

Abstract
Recently, crosstalk between sphingolipid signaling pathways and steroid hormones has been illuminated as a possible therapeutic target. Sphingosine kinase (SK), the key enzyme metabolizing pro-apoptotic ceramide to pro-survival sphingosine-1-phosphate (S1P), is a promising therapeutic target for solid tumor cancers. In this study, we examined the ability of pharmacological inhibition of S1P formation to block estrogen signaling as a targeted breast cancer therapy. We found that the Sphk1/2 selective inhibitor (SK inhibitor (SKI))-II, blocked breast cancer viability, clonogenic survival and proliferation. Furthermore, SKI-II dose-dependently decreased estrogen-stimulated estrogen response element transcriptional activity and diminished mRNA levels of the estrogen receptor (ER)-regulated genes progesterone receptor and steroid derived factor-1. This inhibitor binds the ER directly in the antagonist ligand-binding domain. Taken together, our results suggest that SKIs have the ability to act as novel ER signaling inhibitors in breast carcinoma.
Introduction
Breast cancer is the second leading cause of cancer death in women today, with one in eight women diagnosed in her lifetime, and drug resistance remains the leading cause of breast cancer treatment failure. Approximately 70% of breast cancer diagnoses are estrogen receptor (ER)-positive, and selective ER modulators (SERMs), such as tamoxifen and fulvestrant, are the first-line therapies for these cancers (Chu & Anderson 2002, Burstein et al. 2004, Gail et al. 2007, Punglia et al. 2007). Unfortunately, resistance to all endocrine therapies has been documented in both laboratory and clinical experiments, thereby necessitating novel strategies for targeting ER signaling to combat ER-positive breast cancer drug resistance (Dowsett 1996, Kurebayashi 2005).
Over the past several years, much research has been dedicated to the role of lipid signaling in chemotherapy, endocrine therapy and radiation resistance. Alterations in the metabolism of bioactive sphingolipids have been implicated in cancer tumorigenesis and treatment failure (Meacham et al. 2009). Targeting the ceramide (CER)–sphingosine-1-phosphate (S1P) rheostat is of particular interest. The conversion of proapoptotic CER into pro-survival S1P is a proposed ‘switch’ for cells in an anti-proliferative state to transition to a proliferative state (Cuvillier et al. 1996). The enzyme sphingosine kinase (SK) is the primary regulator for the conversion of CER to S1P, which is a potent inducer of proliferation, tumorigenesis and survival in many solid tumor cancers, including breast and prostate (Taha et al. 2006, Hannun & Obeid 2008).
Recent studies on the role of SK/S1P in breast cancer suggest this signaling pathway is important in promoting tumorigenesis, endocrine therapy resistance and chemoresistance. SK signaling is a mechanism of estrogen-induced proliferation and S1P is known to increase levels of circulating estrogen in the system (Doll et al. 2007, Lucki & Sewer 2008). The role of S1P in endocrine resistance was first characterized by the Xia laboratory, which showed that overexpressing S1P confers tamoxifen resistance to ER-positive MCF-7 cells. Furthermore, it has been shown that growing MCF-7 cells in increasing concentrations of tamoxifen until resistance is established produces an overexpression of SK-1. These results provide evidence that SK/S1P signaling is involved in endocrine drug resistance (Sukocheva et al. 2009). SK can also increase estrogen-dependent tumorigenesis and promote motility and survival of ER-positive breast cancer (Wang et al. 1999, Nava et al. 2002, Sankala et al. 2007). In a microarray analysis of human breast cancer biopsies, SK mRNA levels directly correlated with a negative prognostic outcome (Ruckhaberle et al. 2008). Given this evidence, the SK/S1P signaling pathway may be a possible therapeutic target to ablate ER-mediated breast cancer proliferation.
Though the CER–S1P has been well characterized over the past several years, there are few pharmacological inhibitors targeting this pathway. The first, and most commonly published, selective inhibitor of SK is SKI-II. This drug has been commercialized and is now readily available from a number of pharmaceutical companies (French et al. 2003). For this reason, SKI-II is the most widely used pharmacological inhibitor of SK and has been utilized in a wide range of studies including those involving cardiology, immunology and oncology (French et al. 2006, Yan et al. 2008, Trifilieff et al. 2009).
SKI-II has been previously studied in the context of lung, brain, breast and prostate cancers (Leroux et al. 2007, Ader et al. 2008, Guillermet-Guibert et al. 2009). In murine models of breast cancer, SKI-II was shown to have an anti-tumor effect with little effect on animal body weight (French et al. 2006). These studies demonstrated that SKI-II has several properties advantageous to drug development, such as high oral bioavailability and limited toxicity (French et al. 2003, 2006). However, there is little in vitro data on SKI-II in ER-positive breast cancer. Furthermore, to date, there are no published studies on the effect of SKI-II on estrogen and ER signaling. Recently, we showed that the Sphk2 selective inhibitor ABC294640 could alter ER signaling in MCF-7 cells (Antoon et al. 2010). In the present study, we demonstrate the ability of SKI-II to abrogate ER signaling, possibly through direct binding to the ER, similar to tamoxifen. SKI-II has potential as a therapeutic agent in breast cancer, as it can diminish survival, viability and proliferation of breast carcinoma cells without similar effects on normal breast epithelial cells. With resistance to first-line drugs such as SERMs on the rise, novel agents targeting ER signaling in human breast cancer are of growing importance.
Materials and methods
Reagents
SKI-II (4-[4-(4-chloro-phenyl)-thiazol-2-ylamino]-phenol, CAS 312636-16-1) was purchased from Sigma–Aldrich. Dimethyl sulfoxide (DMSO) and 17β-estradiol (E2) were purchased from Fisher Scientific (Waltham, MA, USA).
Cell culture
ER-positive MCF-7 cells were cultured as previously described (Antoon et al. 2009). Briefly, the MCF-7 cell line used is a subclone of MCF-7 cells obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), which was generously provided by Louise Nutter (University of Minnesota, MN, USA; Burow et al. 1998). The culture flasks were maintained in a tissue culture incubator in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. For estrogen studies, cells were washed with PBS three times and grown in phenol red-free DMEM supplemented with 5% dextran-coated, charcoal-treated fetal bovine serum (5% CS-FBS) for 72 h before plating for each particular experiment. MCF10A cells were obtained from ATCC and cultured in mammary epithelial cell medium (Cambrex, San Diego, CA, USA) supplemented with bovine pituitary extract.
Real-time reverse transcription-PCR
Real-time reverse transcription (RT)-PCR was performed similar to previously reported studies (Boue et al. 2009, Rhodes et al. 2009). In brief, total cellular RNA was extracted using the RNeasy mini column (Qiagen), following the manufacturer’s instructions. The concentration of RNA was determined using a u.v. spectrophotometer. RT was performed using the Super-Script First-Strand Synthesis System for RT-PCR (Invitrogen). The level of steroid derived factor-1 (SDF-1) and progesterone receptor (PGR) transcripts was determined using the iQ5 real-time quantitative PCR detection system (Bio-Rad, Inc.). Primers for PCR were designed to span intron–exon junctions to minimize amplification of residual genomic DNA. The primer sequences for PGR, SDF-1 and ER are (sense and antisense, respectively): PGR (5′-TACCCGCCCTATCTCAACTACC-3′; 5′-TGCTTCATCCCCACAGATTAAACA-3′), SDF-1 (5′-AGTCAGGTGGTGGCTTAACAG-3′; 5′-AGAGGAGGTGAAGGCAGTGG-3′), ER (5′-GCGATGGTGGAGATCTTCGA-3′; 5′-CCTCTCCCTGCAGATTCATCA-3′). PCR mix contained optimal concentrations of primers, cDNA and SYBR Green PCR Master Mix (Bio-Rad Lab.). Quantification and relative gene expression were calculated with internal controls. The ratio between these values obtained provided the relative gene expression levels.
Lipidomics analysis
Endogenous lipid levels were quantified by mass spectrometry (Lipidomics Core, Medical University of South Carolina), according to published methods (Bielawski et al. 2006). Briefly, cells were collected, fortified with internal standards and extracted with ethyl acetate/isopropyl alcohol. Electrospray ionization, followed by tandem mass spectrometry (ESI/MS/MS) analyses of sphingoid bases, sphingoid base 1-phosphates, CERs and sphingomyelins were performed.
