

Abstract
We investigate the hypothesis that oxidative damage of the cerebral vascular barrier interface (the blood brain barrier, BBB) causes the development of mild traumatic brain injury (mTBI) during primary blast wave spectrum. The underlying biochemical and cellular mechanisms of this vascular layer-structure injury are examined in a novel animal model of shock tube. We first established that low frequency (123 kPa) single or repeated shock wave causes BBB/brain injury through biochemical activation by acute mechanical force that occurs at 6–24 hrs after the exposure. This biochemical damage of the cerebral vasculature is initiated by the induction of free radical generating enzymes NADPH oxidase (NOX1) and inducible nitric oxide synthase (iNOS). Induction of these enzymes by shock wave exposure correlated well with the signatures of oxidative and nitrosative damage (4HNE/3NT) and reduction of the BBB tight junction (TJ) proteins occludin, claudin-5 and zonula occluden 1 (ZO-1) in the brain microvessel. In parallel with TJ protein disruption, the perivascular unit was significantly diminished by single or repeated shock wave exposure coinciding with the kinetic profile. Loosening of the vasculature and perivascular unit was mediated by oxidative stress-induced activation of matrix metalloproteinases and fluid channel aquaporin-4, promoting vascular fluid cavitation/edema, enhanced leakiness of the BBB and progression of neuroinflammation. The BBB leakiness and neuroinflammation were functionally demonstrated in an in vivo model by enhanced permeability of Na-Fl/EB low molecular weight tracers and the infiltration of immune cells across the BBB. The detection of brain cell matters NSE/S100β in the blood samples validated the neuro-astroglial injury in shock wave TBI. Our hypothesis that cerebral vascular injury occurring prior to the development of neurological disorders in mild TBI was further confirmed by the activation of caspase-3 and cell apoptosis mostly around the perivascular region. Thus, induction of oxidative stress and MMPs activation by shock wave underlies the mechanisms of cerebral vascular BBB leakage and neuroinflammation.
Keywords: Mild Traumatic Brain Injury, Blood-brain barrier, Oxidative stress, Perivascular unit, Neuroinflammation, Primary blast
INTRODUCTION
Traumatic brain injury (TBI) is characterized by physical brain injury as a result of acceleration-deceleration, resulting frequently from impact with an immobile object that often leads to cognitive deficits and impairment of behavior. Unlike casualties suffering from moderate to severe TBI, victims diagnosed with mild TBI remain conscious, and typical symptoms include headache, confusion, dizziness, memory impairment, and behavioral changes. U.S. soldiers exposed to blast wave pressure and combat experiences without any physical brain injury during Middle-east wars are commonly diagnosed with mild TBI and post-traumatic stress disorder (1). Mild TBI is the most frequent form of trauma among deployed military populations (2). In recent military conflicts, the repeated exposure of low levels of blast overpressure from improvised explosive devices (IEDs) is believed to account for majority of the mTBI. Ironically, most of these soldiers exposed to low intensity blast remain conscious, and many of these soldiers are frequently re-deployed in the war zone without proper diagnosis. This in turn endangers these military personnel to experience consecutive multiple blast exposures aggravating already existing medical condition (3, 4). These subjects undergo severe mental stress and often develop a misuse of alcohol and other drug of abuse (5, 6), thereby the chances of mental health complications such as post-traumatic stress disorder (PTSD) increases in the long-term (7). It appears that there is strong overlap between symptoms of chronic mTBI and PTSD among many veterans. Re-exposed to multiple blasts wave pressure, which has become a major challenge for military healthcare system. Psychological and physiological stress by repeated blast wave exposure is believed to contribute significantly for the development of PTSD in chronic mTBI resulting in alterations of cognitive behaviors (4, 8). These findings are further supported by recent demonstration in animal model that low level of shock wave can cause cognitive deficits in short-term learning and memory (9).
Intriguingly, epidemiological findings indicated that disruption of the blood-brain barrier (BBB) is involved in shock wave-induced mTBI and neurological disorders like PTSD (10). However, such cohort studies lack the understanding of the underlying mechanisms. Thus, uncovering the molecular, biochemical and cellular mechanisms of shock wave-brain interactions leading to mTBI require a careful investigation. We have shown that cerebral vascular integrity (the BBB) is very sensitive to oxidative stress during substance of abuse (11, 12). Ensuing oxidative damage of the BBB leads to neuroinflammation and neuronal degeneration (13–15). Understanding of underlying molecular and biochemical mechanisms will help define the characteristic biomarkers of cerebral vascular injury and be able to formulate a preventive strategy to mitigate the adverse acute effects of blast exposure and related chronic neurological complications. This includes the identification of blast simulated shock wave range (shock wave frequencies) in causing such physiological deficits and the types of mechanical or biochemical injury.
