment, clinical trials of erythropoietin for stroke have had disappointing outcomes (Ehrenreich et al., 2009), possibly due to untoward side effects produced by the high dosages of erythropoietin required to deliver sufficient amounts to the brain parenchyma. In contrast, pyruvate readily traverses the BBB by simple diffusion or via MCT2, which has a high affinity for pyruvate (Halestrap and Wilson, 2012). In this study hypoxia stimulated MCT2 expression, and addition of pyruvate after OGD increased the expression of MCT2 in HT22 cells. Increased expression and synthesis of MCT2 may serve to maximize the available energy sources to the oxygen depleted cells. Notably, pyruvate treatment activated MCT2 synthesis even further, which could increase delivery of this energy substrate and antioxidant to the hypoxia-challenged neurons. By facilitating pyruvate entry into the brain parenchyma, increased expression of MCT2 enhances pyruvate’s cerebroprotective capabilities.
Although rtPA is the sole FDA approved treatment for ischemic stroke, because of its limited therapeutic window, few patients benefit from rtPA treatment. Pyruvate’s favorable effects prompted us to test whether or not pyruvate can extend rtPA’s therapeutic window. The HT22 neuronal cell model replicated the detrimental effects of delayed rtPA treatment after 6 h OGD. In this model, pyruvate activated the anti-apoptotic protein kinases p70s6 kinase, Erk and, most strikingly, Akt. Activated Akt activates mTOR followed by phosphorylation of ribosomal protein S6 kinase (S6K)(Xie et al., 2012). Phosphorylation of S6K is considered a positive regulator of translation of mRNA with 5′-terminal oligopyrimidine tracts. A serine-threonine protein kinase, mTOR functions as a sensor of cellular metabolic resources and adjusts the anabolic and metabolic processes accordingly (Swiech et al., 2008). Recently, a neuroprotective role of mTOR has been reported (Chen et al., 2012; Swiech et al., 2008; Xie et al., 2012). Pyruvate but not rtPA cotreatment increased Akt phosphorylation 3 h after OGD. Accordingly, we examined the impact of pyruvate and/or rtPA on the mTOR signaling pathway after 3 and 6 h OGD. Pyruvate and rtPA, alone or in combination, intensified phosphorylation of mTOR and its downstream protein kinase, p70S6 kinase, after 3 or 6 h OGD. HT22 cells were not injured by rtPA treatment after 3 h OGD, but when rtPA treatment was delayed until 6 h OGD, viability was compromised, and phosphorylation of p70S6 kinase at 24 h reoxygenation was sharply decreased. In contrast, pyruvate alone or combined with rtPA activated mTOR and p70S6 kinase after 6 h OGD and 24 h reoxygenation. Collectively, these results suggest the toxic effects of delayed rtPA treatment might be linked to impaired mTOR signaling in response to hypoxia-reoxygenation associated stress.
In summary, rtPA administration within its treatment window did not compromise viability of OGD-challenged neuronal-lineage cells. However, delayed rtPA administration beyond its therapeutic window dramatically reduced cell viability. Increased ROS and inactivation of the mTOR-mediated signaling response to stress may have contributed to the toxicity of rtPA. On the other hand, cotreatment with the potent antioxidant and energy substrate pyruvate could extend rtPA’s therapeutic window by minimizing the detrimental side effects of delayed rtPA treatment and restoring the rtPA-impaired mTOR signaling pathway.
Limitations of the experimental model
The current study was conducted in vitro to examine the direct neuroprotective effects of pyruvate against OGD-reoxygenation stress and delayed rtPA treatment, and yielded fundamental information toward understanding the mechanisms of rtPA toxicity and of pyruvate-induced neuroprotection. However, extrapolation of these results in isolated cells of a single type to the in vivo setting, in which multiple cell types interact in a highly specialized, 3-dimensional architecture, may be premature. The O2 concentration applied to the in vitro system is higher than the O2 concentrations to which neurons are normally exposed in the intact brain. Because the trophic support of neurons afforded by astrocytes in vivo was absent in the cell culture model, it was necessary to optimize the basal environment for neuronal cells to maintain cellular homeostasis by providing sufficient energy substrate and optimizing ambient O2 supply and pH.