Western blot analysis
Protein analysis was performed as described in Burow et al. (1998). Briefly, cells were plated at 50–60% confluency in 10 cm2 culture flasks in 5% DMEM for 48 h. Cells were then treated with DMSO or SKI-II (1–4 μM) and 10% DMEM added at 30, 60 or 120 min. Following treatment, cells were detached with PBS–EDTA and centrifuged. After removing supernatant, cells were lyzed in 60–100 μl lysis buffer (Mammalian Protein Extraction Reagent, MPER, and Halt protease inhibitor, Pierce, Rockford, IL, USA; and PhoSTOP phosphatase inhibitor, Roche). Lysed cells were centrifuged for 10 min at 12 000 g at 4 °C to separate protein from cell debris. The supernatants were combined with loading buffer (5% 2-mercaptoethanol in 4× LDS Loading Buffer, Invitrogen), boiled for 5 min, and loaded onto a 4–12% Bis Tris Polyacrylamide Gels (Invitrogen) followed by PAGE at 150 V for 1·25 h. Protein was transferred to nitrocellulose membranes using the iBlot (Invitrogen) transfer unit. Nitrocellulose membranes were blocked in 5% milk (Bio-Rad Lab.) Tris buffered saline–Tween 20 (TBS–T) for 1 h at room temperature. Cells were washed briefly with 1× TBS–T (USB, Cleveland, OH, USA) and primary antibodies were diluted in 5% BSA (Sigma–Aldrich) TBS–T according to manufacturer’s recommended dilutions. Antibodies for tubulin, AKT and phospho-AKT were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA). Membranes were incubated in primary antibody overnight at 4 °C with gentle agitation. Secondary infrared conjugated antibodies (LI-COR Biosciences, Lincoln, NE, USA) were diluted in 5% milk–TBS–T solution at 1:10 000 ratio and the membranes were incubated for 1 h under gentle agitation at room temperature. Membranes were scanned using the LI-COR Odyssey imager and software (LI-COR Biosciences) to detect total and phosphorylated protein levels in cell lysates. Protein levels were quantified using densitometry analyses.
Clonogenic survival assay
Colony assays were performed similar to previously published methods (Struckhoff et al. 2004). MCF-7 cells were plated in six-well plates at a density of 1000 cells/well in full DMEM media. Twenty-four hours later, the cells were treated with SKI-II (0·1–10 μM) and then monitored for colony growth. Ten days later, the cells were fixed with 3% glutaraldehyde. Following fixation for 15 min, the plates were washed and stained with a 0·4% solution of crystal violet in 20% methanol for 30 min, washed with PBS and dried. Colonies of ≥30 cells were counted as positive. Results were normalized to DMSO vehicle-treated control cells. Statistical analysis of IC50 values were calculated from concentration–response curves, using GraphPad Prism 5.0 (Graphpad Software, San Diego, CA, USA), by applying the equation: Y = Bottom + (Top − Bottom)/1 + 10LogEC50 − X.
Cell proliferation immunofluorescence assay
Cells were plated at a density of 10 000 cells/well in a 96-well plate containing 10% DMEM media and allowed to attach overnight. The following day, cells were treated with DMSO or SKI-II for 24 h. At endpoint, cells were fixed using 100 μl of 3·7% formaldehyde in PBS for 10 min. Formaldehyde was removed and the cells were permeabilized using cold methanol for 5 min at room temperature and washed twice with PBS. One hundred microliters of 3% FBS in PBS blocking buffer was then added. After 30 min, blocking buffer was removed and cells were incubated for 1 h with Ki-67 (BD Pharmingen, San Diego, CA, USA) antibody. Cells were then washed with PBS and stained with DAPI nuclear stain for 5 min before imaging. For staining quantification, numbers of positively stained cells were expressed as a percentage of the total number of cells/field of view/image. The vehicle control was then set to 1 for comparison with SKI-II treatment.
Cell viability assay
Viability assays were performed as previously described (Struckhoff et al. 2004). Briefly, cells were plated at a density of 7·5×105 cells/well in a 96-well plate containing phenol-free DMEM supplemented with 5% FBS and allowed to attach overnight. Cells were then treated with SKI-II (ranging from 10 nM to 100 μM) for 24 h. Following treatment, 20 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 5 mg/ml) reagent was incubated in each well for 4 h. Cells were lyzed with 20% SDS in 50% dimethylformamide. The pH and absorbances were read on an ELx808 Microtek plate reader (Bio-Tek Instruments, Winooski, VT, USA) at 550 nm, with a reference wavelength of 630 nm.
Estrogen response element-luciferase assay
As previously described (Boue et al. 2009, Bratton et al. 2009), the cells were seeded in 24-well plates at a density of 5×105 cells/well in the same media and allowed to attach overnight. After 18 h, cells were transfected for 5 h in serum-free DMEM with 300 μg pGL2-ERE2XTK-luciferase plasmid, using 6 μl of Effectene (Qiagen) per μg of DNA. After 5 h, the transfection medium was removed and replaced with phenol red-free DMEM supplemented with 5% CS-FBS containing vehicle, E2, SKI or E2 + SKI, and incubated at 37 °C. After 18 h, the medium was removed and 100 μl of lysis buffer was added per well and then incubated for 15 min at room temperature. Cell debris was pelleted by centrifugation at 15 000 g for 5 min. Cell extracts were normalized for protein concentration using reagent according to the manufacturers protocol (Bio-Rad Lab.). Luciferase activity for the cell extracts was determined using luciferase substrate (Promega Corp.) in an Autoluminat Plus luminometer (Berthhold Technologies, Bad Wildbad, Germany).
ERα binding assays
Receptor-binding assays were performed as previously described (Boue et al. 2009). In this method, recombinant ER is in equilibrium with a fluorescent ligand (ES2) and a concentration of the competitor (SKI-II). The relative displacement of the ES2 is measured as a change in polarization anisotropy. Serial dilutions of competitors (SKI-II and E2) were prepared from DMSO stock solutions in screening buffer at the desired concentrations. The ER and ES2 were combined with each competitor aliquot to a final concentration of 2 nMER and 3 nM ES2, respectively. In addition, both a no-binding control (ER + ES2, equivalent to 0% competitor inhibition) and a 100% binding control (only free ES2, no ER, equivalent to 100% competitor inhibition) were prepared. All competitors and controls were prepared in duplicate within a binding experiment. After a 2 h incubation at room temperature, the anisotropy value for each sample and control were measured using the Beacon 2000. Anisotropy values were converted to percent inhibition using the following formula: I% = (A0 − A)/(A0 − A100) × 100, where I% is the percent inhibition, A0 is 0% inhibition, A100 is 100% inhibition and A represents the observed value. This conversion to percent inhibition makes the data more intuitive and normalizes the experiment-to-experiment differences in the range of anisotropy values. The percent inhibition versus competitor concentration curves were analyzed by nonlinear least-squares curve fitting (Prism 5.0a, GraphPad Software) to yield IC50 values (the concentration of competitor needed to displace half of the bound ligand). To compare binding affinities of the test compounds to those reported in the literature, IC50 values were converted to relative binding affinities (RBA) using E2 as a standard. The E2 RBA was set equal to 100 RBA = (IC50/IC50 of E2)×100.
Molecular modeling
The structures of SKI-II were converted to unique SMILE strings with ChemDraw (CambridgeSoft, Cambridge, MA, USA) and then converted to 3D structures using MOE 2008.10 (Chemical Computing Group, Montreal, Quebec, Canada). The initial 3D models were then optimized in MOE using the MMFF94 force field with the conjugated gradient method using a termination of 0·005 kcal/mol. The SKI-II model was added to a database containing an optimized model of 4-hydroxytamoxifen (4-OH-Tam). Docking and scoring of SKI-II was performed using the crystal structure of the human ERα ligand-binding domain in complex with 4-OH-Tam (pdb: 3ERT, antagonist configuration) and the docking function of MOE. The A chain, including associated waters, was extracted from the 3ERT crystal structure and then hydrogens were added and optimized for docking with the Protonate 3D function of MOE. The models of SKI-II and 4-OH-Tam were then docked into the 3ERT Chain A model with MOE dock using the Triangle Matcher placement (default settings), London dG rescoring 1, Forcefield refinement (default settings) and London dG rescoring 2. This docking procedure was previously defined in this study as the MOE docking method that produced the best RMSD (0·758) replacement of the 4-OH-Tam model into this crystal structure.