Blast injuries are classified as primary (pure blast), secondary (interaction with shrapnel or fragments), tertiary (impact with environmental structures) and/or quarternary (toxic gases) (16–18). The 15-point Glascow Coma Scale (GCS) (19) defines severity of traumatic brain injury as mild TBI (13–15), moderate TBI (9–12), severe TBI (3–8), and vegetative state TBI (3). Mild TBI (mTBI) considered in this work is defined as loss of consciousness for less than 24 hours (20). At the initial stage of the present study we tentatively identified the pressure ranges of 90 – 150 kPa, 150 – 230 kPa, and 230 – 350 kPa as corresponding to the mild, moderate, and severe TBI in a rodent model. All tests were conducted using 9 inch square cross section shock tube at the US Army-UNL Center for Trauma Mechanics facility (Figure 1). The detailed description of the shock wave generator, the blast wave spectral content, the numerical models of skull and brain responses to blast loading and corresponding transition of blast energy are provided elsewhere (21–23). We hypothesize that induction of oxidative stress by single (only one time shock wave pressure exposure) and repeated (more than one time shock wave pressure exposure on the same animal) exposure to low intensity blast overpressure initiates the cerebral vascular injury (the BBB damage) and neuroinflammation, which is accompanied by the release of mTBI-specific biomarkers to the blood circulation. Thus, the disruption of the barrier interface (BBB) is a key event in the acute phase of mTBI development. We demonstrate the induction of free radical generating enzymes, oxidative damage markers, BBB leakage, perivascular regulated by matrix metalloproteinases and fluid channel activator aquaporin-4 ultimately lead to neuroinflammation. These pathological processes can be manifested as long-term neurological disorders.
Figure 1. Blast wave simulation and testing facility at University of Nebraska, Lincoln.
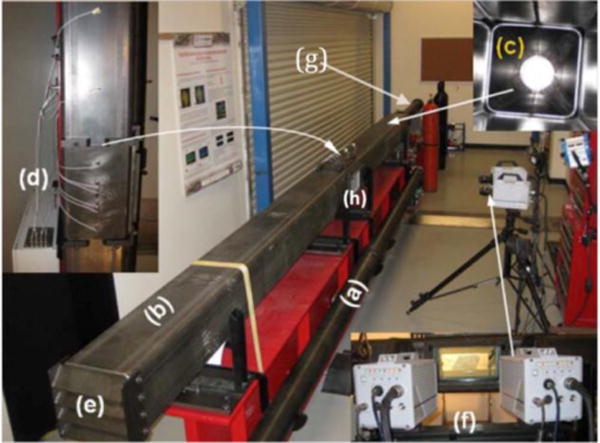
(a) 4″ cylindrical shock tube; (a) 9″ square shock tube; (c) transition section; (d) pressure sensor array; (e) adjustable end reflector; and (f) Photron SA1.1 high speed cameras, (g) driver section (h) test section.
MATERIALS AND METHODS
Reagents
Primary antibodies of rabbit anti-NOX1, anti-iNOS, anti-4HNE, anti-Claudin-5, anti-MMP-3, anti-MMP-9, anti-Aquaporin-4 (AQP-4), anti-Iba1, anti-caspase3; mouse anti-GLUT1, anti-3NT, anti-MMP-2; sheep anti-von Willibrand factor (vWF); and goat anti-Iba1 were purchased from Abcam (Cambridge, MA). Antibodies of mouse anti-Occludin was purchased from Invtrogen (Carlsbad, CA, USA); rabbit anti-ZO-1 was from US Biological (Massachusetts, MA); mouse anti-β-actin was from Millipore (Billerica, MA); rabbit anti-XOX was from Santa Cruz Biotechnology (Santa Cruz, CA, USA.) and mouse anti-PDGFR-β was from eBiosciences (San Diego, CA, USA). All secondary Alexa Fluor conjugated antibodies and Fluo-3 were purchased from Invitrogen. The ELISA kits for Neuron-specific enolase (NSE) and S100β were from Alpha Diagnostic (San Antonio, Texas, USA) and Abnova (Walnut, CA, USA) respectively. Evans Blue (EB) and Sodium Fluorescein (NaFl) were purchased from Sigma-Aldrich (St. Louis, MO).