4. Experimental procedure
4.1. Cell culture
Mouse neuronal HT22 cells, a generous gift from Dr. David Schubert, Salk Institute, San Diego, CA, were maintained in DMEM supplemented with 10% fetal bovine serum and penicillin (10,000 units/ml)-streptomycin (10,000 μg/ml). Cells under 20 passages were used in this study. Primary rat microvascular endothelial cells (MVEC) harvested from Sprague-Dawley rats were purified with Dynabeads (Invitrogen, Carlsbad, CA) coated with mouse anti-CD31, and then cultured in EGM-2-MV media. All procedures for MVEC harvest were conducted in accordance with EC Directive 86/609/EEC for animal experiments, and were approved by the Institutional Animal Care and Use Committee of the University of North Texas Health Science Center, Fort Worth, TX, USA.
4.2. Hypoxia-reoxygenation protocol
For OGD treatment, HT22 cells were washed with PBS to remove residual glucose and FBS, maintained in DMEM without glucose, pyruvate, and FBS, and then subjected to the protocols summarized in Fig. 7. The cells were placed in auto-controlled hypoxia chambers (BioSpherix, Lacona, NY, USA) housed in CO2 incubators for 3 or 6 h at 0.5% O2 and 37 °C, followed by 4 or 24 h reoxygenation in normoxic CO2 incubators at 37 °C. During reoxygenation, media contained 11 mmol/L D-glucose alone or with rtPA (10 μg/ml; Genentech, South San Francisco, CA, USA), pyruvate (8 mmol/L; tissue culture grade, Sigma, St. Louis, MO, USA), or both pyruvate and rtPA.
Fig. 7.
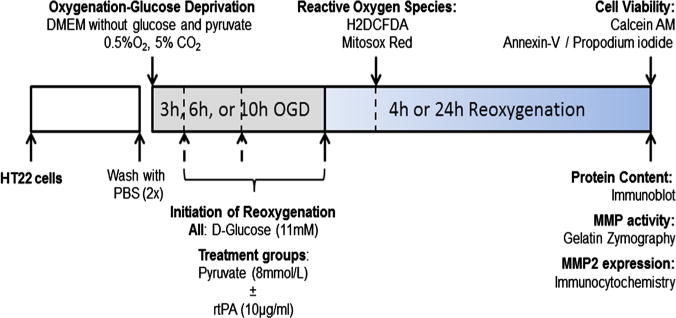
Experimental protocol. HT22 cells were subjected to oxygen-glucose deprivation by incubation in severely hypoxic (0.5% O2), glucose-free culture medium for 3 or 6 h, then reoxygenated with media containing 21% O2 and 11 mmol/L D-glucose for 4 or 24 h in the absence or presence of 10 μg/ml rtPA and/or 8 mmol/L pyruvate. Reactive oxygen species were measured at 4 h reoxygenation, and cell viability, contents of signaling proteins, and matrix metalloproteinase (MMP) activities and contents were measured at 24 h reoxygenation.
4.3. Cell viability assay
After 3 or 6 h OGD and 24 h reoxygenation, cells were washed with PBS and incubated with Calcein AM (1 μmol/L) at 37 °C for 20 min. Fluorescence intensity was measured at 485 nm excitation and 530 nm emission wavelengths. Treatment groups consisted of six independent trials, each in sextuplicate. After 6 h OGD and 24 h reoxygenation, apoptotic cell death was confirmed with flow cytometry (BD LSR II, San Jose, CA, USA) with annexin-V and propidium iodide stain.
4.4. Reactive oxygen species measurements
Reactive oxygen species were detected with two techniques. (1) MitoSox red (Invitrogen, Eugene, OR) was used to detect primarily superoxide generated in mitochondria. HT22 cells were washed with PBS and incubated with Mitosox red (5 μmol/L) for 10 min at 37 °C. The fluorescent signal was visualized and photographed on a Zeiss fluorescence microscope. (2) ROS also were assessed by a fluorometric assay using H2DCFDA (Molecular probes, Eugene, OR, USA). After 3 h OGD and 4 h reoxygenation, cells were loaded with 10 μmol/L H2DCFDA for 30 min at 37 °C, and fluorescence was measured at 530 nm emission and 495 nm excitation wavelengths.
4.5. NADPH/NADP+ ratio and ATP
After 3 h OGD and 24 h reoxygenation, NADP+ and NADPH were measured with an enzymatic cycling assay kit (BioVision, Mountain View, CA, USA). Briefly, HT22 cells were homogenized and NADP+ and NADPH were extracted. After total NADP (NADPt) was measured, NADP+ was decomposed by heating the extract at 60 °C for 30 min, and then NADPH was measured. NADPt and NADPH were detected at 450 nm and quantified with a standard curve. NADP+ was taken as the difference between NADPt and NADPH; thus, NADPH/NADP+ ratio equaled NADPH/(NADPt-NADPH). ATP was extracted and measured with a commercial assay kit (Invitrogen, Eugene, OR, USA). ATP concentrations normalized to the protein concentration of corresponding samples were expressed as percentage of the mean value of normoxic control experiments.