Results
SKI-II blocks SK/S1P signaling in MCF-7 cells
It has been previously reported that SK1 mRNA is overexpressed in human breast cancer tissue. Therefore, we chose to examine the expression of SK1 mRNA levels in MCF10A cells and compare them to MCF-7 breast cancer cells. MCF10A are nontransformed, noncancerous breast epithelial cell cells commonly used as controls for MCF-7 ductal carcinoma cells. Our RT-PCR results demonstrate that MCF-7 cells have an 8·01±1·24 (P<0·05) fold increase in SK1 mRNA expression compared with MCF-10A (Fig. 1A). This overexpression of SK suggests that as cells progress to a cancer phenotype, there is an increase in SK1, which may provide a potential therapeutic target for MCF-7 breast cancer cells.
Figure 1.
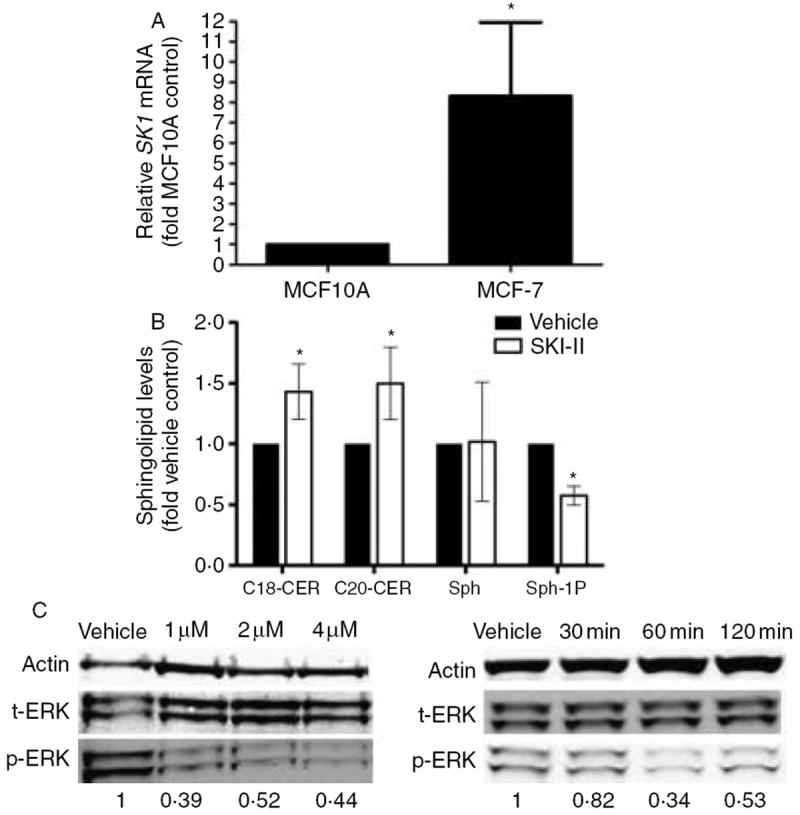
SKI-II blocks downstream SK signaling. (A) PCR analysis for endogenous expression of SK1. Data are expressed as fold-change relative to MCF10A cell control as normalized to internal β-actin±s.e.m. Data points and error bars represent the mean±s.e.m. of three independent experiments. (B) MCF-7 cells were treated with either vehicle control or SKI-II (10 μM) for 24 h and measured for cellular levels of C18-ceramide, C20-ceramide, sphingosine and sphingosine-1-phosphate using ESI/MS/MS. Data points and error bars represent the mean±s.e.m. of two independent experiments (*P<0·05). (C) MCF-7 cells were treated with either vehicle control or SKI-II analyzed by western blot for phosphorylated and total ERK proteins with densitometry analysis (control set to 1). Phosphorlyated ERK levels were normalized to corresponding β-tubulin and vehicle control set to 1. Blots are representative of three independent experiments.
It was previously reported that SKI-II decreased S1P formation and MAPK signaling in JC murine adenocarcinoma cells (French et al. 2003). However, SKI-II has yet to be examined in ER-positive human breast cancer. Therefore, we examined whether SKI-II could block formation of S1P and increase levels of endogenous long-chain CERs in the ER-positive MCF-7 cell system. Since SK blocks the conversion of proapoptotic CER into pro-survival S1P, the concomitant increase in CER and decrease in S1P would be advantageous as a cancer treatment. Using ESI/MS/MS analyses, we determined levels of S1P, C18-CER and C20-CER in MCF-7 cells with SKI-II treatment. As seen in Fig. 1B, SKI-II treatment diminished formation of S1P by half compared with vehicle control at 24 h, while endogenous levels of C18-CER and C20-CER were increased by 43·5 and 49·1%, respectively. There was no statistically significant reduction in the protein levels of the intermediate lipid sphingosine.
We further examined the ability of SKI-II to affect downstream S1P signaling. S1P is known to activate MAPK signaling pathways as a mechanism of its proliferative effects. In particular, the ERK/MAPK pathway is phosphorylated upon stimulation of S1P signaling (Nava et al. 2002). The ERK/MAPK pathway has been shown to be critical in the promotion of breast cancer proliferation and progression to endocrine therapy resistance (Weldon et al. 2002, Geffroy et al. 2005, Thomas et al. 2008). Therefore, we examined the effect of SKI-II on the phosphorylation of the ERK/MAPK protein (Fig. 1C). As expected, SKI-II decreased ERK phosphorylation in both time-course and dose–response analyses. These results provide proof of principle that SKI-II inhibits S1P signaling in the ER-positive MCF-7 breast carcinoma cell system.
SKI-II decreases breast cancer viability, survival and proliferation in vitro
Given that SKI-II blocks S1P formation and downstream ERK phosphorylation in breast epithelial cells, we next examined the ability of SKI-II to selectively target normal breast epithelial cells compared with breast cancer cells. Using the MTT assay, we determined the effect of SKI-II on MCF-7 and MCF10A viability both in short- and long-term treatments (Fig. 2). SKI-II diminished MCF-7 cell viability while having little affect on MCF10A cells. At the highest dose of SKI-II tested, 100 μM, 24 and 48 h MCF10A cell viability decreased by 7·6 and 32·7% (P<0·01), respectively. In comparison, MCF-7 cells viability decreased by 82·9% (P<0·001) and 78·6% (P<0·01) at the 24 and 48 h time-points. Taken together, these data suggest that SKI-II preferentially affects abnormal breast epithelial cells and is selective for ER-positive breast cancer.
Figure 2.
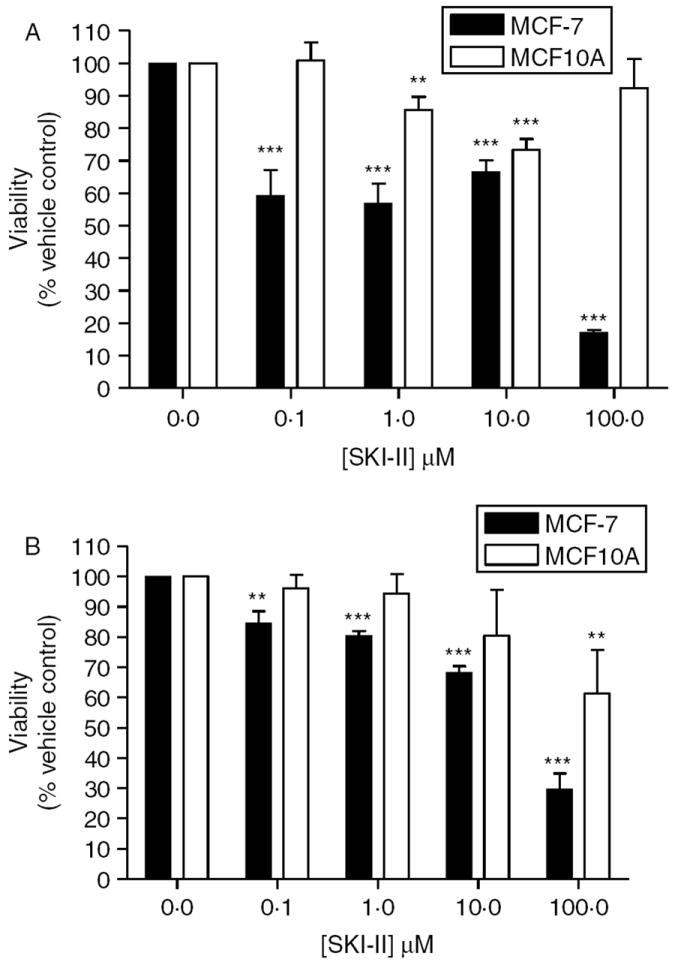
SKI-II decreases MCF-7 viability. MCF10A and MCF-7 cells were treated with SKI-II for (A) 24 h or (B) 48 h. Cell viability was determined using the MTT assay as described in the Materials and methods section. Data are presented as percent viability of vehicle-treated control cells. Mean values±s.e.m. of three different experiments in triplicate are reported (***P<0·001, **P<0·01).