Primary blast wave exposure to animals
Nine-weeks old male Sprague-Dawley rats were purchased and maintained in sterile cages under pathogen-free conditions in accordance with National Institutes of Health (NIH) ethical guidelines for care of laboratory animals, and the Institutional Animal Care Use Committee at animal facility of University of Nebraska Lincoln. First, we determined the effects of various intensity blasts at 123, 190, 230 and 250 kPa on the severity of brain injury. For this, we exposed 11th week old rats (6 animals per intensity wave) to these range of intensity blasts and we evaluated the blast induced brain pathology. Second, since 123 kPa (mTBI) intensity did cause any visible brain injury, we determined the kinetic profile of one time 123 kPa intensity on the underlying mechanisms of cerebral vascular and brain injuries at 1, 6, 24, 48 hours, and 8 days of post-exposure in same age group animals (11 weeks old rat). Third, after establishing the time-dependent effect at 6–24 hours following 123 kPa exposure, we then examined the possible exacerbated effect of repeated exposure of 123 kPa intensity at 24 hour. That is, eleven week-old rats (12 rats) were exposed to 123 kPa intensity and then 24 hours after the first exposure, six animals out of the 12 rats were re-exposed to 123 kPa intensity and left them for another 24 hours, which is termed here as repeated exposure or 24 hrR group. Animals consisting of control (unexposed), 24 hr exposure, and 24 hrR (6 each) were euthanized with ketamine/xylazine mixture. Brain tissues were dissected out, embedded in OCT (optimal cutting temperature) and kept frozen until we analyzed the mechanisms of injuries by immunohistochemical staining and Western blotting.
Reactive Oxygen Species (ROS) detection
Brain cortical tissues from the various time points of 123 kPa blasts were washed in KDD (Krebs Hepes buffer and the chelators DF (25 μM) and DETC (5 μM)) buffer and Krebs Hepes buffer contains 99mM NaCl, 4.69mM KCl, 2.5mM CaCl2·2H2O, 1.2 mM MgSO4·7H2O, 25mM NaHCO3, 1.03mMKH2PO4, 5.6 mM d-(+)-glucose, and 20mM Na-Hepes. The tissue samples were cut into small pieces with nonmetallic (plastic) scalpel, and suspended (approximately 100 mg/sample) in spin-probe solution for detection of reactive oxygen species (ROS). Tissues were totally submerged in the spin probe solution (1.0 ml) in 12 well plates before incubation at 37°C for 30 min. Samples aspirated in a 1.0 ml syringe were snap-frozen in liquid nitrogen until detection of ROS by electron paramagnetic resonance (EPR; Bruker Escan E-box tabletop model, Serial 0264). The EPR was set at field sweep in lagtime that read 10°-10 scans for an accumulative, or a “time course” protocol: microwave bridge → attenuator 4.0 for liquid samples, 17 for frozen samples; number of scans per sample 10; hall→center field 3450.189 Gauss (G), sweep width 60 G, static field 3451.189 G. We used the spin-probe solution CMH (Noxygen, NOX-2.3–100mg; Axxora ALX-430-117-M010) in KDD buffer, which reacted with intracellular superoxide. Data are presented as cumulative detection of both the hydroxyl and the superoxide free radicals and values are expressed as amplitude of signal per mg tissue weight.
Immunofluorescence and microscopy
Intact external cerebral capillary vessels were surgically removed from the brain and smeared onto the slides. Adhesion and migration of Fluo-3 labeled immune cells were detected in these vessels directly under fluorescent microscope. Brain tissue sections (8 μm thickness) containing the external and internal capillaries were used for immunofluorescence staining. Tissue sections on glass slides were washed with PBS, fixed in acetone-methanol (1:1 v/v) fixative (10 minutes at 95°C), and incubated for 10 minutes at 25°C in 3% formaldehyde PBS. Washed tissue slides were then blocked with 3% bovine serum albumin at 25°C for 1 hr, in the absence of Triton X-100, for occludin, claudin-5 and ZO-1 staining or in the presence of 0.1% Triton X-100 for all other antibodies, followed by overnight incubation at 4°C with respective primary antibodies. After washing with PBS, tissue slides were incubated with Alexa Fluor 488 or 594 conjugated with anti-mouse, anti-rabbit or anti-sheep immunoglobulin G (IgG) for 1 hr, mounted with immunomount containing DAPI (Invitrogen), and microphotographs were captured using fluorescent microscope Eclipse TE2000-U (Nikon, Melville, NY).