4.6. FITC-dextran leakage assay
Microvascular endothelial cells were seeded on collagen-coated transwell membrane (6.5 mm diameter, 0.4 μm pore size) at a concentration of 25,000 cells/membrane (Corning, Lowell, MA, USA). Once MVEC reached 80% confluency, primary rat astrocytes (50,000 cells/transwell membrane) were seeded on the other side of the membrane. After a confluent monolayer of MVEC formed, cells were exposed to 3 h OGD and 24 h reoxygenation, and then culture medium in the upper chamber was replaced with Hanks Balanced Salt Solution containing 0.5 mg/ml of FITC-Dextran for 2 h at 37 °C. The leakage of FITC-Dextran to the bottom chamber was measured by fluorometry at 485 nm excitation and 538 nm emission wavelengths.
4.7. Gelatin zymography for MMP2 activity
Matrix metalloproteinase-2 activities were measured in culture medium after subjecting HT22 cells to 3 h OGD and 24 h reoxygenation. After electrophoretic separation of proteins in 10% SDS-PAGE gels containing 0.3% gelatin, the gels were washed with buffer containing 2.5% triton X-100 for 4 h at 37 °C, followed by a second wash with buffer containing 1% Triton X-100 overnight at 37 °C. Gels were stained with coomassie blue in 45% methanol and 10% acetic acid for at least 2 h. After destaining, the gel was photographed and the intensities of bands were analyzed by UVP image system (Ultraviolet Products, Upland, CA, USA). Values were normalized to actin contents on the same gels measured by immunoblot.
4.8. Immunoblot analysis of signaling proteins
Contents of Akt, phosphorylated Akt (pAkt), Erk, phosphorylated Erk (pErk), mammalian target of rapamycin (mTOR), phosphorylated p70S6 kinase and actin were analyzed by immunoblotting of cell lysates. Protein concentrations were measured with a kit (Thermo Scientific, Rockford, IL, USA) at 660 nm to ensure equal loading of 20 μg/lane. Primary antibodies for mTOR signaling components and Akt/pAkt were from Cell Signaling Technology (Danvers, MA, USA). Goat anti-mouse and rabbit secondary antibodies were obtained from Jackson Immunoresearch (West Grove, PA, USA). Protein bands were quantified (Ultraviolet Products, Upland, CA, USA) and normalized to actin.
4.9. Statistical analysis
Data are expressed as mean values±SEM. Multiple comparisons between OGD and normoxic groups were accomplished by two-factor (treatment, protocol) analysis of variance followed by Tukey’s multiple comparison test to identify statistically significant effects. Statistical significance was assumed at P values<0.05.
Acknowledgments
This work was supported by National Institutes of Health grants R01NS054687 (SY), R01NS054651 (SY), R01NS076975 (RM), T32 AG020494 (MR), and RI6055 (MR).
Abbreviations
- BBB
blood–brain barrier
- MCT
monocarboxylate transporter
- MMP
matrix metalloproteinase
- OGD
oxygen-glucose deprivation
- ROS
reactive oxygen species
- rtPA
recombinant tissue plasminogen activator
- TIMP
tissue inhibitor of metalloproteinase
References
- Alvarez G, Ramos M, Ruiz F, Satrustegui J, Bogonez E. Pyruvate protection against beta-amyloid-induced neuronal death: role of mitochondrial redox state. J Neurosci Res. 2003;73:260–269. doi: 10.1002/jnr.10648. [DOI] [PubMed] [Google Scholar]
- Chen H, Qu Y, Tang B, Xiong T, Mu D. Role of mammalian target of rapamycin in hypoxic or ischemic brain injury: potential neuroprotection and limitations. Rev Neurosci. 2012;23:279–287. doi: 10.1515/revneuro-2012-0001. [DOI] [PubMed] [Google Scholar]
- Ehrenreich H, Weissenborn K, Prange H, Schneider D, Weimar C, Wartenberg K, Schellinger PD, Bohn M, Becker H, Wegrzyn M, Jahnig P, Herrmann M, Knauth M, Bahr M, Heide W, Wagner A, Schwab S, Reichmann H, Schwendemann G, Dengler R, Kastrup A, Bartels C, Stroke Trial EPO. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke. 2009;40:e647–e656. doi: 10.1161/STROKEAHA.109.564872. [DOI] [PubMed] [Google Scholar]