S1P is known to be a pro-survival mediator and can act in both a paracrine and autocrine manner to promote its effects. There is disagreement in the literature as to whether short-term viability assays accurately reflect chemotherapeutic potential, since cancer treatment usually extends over weeks and not hours (Brown & Wouters 1999). Therefore, we determined the effect of SKI-II on long-term breast cancer clonogenic survival. As seen in Fig. 3, SKI-II dose-dependently decreases MCF-7 survival, with an IC50 value of 2·04 μM (P<0·01). The low-micromolar efficacy of SKI-II indicates the potential of the inhibitor as a breast cancer therapeutic agent.
Figure 3.
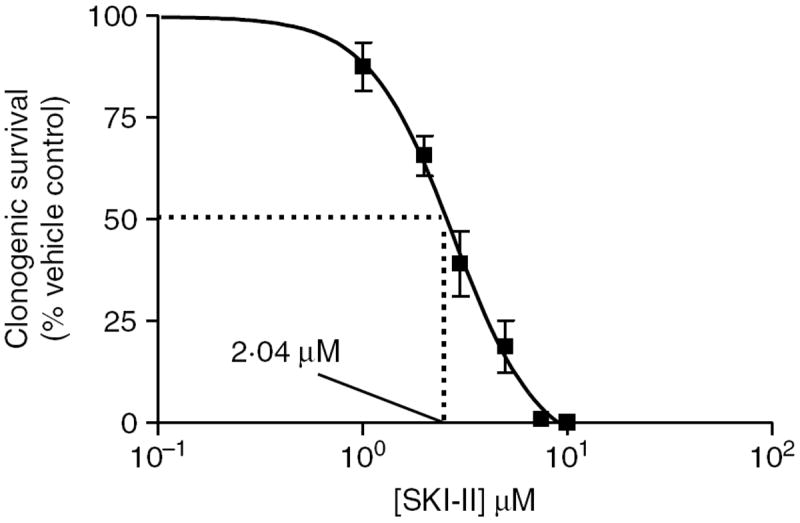
Inhibition of SK diminishes MCF-7 clonogenic survival. MCF-7 cells were plated at 1000 cells/well in six-well plates. Twenty-four hours later cells were treated with DMSO (vehicle) or SKI-II (0·1–10 μM) for 14 days. Colonies of >30 were counted as positive. Results were normalized to percent clonogenic survival of vehicle control cells. Data represented as mean±s.e.m. of three independent experiments.
Given that one of the main actions of S1P is to stimulate proliferation, we next determined whether SKI-II could abrogate the growth effects of S1P. The ability to block both proliferation and survival is an advantageous property in any cancer therapy. Using immunofluorescence staining for Ki-67, we found that SKI-II has potent anti-proliferative properties (Fig. 4A). Ki-67 is a protein expressed exclusively in the nucleus during all active phases of the cell cycle and is a known proliferative and prognostic marker for human breast cancer, both in the clinic and in the laboratory (Gaglia et al. 1993, Starborg et al. 1996, Beresford et al. 2006). Treatment with 10 μM of SKI-II decreased Ki-67 staining by 86·5% (P<0·01; Fig. 4B). Taken together, these data suggest that pharmacological inhibition of SK has therapeutic potential in treating ER-positive breast cancer.
Figure 4.
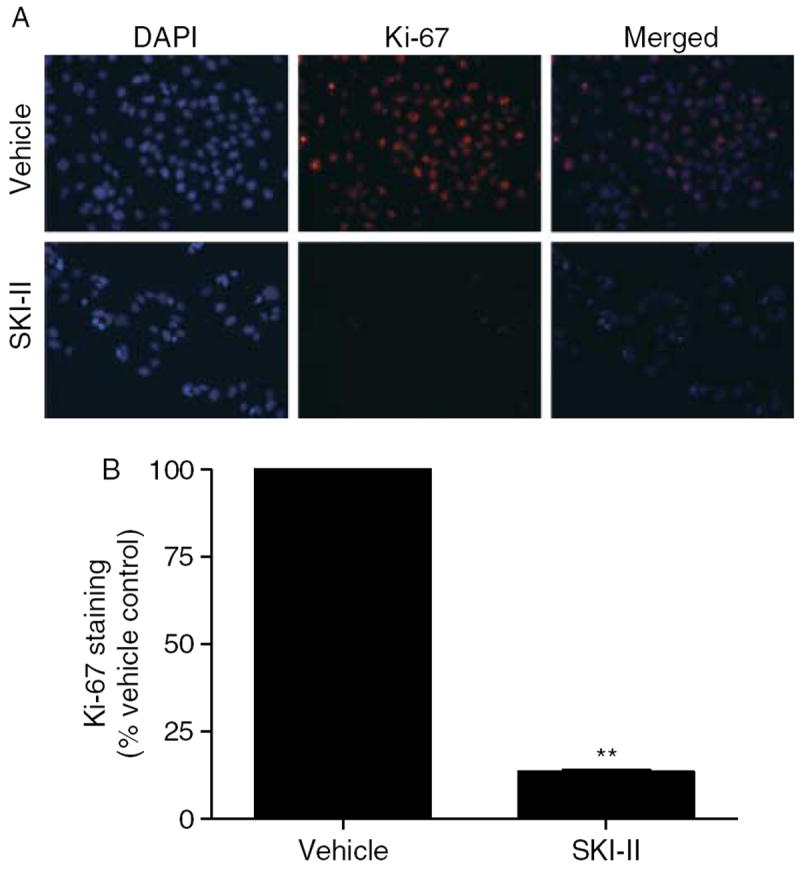
SKI-II blocks MCF-7 proliferation. (A) MCF-7 cells treated with vehicle or SKI-II for 48 h were fixed using 3·7% formaldehyde in PBS permeabilized using cold methanol and incubated with anti-Ki-67 antibody (red). Cells were washed (with PBS) and DAPI (blue) nuclear stained before imaging. Three independent experiments were performed with representative pictures shown here. (B) Quantitation of Ki-67 staining was determined as a percentage of total positive cells/image. The vehicle control was set to 1 for comparison with SKI-II treatment (**P<0·001).
Inhibition of SK diminishes ER signaling in vitro
ER signaling is an essential mediator of breast cancer proliferation and tumorigenesis (Fuqua & Cui 2002, Katzenellenbogen & Frasor 2004). Given that S1P can promote estrogen-dependent tumorigenesis, as well as increase systemic estrogen levels, we examined the estrogen signaling pathway as a possible mechanism of the potent anti-proliferative and anti-survival properties of SKI-II (Nava et al. 2002, Lucki & Sewer 2008). The effect of SKI-II on ER transcriptional activity was determined using the estrogen response element (ERE)-luciferase assay. Using the endogenously ER-negative HEK293 cell line, we transiently transfected in both the ER and an ERE-luciferase promoter and treated with increasing concentrations of SKI-II. As seen in Fig. 5A, SKI-II treatment dose-dependently decreased ERE-transcriptional activity. In order to verify that these results occur in an ER-positive system, this experiment was repeated in MCF-7 cells transfected with the same ERE-luciferase construct (Fig. 5B). SKI-II treatment also decreased ERE activity in the MCF-7 cell system, suggesting that this inhibitor can decrease ER activity in vitro.