Western blotting
Cortical brain tissues and brain microvessels were lysed with CellLytic-M (Sigma) for 30 min at 4°C, centrifuged at 14000 × g, and then homogenate protein concentrations were estimated by bicinchoninic acid (BCA) method (Thermo Scientific, Rockford, IL). Protein load was 20 μg/lane in 4–15% SDS-PAGE gradient gels (Thermo Scientific). Molecular size separated proteins were then transferred onto nitrocellulose membranes, blocked with superblock (Thermo Scientific), and incubated overnight with respective primary antibodies at 4 °C, followed by incubation with horse-radish peroxidase conjugated secondary antibodies for 1 hr. Immunoreactive bands were detected by West Pico chemiluminescence substrate (Thermo Scientific). Data were quantified as arbitrary densitometry intensity units using the ImageJ software package.
Zymography of MMP Activity
Zymography was performed to determine MMPs activities in the rat brain cortical tissue lysates using a method similar to that previously described (15). For gelatin or casein zymography, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed by loading 40 μg protein in a 10% polyacrylamide gel containing 0.1% gelatin or a 12% gel containing 0.1% casein (Bio-rad, Hercules, CA) at 125 V for 90 minutes at 4°C. The gels were soaked in renaturing buffer (Invitrogen) for 30 minutes at room temperature and incubated in developing buffer (Invitrogen) for 30 minutes at room temperature and for overnight at 37°C. Then the gels were stained with 0.5% Coomassie brilliant blue R-250 in 40% methanol and 10% acetic acid for 1 hour after rinsed in distilled water. For destaining, 40% methanol and 10% acetic acid solution was used. The MMP activities show as clear bands of lysis against a dark background of stained gelatin or casein.
Quantitative RT-PCR
Total RNA from cortex was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA). RNA yield and quality were checked using a NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE); and for all samples, absorbance ratio of 260/280 was 1.9 to 2.0. For each sample, cDNA was generated with random hexamers and Moloney murine leukemia virus reverse transcriptase (Invitrogen). Real-time quantitative PCR was performed with cDNA using StepOnePlusTM Real-Time PCR Systems (Applied Biosystems) by employing StepOneTM software v2.0 detection system. Rat NOX1, iNOS and GAPDH expression were analyzed using TaqMan gene expression assays and gene quantification was performed using the standard curve method as described in the software user manual. All PCR reagents and primers were obtained from Applied Biosystems and primer IDs were as follows: NOX1: Rn00583793_m1; and iNOS: Rn00586652_m1. For endogenous control, each gene expression was normalized to GAPDH (Rn01775763_g1).
In vivo cell infiltration into the BBB
Rat femurus bone marrow cells were isolated under sterile conditions, differentiated to monocytes with specific cell differentiating media containing MCSF (macrophage colony stimulating factor), and labeled with Fluo-3. Labeled cells were infused into the right common carotid artery (2 × 106 cells per rat) using 27.5 G needle (see Alikunju et al., (13), for the detailed protocol). Adhesion and infiltration of these cells were detected in intact brain microvessels under fluorescent microscope.
BBB permeability assay
The effect of blast exposure (123 kPa peak overpressure) on BBB permeability was examined by sodium fluorescein (NaFl) and Evans Blue (EB) tracer dye mixtures (5 μM each) using our animal model of infusion into the common carotid artery (13, 24). Two hours after the infusion of NaFl/EB mixtures directly into right common carotid artery (CCA), animals were decapitated, brains removed, dissected, weighed, and homogenized in 600 μl 7.5% (w/v) tri-chloro acetic acid (TCA). Resulting suspensions were divided into two 300 μl aliquots. One aliquot was neutralized with 50 μl of 5 N NaOH and fluorescence was measured on a GENios microplate reader (excitation 485 nm, emission 535 nm) to determine NaFl concentration. The second aliquot was centrifuged for 10 min at 10,000 rpm and 4°C, and the EB concentration in the supernatant was measured by absorbance spectroscopy at 620 nm. Standard curve was generated using a serial dilutions of EB/NaFl solution in 7.5% TCA.