- Florczak-Rzepka M, Grond-Ginsbach C, Montaner J, Steiner T. Matrix metalloproteinases in human spontaneous intracerebral hemorrhage: an update. Cerebrovasc Dis. 2012;34:249–262. doi: 10.1159/000341686. [DOI] [PubMed] [Google Scholar]
- Fukushima M, Lee SM, Moro N, Hovda DA, Sutton RL. Metabolic and histologic effects of sodium pyruvate treatment in the rat after cortical contusion injury. J Neurotrauma. 2009;26:1095–1110. doi: 10.1089/neu.2008.0771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Yebenes I, Sobrado M, Zarruk JG, Castellanos M, Perez de la Ossa N, Davalos A, Serena J, Lizasoain I, Moro MA. A mouse model of hemorrhagic transformation by delayed tissue plasminogen activator administration after in situ thromboembolic stroke. Stroke. 2011;42:196–203. doi: 10.1161/STROKEAHA.110.600452. [DOI] [PubMed] [Google Scholar]
- Gillum RF. New considerations in analyzing stroke and heart disease mortality trends: the year 2000 age standard and the international statistical classification of diseases and related health problems, 10th revision. Stroke. 2002;33:1717–1721. doi: 10.1161/01.str.0000016925.58848.ea. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Falcon A, Candelario-Jalil E, Garcia-Cabrera M, Leon OS. Effects of pyruvate administration on infarct volume and neurological deficits following permanent focal cerebral ischemia in rats. Brain Res. 2003;990:1–7. doi: 10.1016/s0006-8993(03)03378-x. [DOI] [PubMed] [Google Scholar]
- Green AR. Pharmacological approaches to acute ischaemic stroke: reperfusion certainly, neuroprotection possibly. Br J Pharmacol. 2008;153(Suppl 1):S325–38. doi: 10.1038/sj.bjp.0707594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurjar MV, Deleon J, Sharma RV, Bhalla RC. Role of reactive oxygen species in IL-1 beta-stimulated sustained ERK activation and MMP-9 induction. Am J Physiol Heart Circ Physiol. 2001;281:H2568–H2574. doi: 10.1152/ajpheart.2001.281.6.H2568. [DOI] [PubMed] [Google Scholar]
- Hacke W, Kaste M, Bluhmki E, Brozman M, Davalos A, Guidetti D, Larrue V, Lees KR, Medeghri Z, Machnig T, Schneider D, von Kummer R, Wahlgren N, Toni D, ECASS Investigators Thrombolysis with alteplase 3 to 4.5 h after acute ischemic stroke. N Engl J Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Wilson MC. The monocarboxylate transporter family – role and regulation. IUBMB Life. 2012;64:109–119. doi: 10.1002/iub.572. [DOI] [PubMed] [Google Scholar]
- Jagtap JC, Chandele A, Chopde BA, Shastry P. Sodium pyruvate protects against H(2)O(2) mediated apoptosis in human neuroblastoma cell line-SK-N-MC. J Chem Neuroanat. 2003;26:109–118. doi: 10.1016/s0891-0618(03)00037-1. [DOI] [PubMed] [Google Scholar]
- Jia L, Chopp M, Zhang L, Lu M, Zhang Z. Erythropoietin in combination of tissue plasminogen activator exacerbates brain hemorrhage when treatment is initiated 6 hours after stroke. Stroke. 2010;41:2071–2076. doi: 10.1161/STROKEAHA.110.586198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R, Yang G, Li G. Molecular insights and therapeutic targets for blood–brain barrier disruption in ischemic stroke: critical role of matrix metalloproteinases and tissue-type plasminogen activator. Neurobiol Dis. 2010;38:376–385. doi: 10.1016/j.nbd.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CC, Hsieh HL, Shih RH, Chi PL, Cheng SE, Chen JC, Yang CM. NADPH oxidase 2-derived reactive oxygen species signal contributes to bradykinin-induced matrix metalloproteinase-9 expression and cell migration in brain astrocytes. Cell Commun Signal. 2012;10 doi: 10.1186/1478-811X-10-35. 35-811X-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet RT, Sun J. Antioxidant properties of myocardial fuels. Mol Cell Biochem. 2003;253:103–111. doi: 10.1023/a:1026009519783. [DOI] [PubMed] [Google Scholar]