Figure 5.
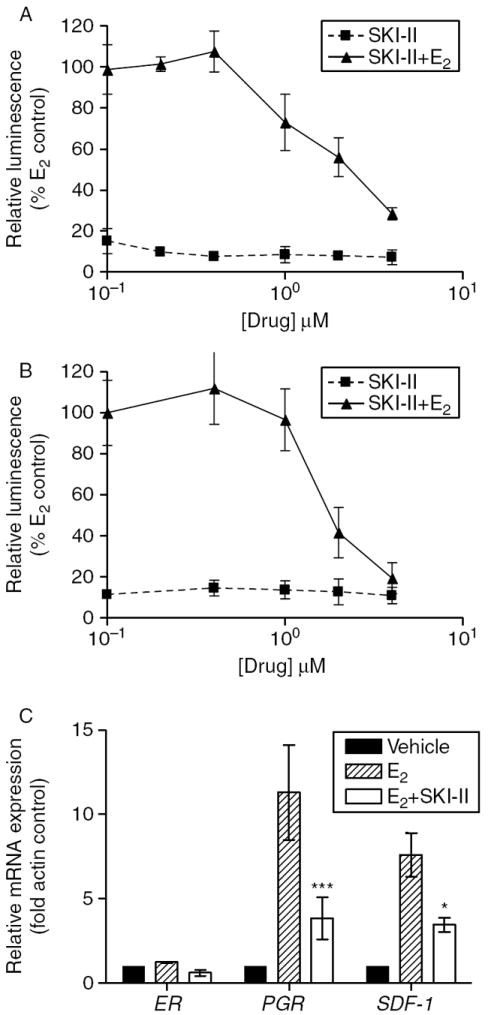
Inhibition of SK decreases estrogen signaling in vitro. HEK293 (A) or MCF-7 (B) cells were transiently transfected with pGl2-ERE2X-TK-luciferase plasmid. HEK293 cells were additionally transfected with pcDNA3.1B-ERα plasmid. Subsequently, the cells were treated with DMSO (control), estrogen (E2) or SKI-II. Cells treated with E2 were set to 1. (C) Effect of SKI-II (10 μM) on ERα, PGR and SDF-1 gene expression. Statistical analysis was performed comparing treatment groups with E2 control (***P<0·001, *P<0·05).
We further examined whether SKI-II could alter downstream ER-regulated gene expression. SDF-1 (Hall & Korach 2003) and PGR (Horwitz et al. 1978), two differentially expressed ER-mediated genes, were analyzed using qRT-PCR. Both of these genes are known to transmit the mitogenic and tumorigenic effects of estrogen in breast cancer. As seen in Fig. 5C, treatment with SKI-II blocked estrogen-stimulated mRNA expression of SDF-1 and PGR. PGR mRNA expression was decreased by 66·1% (P<0·001), while SDF-1 expression was decreased by 54·6% (P<0·05) compared with estrogen controls. Taken together, these results indicate that SKI-II can block ER signaling in vitro.
SKI-II directly binds the ER
Given the decrease in ERE-transcriptional activity, we next determined possible mechanisms of the anti-estrogenic effect of SKI-II. Using exogenous competitive binding assays, we found that SKI-II directly binds ERα, but not ERβ (Fig. 6). Interestingly, SKI-II displays an ERα binding IC50 of 85·8 nM (P<0·05), while estrogen has an IC50 of 0·44 nM (P<0·05). This nanomolar IC50 value was surprising compared with the micromolar anti-survival IC50 of this compound. While the tumorigenic and proliferative role of ERα in the promotion of breast cancer is well characterized, it is believed that ERβ may have opposing effects to that of ERα (Hartman et al. 2009). Therefore, preferential binding to ERα may be advantageous in the treatment of ER-positive breast cancer.
Figure 6.
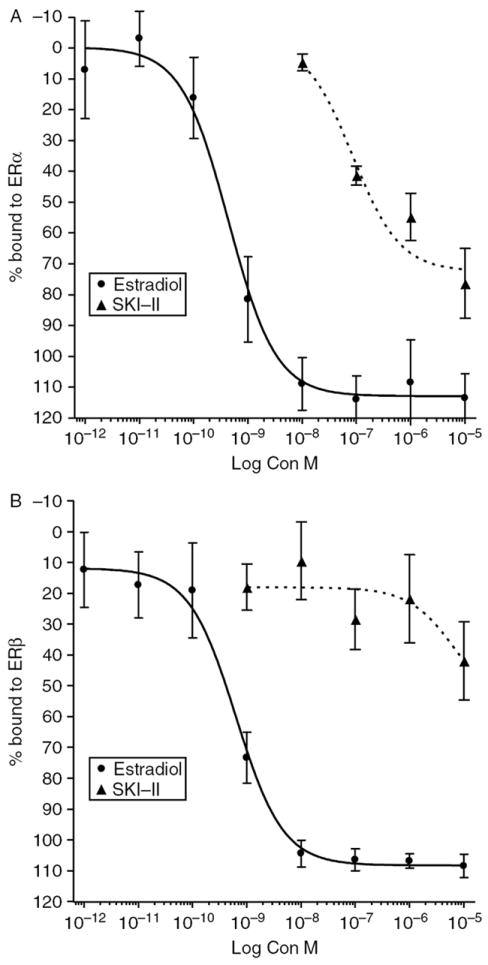
SKI-II directly binds the ER. Binding affinity of SKI-II was compared with E2. Data points and error bars represent the mean±s.e.m. of seven independent experiments for SKI-II and E2 treatment for each concentration tested.
Molecular modeling, docking and scoring were utilized in order to better understand the binding of SKI-II to ERα and ERβ. Figure 7 depicts docking simulations of potential SKI-II binding modes in the cavity of ERα (panels A and B) and ERβ (panel C). The ER-binding cavity is depicted using binding pocket residues, including the critical H-bonding amino acids arginine and glutamate on the right. Two different ERα binding modes of SKI-II were equally scored by the docking simulations (A and B). Pose A utilizes the phenolic ring of the SKI-II to bind the same way that the phenolic group of tamoxifen binds to ERα. In this binding mode, the ligand stabilizing H-bonding network between Arg 395, Glu 353 and water can be completed with the phenolic ring of SKI-II. At the same time, the ‘rest’ of SKI-II occupies the channel leading to the outside of the ERα ligand-binding domain in a similar fashion as the aryl-amino group of 4-hydroxytamoxifen is known to induce the antagonist configuration of ER (Fig. 7A) Pose B utilizes the halogenated phenyl ring of SKI-II to bind ERα in the same binding site location as the phenolic ring of tamoxifen. This halogen may form pseudo H-bonds that are much weaker than those of a hydroxyl group. At the same time, the pose B binding mode of SKI-II places the phenolic OH in proximity for H-bond interactions with Asp 351 of ERα, the same residue that stabilizes the tertamino of tamoxifen with a charge interaction (Fig. 7B). The docking simulation of SKI-II with ERβ is shown in panel C with a binding mode similar to that of the ERα A pose described above and depicted in panel A. In this suggested ERβ ligand interaction, the phenolic ring binding of SKI-II mimics the phenolic ring interactions of tamoxifen bound to ERβ (yellow) with the ‘rest’ of SKI-II occupying the channel leading to the outside of the ERβ ligand-binding domain as does the aryl-amino group of 4-hydroxy-tamoxifen (Fig. 7C) SKI-II did not present with additional or alternative binding modes in the docking simulations with ERβ.
Figure 7.