Enzyme-linked immunosorbent assay (ELISA)
In order to determine the cerebral vascular BBB leakage as well as neuronal damage by shock wave, we analyzed the neuronal and astrocyte specific marker proteins in blood serum samples from control and animals exposed to the blast with 123 kPa peak overpressure at different time points. These experiments were performed using the neuron specific enolase (NSE) (Alpha Diagnostic, San Antonio, Texas, USA) and S100β ELISA kits (Abnova, Walnut, CA, USA) following manufacturer’s instructions.
TUNEL Assay
Using the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) (Roche diagnostics, IN) assay kit, cell apoptosis was determined in tissue sections per manufacturer’s instructions.
Data analysis
All results values are expressed as the mean ± SEM. Statistical analysis of the data was performed using Graphpad Prism V5 (Sorrento Valley, CA). Comparisons between samples were performed by one-way ANOVA with Dunnett’s post-hoc tests. Differences were considered significant at p < 0.05.
RESULTS
Determination of shock wave range and exposure time
The objective of the present study was to establish the underlying biochemical mechanisms of mTBI caused by low frequency shock wave exposure. To rule out the role of direct mechanical injury for development of mTBI, we determine the nature of cerebral vascular and the brain injury following exposure to various frequencies of 123, 190, 230 and 250 kPa shock wave peak overpressure. We found that there was no visible physical injury of the brain or the cerebral vasculature with the low frequencies of shock wave exposure (Figure 2). The higher shock waves appear to cause minor injury at the 3rd ventricle. Based on this establishment, we determined the kinetic profile of the cerebral vascular and brain injuries at 1, 6, 24 and 48 hours, and 8 days post-exposure in animals subjected to a single blast with 123 kPa peak overpressure. Repeated injury scenario was tested in one group of rats exposed to 123 kPa blast with 24 hours of interval twice (24 hrR group).
Figure 2. Mechanical cerebrovascular injury by different blasts exposure.
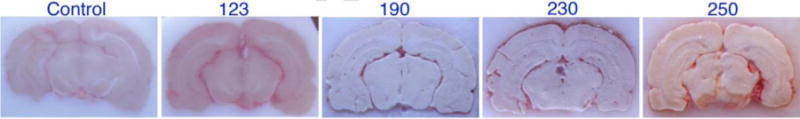
The figure shows neurovascular or brain damage due to different blast exposure of mTBI at 123, 190, 230 and 250 kPa after 24 hr of post-exposure.
Oxidative stress has critical role in brain damage mTBI
Initially we focused on the induction of free radical generating enzymes, i.e. NADPH oxidase (NOX1) and inducible nitric oxide synthase (iNOS). Induction of these enzymes in brain microvessel was compared with the corresponding oxidative/nitrosative damage markers, 4-hydroxynonenal (4HNE) and 3-nitrotyrosine (3NT). Our data indicated that maximum level of oxidative damage signature, 4HNE was observed within the time frame of 6 – 24 hrs after single 123 kPa shock wave exposure (Figure 3 A–E). However, induction of NOX1 persisted perhaps beyond 48 hr and appeared to be tapered down at 8 days similar to the effect seen at 1 hr after the blast exposure. The mechanism of this interesting discrepancy results is not understood in the present study. We can only postulate that even if NOX1 induction persisted beyond 48 hr, an active repair mechanism of the oxidative damage may play a role in manifesting this discrepancy result. On the other hand, induction of iNOS paralleled the extent of nitrosative damage marker 3NT in all the time points studied, indicating a maximum increase levels within 6 – 24 hrs after the 123 kPa shock wave exposure (Figure 3 A–E).
Figure 3. mTBI induces free radical adducts in rat brain.