- Mallet RT. Pyruvate: metabolic protector of cardiac performance. Proc Soc Exp Biol Med. 2000;223:136–148. doi: 10.1046/j.1525-1373.2000.22319.x. [DOI] [PubMed] [Google Scholar]
- Morancho A, Rosell A, Garcia-Bonilla L, Montaner J. Metalloproteinase and stroke infarct size: role for anti-inflammatory treatment? Ann N Y Acad Sci. 2010;1207:123–133. doi: 10.1111/j.1749-6632.2010.05734.x. [DOI] [PubMed] [Google Scholar]
- O’Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1026 experimental treatments in acute stroke. Ann Neurol. 2006;59:467–477. doi: 10.1002/ana.20741. [DOI] [PubMed] [Google Scholar]
- Pan R, Rong Z, She Y, Cao Y, Chang LW, Lee WH. Sodium pyruvate reduces hypoxic-ischemic injury to neonatal rat brain. Pediatr Res. 2012;72:479–489. doi: 10.1038/pr.2012.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzi E, Ceron CS, Guimaraes DA, Prado CM, Rossi MA, Gerlach RF, Tanus-Santos JE. Temporal changes in cardiac matrix metalloproteinase activity, oxidative stress, and TGF-beta in renovascular hypertension-induced cardiac hypertrophy. Exp Mol Pathol. 2012;94:1–9. doi: 10.1016/j.yexmp.2012.10.010. [DOI] [PubMed] [Google Scholar]
- Ryou MG, Liu R, Ren M, Sun J, Mallet RT, Yang SH. Pyruvate protects the brain against ischemia-reperfusion injury by activating the erythropoietin signaling pathway. Stroke. 2012;43:1101–1107. doi: 10.1161/STROKEAHA.111.620088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryou MG, Flaherty DC, Hoxha B, Sun J, Gurji H, Rodriguez S, Bell G, Olivencia-Yurvati AH, Mallet RT. Pyruvate-fortified cardioplegia evokes myocardial erythropoietin signaling in swine undergoing cardiopulmonary bypass. Am J Physiol Heart Circ Physiol. 2009;297:H1914–H1922. doi: 10.1152/ajpheart.01213.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland BA, Minnerup J, Balami JS, Arba F, Buchan AM, Kleinschnitz C. Neuroprotection for ischaemic stroke: translation from the bench to the bedside. Int J Stroke. 2012;7:407–418. doi: 10.1111/j.1747-4949.2012.00770.x. [DOI] [PubMed] [Google Scholar]
- Swiech L, Perycz M, Malik A, Jaworski J. Role of mTOR in physiology and pathology of the nervous system. Biochim Biophys Acta. 2008;1784:116–132. doi: 10.1016/j.bbapap.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Takamiya M, Miyamoto Y, Yamashita T, Deguchi K, Ohta Y, Abe K. Strong neuroprotection with a novel platinum nanoparticle against ischemic stroke- and tissue plasminogen activator-related brain damages in mice. Neuroscience. 2012;221:47–55. doi: 10.1016/j.neuroscience.2012.06.060. [DOI] [PubMed] [Google Scholar]
- Toba H, Kojima Y, Wang J, Noda K, Tian W, Kobara M, Nakata T. Erythropoietin attenuated vascular dysfunction and inflammation by inhibiting NADPH oxidase-derived superoxide production in nitric oxide synthase-inhibited hypertensive rat aorta. Eur J Pharmacol. 2012;691:190–197. doi: 10.1016/j.ejphar.2012.07.018. [DOI] [PubMed] [Google Scholar]
- Wang X, Perez E, Liu R, Yan LJ, Mallet RT, Yang SH. Pyruvate protects mitochondria from oxidative stress in human neuroblastoma SK-N-SH cells. Brain Res. 2007;1132:1–9. doi: 10.1016/j.brainres.2006.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie R, Li X, Ling Y, Shen C, Wu X, Xu W, Gao X. Alpha-lipoic acid pre- and post-treatments provide protection against in vitro ischemia-reperfusion injury in cerebral endothelial cells via Akt/mTOR signaling. Brain Res. 2012;1482:81–90. doi: 10.1016/j.brainres.2012.09.009. [DOI] [PubMed] [Google Scholar]
- Yi JS, Kim TY, Kyu Kim D, Koh JY. Systemic pyruvate administration markedly reduces infarcts and motor deficits in rat models of transient and permanent focal cerebral ischemia. Neurobiol Dis. 2007;26:94–104. doi: 10.1016/j.nbd.2006.12.007. [DOI] [PubMed] [Google Scholar]