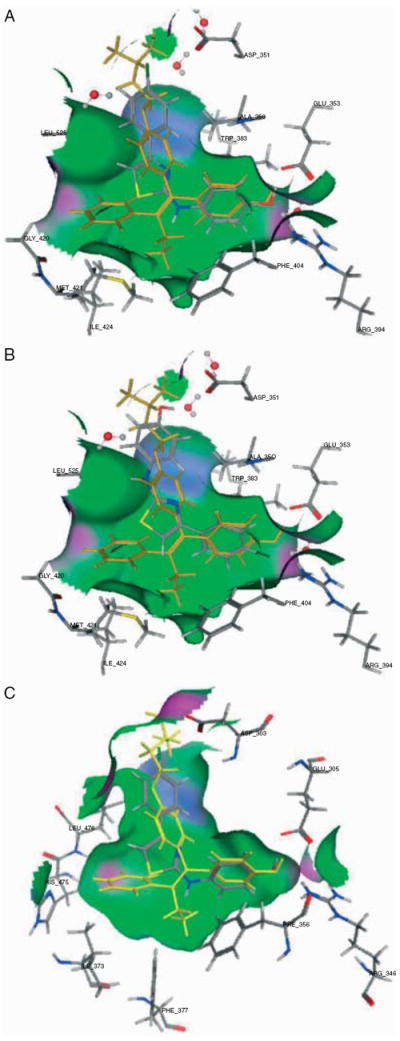
Molecular modeling docking simulations of SKI-II interacting with the ER. SKI-II (atom color) is shown inside the binding cavity of the ERα (panels A and B) and ERβ (panel C) in the context of the 4-OH-Tam (yellow) and key residues of the crystal structures.
SKI-II alters ER domain function
We next set out to determine the mechanism through which SKI-II binding inhibits ER activation. There are two activation regions within ERα, the AF1 and AF2 domains. While these two domains usually act in a synergistic manner, they can also be activated independently of each other (Berry et al. 1990, Tzukerman et al. 1994). In certain contexts (such as differential tissue types), AF1 is the dominant activator, while in others, AF2 is the primary modulator of ER function. Estrogen activates both domains and is a pure ER agonist in all organ systems. The differential activation of AF1 and AF2 accounts for the tissue-type specific effects of tamoxifen, which blocks only the AF2 domain. Therefore, in tissues where AF2 is dominant, such as the breast, tamoxifen acts an antagonist. In tissues where the AF1 domain is dominant, such as endometrial tissues, tamoxifen acts as an agonist (Hall & McDonnell 1999). To determine which ER domain SKI-II affects, we utilized two ER mutant models which preferentially inactivate or knockout either AF1 or AF2. As seen in Fig. 8A and B, SKI-II dose-dependently blocks ER activity of both inactivated AF1 and knocked-out AF2 models. These results suggest that SKI-II binding to the ER creates a global confirmation change that inhibits both the AF1 and AF2 domains.
Figure 8.
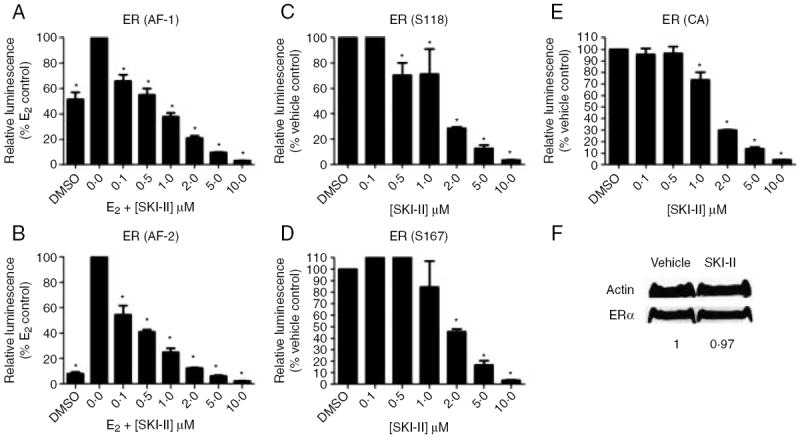
Effect of SKI-II on ER domain function. HEK293 cells were transfected with ER-mutant plasmids containing: (A) AF1 domain knockout (B) AF2 domain knockout, (C) constitutively active SerS118, (D) constitutively active Ser167 or (E) constitutively active ER. Subsequently, the cells were treated with DMSO (control), estrogen (E2) or SKI-II. Cells treated with E2 were set to 100. If no E2 treatment, DMSO control was set to 100. Statistical analysis was performed comparing treatment groups with E2 or DMSO control (*P<0·05). (F) MCF-7 cells were treated with either vehicle control or SKI-II and analyzed by western blot for total ER protein with densitometry analysis (control set to 1). Blots are representative of three independent experiments.
SKI-II may inhibit ER signaling by inhibiting the phosphorylation of ER by kinases that are known to be regulated by S1P, such as AKT and ERK. The ER is phosphorylated and activated at serine 118 (S118) and serine 167 (S167) by ERK and AKT, respectively. Therefore, we used two ER mutants, where serine residues 118 and 167 were changed to glutamic acid residues (S118E and S167E), mimicking the phosphorylated state of the residue. As seen in Fig. 8C and D, SKI-II can inhibit ER activity of each mutant, suggesting that SKI-II acts as an ER antagonist independent of ERK and AKT signaling.
In addition to the aforementioned AF1 and AF2 domain mutant studies, we used a constitutively activated ER to determine whether SKI-II could inhibit ER activity of a tamoxifen-resistant mutant. The constitutively active Y537S ER mutant is an endogenous mutation found in nature that confers resistance to tamoxifen (Zhang et al. 1997). Using this Y537S ER mutant, we found that SKI-II can inhibit a tamoxifen-resistant ER model, suggesting that SKI-II inhibits the ER in a manner distinct from tamoxifen (Fig. 8E). The fact that SKI-II can inhibit the ER in the setting of tamoxifen resistance makes it an attractive therapeutic option.
Discussion
Recent research suggests a critical role for bioactive sphingolipids in the promotion, proliferation and tumorigenesis of ER-positive breast cancer (Meacham et al. 2009). S1P, in particular, has been shown to increase systemic levels of circulating estrogen and plays a role in breast cancer survival and endocrine therapy resistance (Lucki & Sewer 2008, Sukocheva et al. 2009). SK, the main regulator of S1P formation, is an attractive therapeutic target for cancer treatment, though few selective small molecule inhibitors are currently available. The most commonly used inhibitor, SKI-II, is commercially available from multiple sources and has been studied in the context of various cancers, including breast and prostate cancers (French et al. 2006, Leroux et al. 2007).
In this study, we demonstrate, for the first time, the effects of SKI-II on estrogen signaling in ER-positive breast cancer. SKI-II dose-dependently blocks ERE-transcriptional activity, both in ERα-transfected HEK293 cells and endogenously ER-positive MCF-7 cells. Treatment with SKI-II decreased estrogen-induced transcription of the ER-regulated genes PGR and SDF-1. Exogenous competitive binding assays reveal that this inhibitor binds ERα, but not ERβ, with an IC50 value in the high nanomolar range. Molecular modeling, docking and scoring analyses suggest that SKI-II binds the antagonist ligand-binding domain of both ERα and ERβ in a similar fashion as the widely used SERM tamoxifen. Taken together, this data suggest a dual mechanism of action for SKII: inhibition of SK and inhibition of ER signaling.
Our findings that pharmacological inhibition of SK can disrupt estrogen signaling are of therapeutic interest in the development of novel breast cancer therapeutics. Nearly, all SERM- and SERD-treated breast cancers ultimately become drug resistant and there are few second-line therapies available, most of which show systemic toxicity (Dowsett 1996, Kurebayashi 2005). Breast cancer chemotherapeutics, such as doxorubicin and paclitaxel, have a myriad of adverse effects and dose-limiting toxicities. The development of new ER targeting agents to treat ER-positive breast cancer is of growing importance. The S1P signaling pathway is known to crosstalk with various elements of the ER signaling cascade. For example, S1P can increase p38/MAPK, ERK/MAPK and PI3K/AKT activity (Baudhuin et al. 2002, 2004). AKT, ERK and p38 are known to phosphorylate the ER independently of ligand and promote endocrine therapy resistance (Weldon et al. 2002, Park et al. 2003, Thomas et al. 2008). Furthermore, S1P can activate the NF-κB signaling pathway, which is known to mediate superoxide dismutase (SOD). SOD stabilizes the ER complex and enhances ER-DNA binding (Rao et al. 2008). Therefore, the direct binding of SKI-II to the ER may be only one mechanism for the anti-estrogenic action of this drug. It is possible there are other secondary mechanisms for this effect as well. Further study is needed to fully understand the effects of SKI-II on ER signaling.