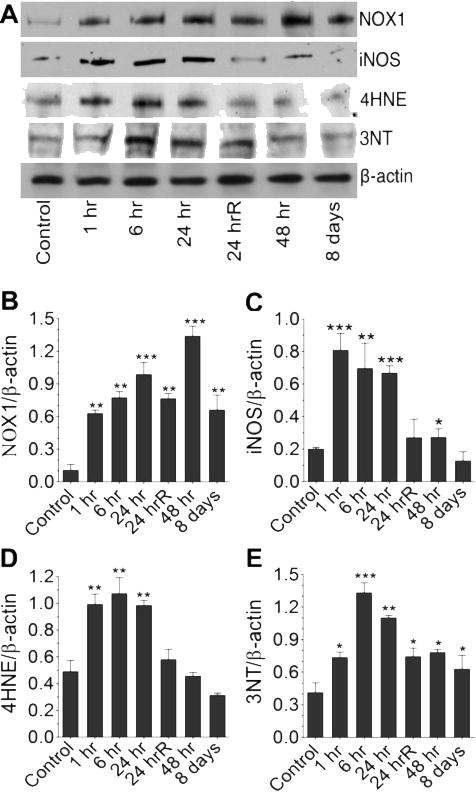
(A) Western blot analyses of NOX1, iNOS, 4HNE and 3NT in the whole rat brain homogenates at different time points after exposure to blast with 123 kPa peak. (B–E) Bar graphs show the results that are expressed as ratio of NOX1/iNOS/4HNE/3NT to that of β-actin band. Values are mean ±SEM (n = 4). *p<0.05; **p<0.01, ***p<0.001 versus control.
These quantitative data on the levels of the enzymes and oxidative/nitrosative damage markers were further validated by the qualitative analyses of the respective proteins and free radical adducted markers as demonstrated by immunofluorescent staining in the brain microvessel (Figure 4A). These data suggest that in mTBI oxidative/nitrosative stress has a critical role in the cerebrovascular inflammatory damage in a single mild TBI shock wave exposure. Our putative conclusion was further verified by direct detection of reactive oxygen species (ROS) levels in the brain cortical region using electron paramagnetic resonance (EPR) method, which confirmed that mTBI significantly enhanced the production of ROS (Figure 4B). We then evaluated the molecular mechanisms of mTBI-induced induction of NOX1 and iNOS by analyzing the changes in mRNA levels (transcription level) of NOX1 and iNOS using quantitative RT-PCR using TaqMan primers. Interestingly, increase in NOX1 mRNA levels by single 123 kPa exposure was further elevated by repeated exposure at 24 hr (Figure 4C), but there was no significant changes in iNOS mRNA level (data not shown). This data suggest that up-regulation of NOX1 gene level and translational stability of its mRNA levels could be responsible for the elevation of protein levels.
Figure 4. Primary blast induced oxidative and nitrosative stress in the rat brain microvessels.
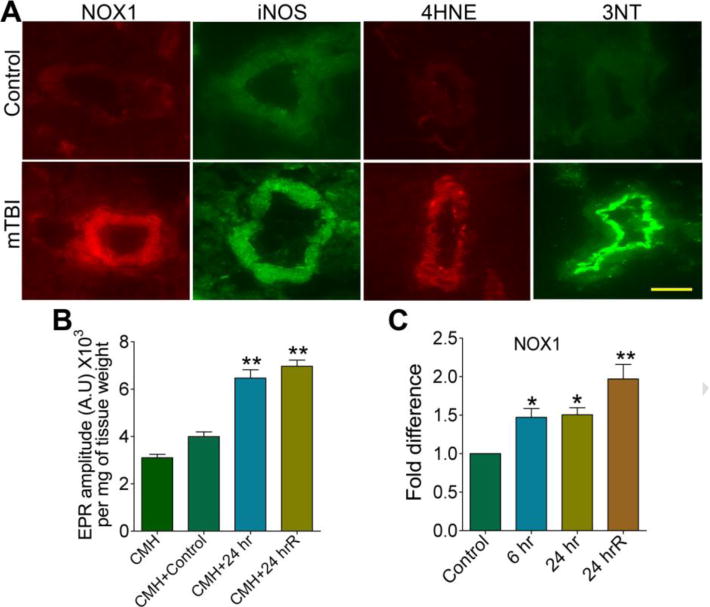
(A) Immunofluorescent staining of NOX1, iNOS, 4HNE, and 3NT in intact brain microvessels of rats subjected to a single blast with 123 kPa peak overpressure, 24 hours post-exposure. (B) ROS generation was detected by electron paramagnetic resonance (EPR) in brain tissue slices from 24 hr and 24 hrR post-exposure of mTBI of 123 kPa blast exposed and compared with control. Results are expressed in EPR amplitude arbitrary units per milligrams of tissue weight. (C) Changes in mRNA level of NOX1 in brain cortical tissues of rats in different time intervals of 123 kPa blast post-exposure by quantitative RT-PCR using TaqMan primers. Values are mean ±SEM (n = 3 in B and C). Statistically significant *p<0.05; **p<0.01 versus control in C; and versus CMH+control in B. Scale bar: 5 μm in all panels of A.
Disruption of cerebral BBB and perivascular units in mTBI
After establishing the kinetics of cerebral vascular and brain injury, we then focus our study in understanding the mechanisms of neurovascular injury within this time frame of single or repeated exposure. To link oxidative damage of the microvessel with that of BBB components, we examined the changes in the expression of the BBB tight junction (TJ) proteins occludin, claudin-5 and zonula occluden 1 (ZO-1) by immunofluorescence staining and Western blotting. Immunofluorescence staining and microscopy analyses revealed that 123 kPa shock wave pressure diminished the expression of TJ proteins in the microvessel within this time frame compared with control (Figure 5A), which is in agreement with the vascular oxidative damage results. In repeated exposure further reduced the expression of TJ proteins. These shock wave-induced changes in TJ protein expressions were validated by the alterations of the TJ proteins levels following SDS-PAGE protein separation, Western blot, and quantification of the TJ protein immunoreactive bands (Figure 5B–C).
Figure 5. Primary blast causes impairment in tight junction proteins.
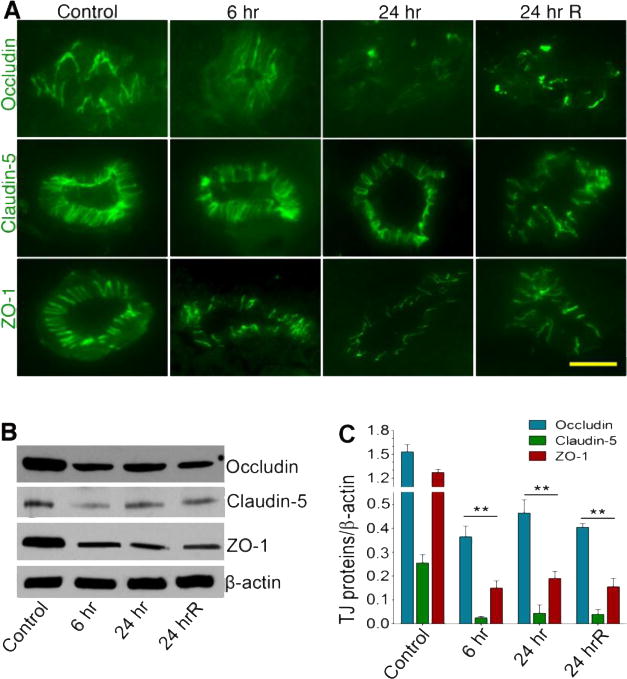
(A) Immunofluorescent staining of tight junction proteins: Occludin, Claudin-5 and ZO-1 in intact brain microvessels of rats at different time periods post-exposure (blast with 123 kPa peak overpressure): 6 hr, 24 hr and 24 hrR (two repeated exposures of every 24 hours). (B) Western blot analysis of Occludin, Claudin-5, ZO-1 and actin in the whole brain tissue homogenates of rats at different time points after exposure to blast (123 kPa peak overpressure). (C) Graph shows the results that are expressed as ratio of occludin/claudin5/ZO-1 to that of β-actin band. Values are mean ±SEM (n = 3). Statistically significant, **p<0.01 versus control in C. Scale bar:5 μm.
Further, we evaluated the alterations of the BBB basement membrane component – the perivascular units that surround the TJ proteins along with the astrocyte end-feet. We used antibody to PDGFR-β as pericyte specific marker to detect the alterations of the perivascular structure. Both von Willebrand factor (vWF) and endothelial specific glucose transporter-1 (GLUT1) are authentic marker for BBB endothelium. In agreement with the disruption of TJ protein, mild shock wave exposed animals showed significant reduction of PDGFR-β expression at 6, 24 hr and 24 hrR compared with controls (Figure 6). These data suggest that disarray of perivascular units is involved in the loss of the BBB integrity, neurovascular leakage and neuroinflammatory process in primary blast mild traumatic brain injury.
Figure 6. Pericytes have significant role in BBB dysfunction in primary blast-induced mTBI.