We have further characterized SKI-II as a potential breast cancer therapeutic agent. SKI-II decreases MCF-7 clonogenic survival, with a low IC50 value of 2·04 μM. The inhibitor has potent anti-proliferative properties, with 10 μM treatment significantly blocking Ki-67 immunostaining, as well as selective toxicity for abnormal breast epithelia, with little effect on normal breast epithelial cell viability. We have also found that SKI-II can be an effective ER antagonist in the setting of tamoxifen resistance, making SKI-II an attractive therapeutic candidate as an endocrine-resistant breast cancer treatment. It has been previously published that SKI-II possesses high oral bioavailability and little systemic toxicity. Our in vitro results here, combined with previous in vivo studies, indicate that SKI-II is a promising therapeutic agent for the treatment of ER-positive breast cancer.
Acknowledgments
Funding
This work was supported by The Center for Bioenvironmental Research at Tulane and Xavier Universities, National Institutes of Health Grant NIDDK and the PhRMA Foundation Paul Calabresi Research Fellowship (J W A).
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Ader I, Brizuela L, Bouquerel P, Malavaud B, Cuvillier O. Sphingosine kinase 1: a new modulator of hypoxia inducible factor 1α during hypoxia in human cancer cells. Cancer Research. 2008;68:8635–8642. doi: 10.1158/0008-5472.CAN-08-0917. [DOI] [PubMed] [Google Scholar]
- Antoon JW, Liu J, Gestaut MM, Burow ME, Beckman BS, Foroozesh M. Design, synthesis, and biological activity of a family of novel ceramide analogues in chemoresistant breast cancer cells. Journal of Medicinal Chemistry. 2009;52:5748–5752. doi: 10.1021/jm9009668. [DOI] [PubMed] [Google Scholar]
- Antoon JW, White MD, Meacham WD, Slaughter EM, Muir SE, Elliott S, Rhodes LV, Hasina AB, Wiese TE, Smith CD, et al. Anti-estrogenic effects of the novel sphingosine kinase-2 inhibitor ABC294640. Endocrinology. 2010;151:5124–5135. doi: 10.1210/en.2010-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudhuin LM, Cristina KL, Lu J, Xu Y. Akt activation induced by lysophosphatidic acid and sphingosine-1-phosphate requires both mitogen-activated protein kinase kinase and p38 mitogen-activated protein kinase and is cell-line specific. Molecular Pharmacology. 2002;62:660–671. doi: 10.1124/mol.62.3.660. [DOI] [PubMed] [Google Scholar]
- Baudhuin LM, Jiang Y, Zaslavsky A, Ishii I, Chun J, Xu Y. S1P3-mediated Akt activation and cross-talk with platelet-derived growth factor receptor (PDGFR) FASEB Journal. 2004;18:341–343. doi: 10.1096/fj.03-0302fje. [DOI] [PubMed] [Google Scholar]
- Beresford MJ, Wilson GD, Makris A. Measuring proliferation in breast cancer: practicalities and applications. Breast Cancer Research. 2006;8:216. doi: 10.1186/bcr1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry M, Metzger D, Chambon P. Role of the two activating domains of the oestrogen receptor in the cell-type and promoter-context dependent agonistic activity of the anti-oestrogen 4-hydroxytamoxifen. EMBO Journal. 1990;9:2811–2818. doi: 10.1002/j.1460-2075.1990.tb07469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielawski J, Szulc ZM, Hannun YA, Bielawska A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography–tandem mass spectrometry. Methods. 2006;39:82–91. doi: 10.1016/j.ymeth.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Boue SM, Tilghman SL, Elliott S, Zimmerman MC, Williams KY, Payton-Stewart F, Miraflor AP, Howell MH, Shih BY, Carter-Wientjes CH, et al. Identification of the potent phytoestrogen glycinol in elicited soybean (Glycine max) Endocrinology. 2009;150:2446–2453. doi: 10.1210/en.2008-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratton MR, Frigo DE, Vigh-Conrad KA, Fan D, Wadsworth S, McLachlan JA, Burow ME. Organochlorine-mediated potentiation of the general coactivator p300 through p38 mitogen-activated protein kinase. Carcinogenesis. 2009;30:106–113. doi: 10.1093/carcin/bgn213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Wouters BG. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Research. 1999;59:1391–1399. [PubMed] [Google Scholar]
- Burow ME, Weldon CB, Tang Y, Navar GL, Krajewski S, Reed JC, Hammond TG, Clejan S, Beckman BS. Differences in susceptibility to tumor necrosis factor α-induced apoptosis among MCF-7 breast cancer cell variants. Cancer Research. 1998;58:4940–4946. [PubMed] [Google Scholar]
- Burstein HJ, Polyak K, Wong JS, Lester SC, Kaelin CM. Ductal carcinoma in situ of the breast. New England Journal of Medicine. 2004;350:1430–1441. doi: 10.1056/NEJMra031301. [DOI] [PubMed] [Google Scholar]
- Chu KC, Anderson WF. Rates for breast cancer characteristics by estrogen and progesterone receptor status in the major racial/ethnic groups. Breast Cancer Research and Treatment. 2002;74:199–211. doi: 10.1023/A:1016361932220. [DOI] [PubMed] [Google Scholar]
- Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind S, Spiegel S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature. 1996;381:800–803. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- Doll F, Pfeilschifter J, Huwiler A. Prolactin upregulates sphingosine kinase-1 expression and activity in the human breast cancer cell line MCF7 and triggers enhanced proliferation and migration. Endocrine-Related Cancer. 2007;14:325–335. doi: 10.1677/ERC-06-0050. [DOI] [PubMed] [Google Scholar]
- Dowsett M. Endocrine resistance in advanced breast cancer. Acta Oncologica. 1996;35(Supplement 5):91–95. doi: 10.3109/02841869609083979. [DOI] [PubMed] [Google Scholar]
- French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Research. 2003;63:5962–5969. [PubMed] [Google Scholar]
- French KJ, Upson JJ, Keller SN, Zhuang Y, Yun JK, Smith CD. Antitumor activity of sphingosine kinase inhibitors. Journal of Pharmacology and Experimental Therapeutics. 2006;318:596–603. doi: 10.1124/jpet.106.101345. [DOI] [PubMed] [Google Scholar]
- Fuqua SA, Cui Y. Targeting the estrogen receptor in clinical breast cancer. Breast Disease. 2002;15:3–11. doi: 10.3233/bd-2002-15102. [DOI] [PubMed] [Google Scholar]
- Gaglia P, Bernardi A, Venesio T, Caldarola B, Lauro D, Cappa AP, Calderini P, Liscia DS. Cell proliferation of breast cancer evaluated by anti-BrdU and anti-Ki-67 antibodies: its prognostic value on short-term recurrences. European Journal of Cancer. 1993;29A:1509–1513. doi: 10.1016/0959-8049(93)90284-M. [DOI] [PubMed] [Google Scholar]
- Gail MH, Anderson WF, Garcia-Closas M, Sherman ME. Absolute risk models for subtypes of breast cancer. Journal of the National Cancer Institute. 2007;99:1657–1659. doi: 10.1093/jnci/djm228. [DOI] [PubMed] [Google Scholar]
- Geffroy N, Guedin A, Dacquet C, Lefebvre P. Cell cycle regulation of breast cancer cells through estrogen-induced activities of ERK and Akt protein kinases. Molecular and Cellular Endocrinology. 2005;237:11–23. doi: 10.1016/j.mce.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Guillermet-Guibert J, Davenne L, Pchejetski D, Saint-Laurent N, Brizuela L, Guilbeau-Frugier C, Delisle MB, Cuvillier O, Susini C, Bousquet C. Targeting the sphingolipid metabolism to defeat pancreatic cancer cell resistance to the chemotherapeutic gemcitabine drug. Molecular Cancer Therapeutics. 2009;8:809–820. doi: 10.1158/1535-7163.MCT-08-1096. [DOI] [PubMed] [Google Scholar]
- Hall JM, Korach KS. Stromal cell-derived factor 1, a novel target of estrogen receptor action, mediates the mitogenic effects of estradiol in ovarian and breast cancer cells. Molecular Endocrinology. 2003;17:792–803. doi: 10.1210/me.2002-0438. [DOI] [PubMed] [Google Scholar]
- Hall JM, McDonnell DP. The estrogen receptor β-isoform (ERβ) of the human estrogen receptor modulates ERα transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology. 1999;140:5566–5578. doi: 10.1210/en.140.12.5566. [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nature Reviews Molecular Cell Biology. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- Hartman J, Strom A, Gustafsson JA. Estrogen receptor β in breast cancer – diagnostic and therapeutic implications. Steroids. 2009;74:635–641. doi: 10.1016/j.steroids.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Horwitz KB, Koseki Y, McGuire WL. Estrogen control of progesterone receptor in human breast cancer: role of estradiol and antiestrogen. Endocrinology. 1978;103:1742–1751. doi: 10.1210/endo-103-5-1742. [DOI] [PubMed] [Google Scholar]
- Katzenellenbogen BS, Frasor J. Therapeutic targeting in the estrogen receptor hormonal pathway. Seminars in Oncology. 2004;31:28–38. doi: 10.1053/j.seminoncol.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Kurebayashi J. Resistance to endocrine therapy in breast cancer. Cancer Chemotherapy and Pharmacology. 2005;56(Supplement 1):39–46. doi: 10.1007/s00280-005-0099-z. [DOI] [PubMed] [Google Scholar]
- Leroux ME, Auzenne E, Evans R, Hail N, Jr, Spohn W, Ghosh SC, Farquhar D, McDonnell T, Klostergaard J. Sphingolipids and the sphingosine kinase inhibitor, SKI II, induce BCL-2-independent apoptosis in human prostatic adenocarcinoma cells. Prostate. 2007;67:1699–1717. doi: 10.1002/pros.20645. [DOI] [PubMed] [Google Scholar]
- Lucki NC, Sewer MB. Multiple roles for sphingolipids in steroid hormone biosynthesis. Sub-Cellular Biochemistry. 2008;49:387–412. doi: 10.1007/978-1-4020-8831-5_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham WD, Antoon JW, Burow ME, Struckhoff AP, Beckman BS. Sphingolipids as determinants of apoptosis and chemoresistance in the MCF-7 cell model system. Experimental Biology and Medicine. 2009;234:1253–1263. doi: 10.3181/0902-MR-77. [DOI] [PubMed] [Google Scholar]
- Nava VE, Hobson JP, Murthy S, Milstien S, Spiegel S. Sphingosine kinase type 1 promotes estrogen-dependent tumorigenesis of breast cancer MCF-7 cells. Experimental Cell Research. 2002;281:115–127. doi: 10.1006/excr.2002.5658. [DOI] [PubMed] [Google Scholar]
- Park MT, Choi JA, Kim MJ, Um HD, Bae S, Kang CM, Cho CK, Kang S, Chung HY, Lee YS, et al. Suppression of extracellular signal-related kinase and activation of p38 MAPK are two critical events leading to caspase-8- and mitochondria-mediated cell death in phytosphingosine-treated human cancer cells. Journal of Biological Chemistry. 2003;278:50624–50634. doi: 10.1074/jbc.M309011200. [DOI] [PubMed] [Google Scholar]
- Punglia RS, Morrow M, Winer EP, Harris JR. Local therapy and survival in breast cancer. New England Journal of Medicine. 2007;356:2399–2405. doi: 10.1056/NEJMra065241. [DOI] [PubMed] [Google Scholar]
- Rao AK, Ziegler YS, McLeod IX, Yates JR, Nardulli AM. Effects of Cu/Zn superoxide dismutase on estrogen responsiveness and oxidative stress in human breast cancer cells. Molecular Endocrinology. 2008;22:1113–1124. doi: 10.1210/me.2007-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes LV, Muir SE, Elliott S, Guillot LM, Antoon JW, Penfornis P, Tilghman SL, Salvo VA, Fonseca JP, Lacey MR, et al. Adult human mesenchymal stem cells enhance breast tumorigenesis and promote hormone independence. Breast Cancer Research and Treatment. 2009;121:293–300. doi: 10.1007/s10549-009-0458-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckhaberle E, Rody A, Engels K, Gaetje R, von Minckwitz G, Schiffmann S, Grosch S, Geisslinger G, Holtrich U, Karn T, et al. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Research and Treatment. 2008;112:41–52. doi: 10.1007/s10549-007-9836-9. [DOI] [PubMed] [Google Scholar]
- Sankala HM, Hait NC, Paugh SW, Shida D, Lepine S, Elmore LW, Dent P, Milstien S, Spiegel S. Involvement of sphingosine kinase 2 in p53-independent induction of p21 by the chemotherapeutic drug doxorubicin. Cancer Research. 2007;67:10466–10474. doi: 10.1158/0008-5472.CAN-07-2090. [DOI] [PubMed] [Google Scholar]
- Starborg M, Gell K, Brundell E, Hoog C. The murine Ki-67 cell proliferation antigen accumulates in the nucleolar and heterochromatic regions of interphase cells and at the periphery of the mitotic chromosomes in a process essential for cell cycle progression. Journal of Cell Science. 1996;109:143–153. doi: 10.1242/jcs.109.1.143. [DOI] [PubMed] [Google Scholar]
- Struckhoff AP, Bittman R, Burow ME, Clejan S, Elliott S, Hammond T, Tang Y, Beckman BS. Novel ceramide analogs as potential chemotherapeutic agents in breast cancer. Journal of Pharmacology and Experimental Therapeutics. 2004;309:523–532. doi: 10.1124/jpet.103.062760. [DOI] [PubMed] [Google Scholar]
- Sukocheva O, Wang L, Verrier E, Vadas MA, Xia P. Restoring endocrine response in breast cancer cells by inhibition of the sphingosine kinase-1 signaling pathway. Endocrinology. 2009;150:4484–4492. doi: 10.1210/en.2009-0391. [DOI] [PubMed] [Google Scholar]
- Taha TA, Hannun YA, Obeid LM. Sphingosine kinase: biochemical and cellular regulation and role in disease. Journal of Biochemistry and Molecular Biology. 2006;39:113–131. doi: 10.5483/bmbrep.2006.39.2.113. [DOI] [PubMed] [Google Scholar]
- Thomas RS, Sarwar N, Phoenix F, Coombes RC, Ali S. Phosphorylation at serines 104 and 106 by Erk1/2 MAPK is important for estrogen receptor-α activity. Journal of Molecular Endocrinology. 2008;40:173–184. doi: 10.1677/JME-07-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifilieff A, Baur F, Fozard JR. Role of sphingosine-1-phosphate (S1P) and the S1P(2) receptor in allergen-induced, mast cell-dependent contraction of rat lung parenchymal strips. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2009;380:303–309. doi: 10.1007/s00210-009-0438-4. [DOI] [PubMed] [Google Scholar]
- Tzukerman MT, Esty A, Santiso-Mere D, Danielian P, Parker MG, Stein RB, Pike JW, McDonnell DP. Human estrogen receptor transactivational capacity is determined by both cellular and promoter context and mediated by two functionally distinct intramolecular regions. Molecular Endocrinology. 1994;8:21–30. doi: 10.1210/me.8.1.21. [DOI] [PubMed] [Google Scholar]
- Wang F, Van Brocklyn JR, Edsall L, Nava VE, Spiegel S. Sphingosine-1-phosphate inhibits motility of human breast cancer cells independently of cell surface receptors. Cancer Research. 1999;59:6185–6191. [PubMed] [Google Scholar]
- Weldon CB, Scandurro AB, Rolfe KW, Clayton JL, Elliott S, Butler NN, Melnik LI, Alam J, McLachlan JA, Jaffe BM, et al. Identification of mitogen-activated protein kinase kinase as a chemoresistant pathway in MCF-7 cells by using gene expression microarray. Surgery. 2002;132:293–301. doi: 10.1067/msy.2002.125389. [DOI] [PubMed] [Google Scholar]
- Yan G, Chen S, You B, Sun J. Activation of sphingosine kinase-1 mediates induction of endothelial cell proliferation and angiogenesis by epoxyeicosatrienoic acids. Cardiovascular Research. 2008;78:308–314. doi: 10.1093/cvr/cvn006. [DOI] [PubMed] [Google Scholar]
- Zhang QX, Borg A, Wolf DM, Oesterreich S, Fuqua SA. An estrogen receptor mutantwith strong hormone-independent activity from a metastatic breast cancer. Cancer Research. 1997;57:1244–1249. [PubMed] [Google Scholar]