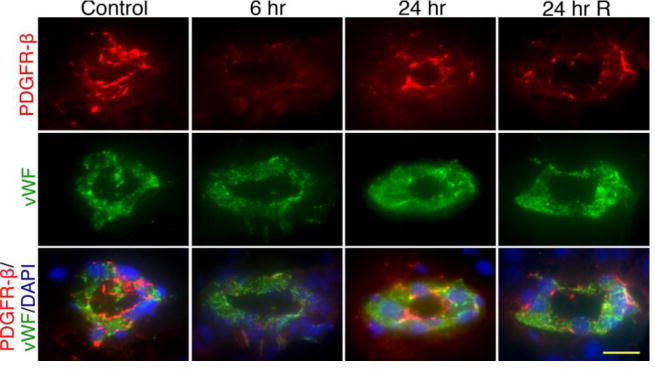
Immunofluorescent staining of pericyte specific marker, PDGFR-β (red) and endothelial marker vWF (green) in intact brain microvessels of rats exposed to primary blast (123 kPa peak overpressure). Cell nuclei were counterstained with DAPI (blue). The expression of PDGFR-β decreased with time (6 and 24 hours, single exposure) and after repeated insult (24 hr R). Scale bar = 5 μm in all panels.
Mechanistic disintegration of BBB and perivascular unit
Since activation of matrix metalloproteinases (MMPs) by oxidative stress is involved in digestion of the tight junction and basement membrane proteins (15, 25), here we examine the role of shock wave mediated activation of MMPs on the degradation of perivascular units and BBB leakiness. We observed that single or repeated mild shock wave exposure elevated the expression of MMP-2, MMP-3 and MMP-9 in brain microvessel (Figure 7A). Up-regulation of MMPs expression was validated by significant increase in respective protein levels in brain tissue homogenates (Figure 7B–C). We noted that the levels of MMP-3/-9 protein increased gradually up to 24 hrs, while up-regulation of MMP-2 seems to be short-lived because MMP-2 gradually decreases after 6 hr. Further, detection of MMPs activity by zymography validated the changes in expression and protein levels of MMPs in rat brain tissues. Thus, we found that mTBI increased the gelatinolytic (for MMP2/9) or stromelysin (for MMP3) activity in brain tissues (Figure 7D). Taken together, these data suggest that MMP-2, MMP-3, and MMP-9 are involved in degradation of perivascular units and TJ proteins, which leads to BBB leakiness and inflammation of cerebral vascular unit.
Figure 7. mTBI activates matrix metalloproteinases (MMPs).
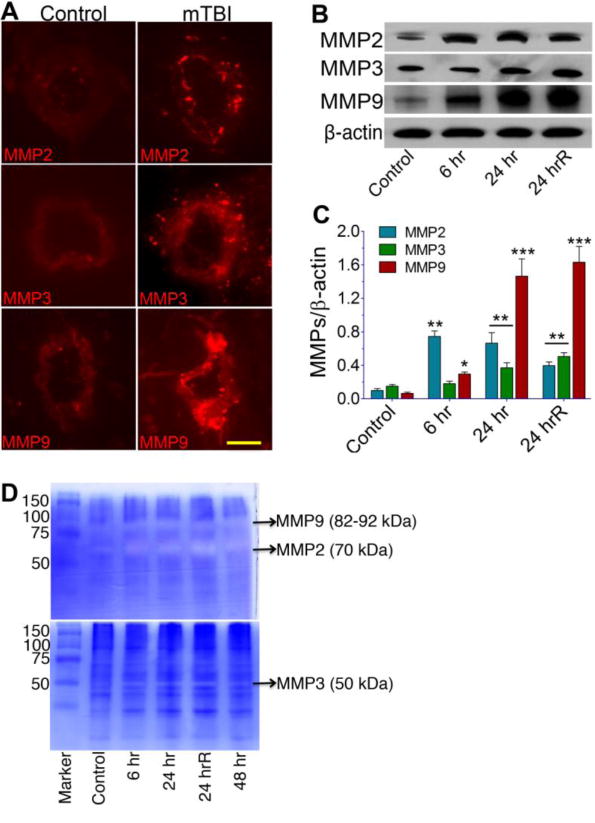
(A) Immunofluorescent staining of MMP-2, MMP-3, and MMP-9 in intact brain microvessels of mTBI exposed and control rats. (B) Western blots analyses of MMP-2, MMP-3, MMP-9, and actin in whole brain tissue homogenates. (C) Graph shows the results that are expressed as ratio of MMP-2/3/9 to that of β-actin band. (D) The gelatinolytic activity of MMP-2/9 (top gel) and caseinolytic activity of MMP-3 (bottom gel) were demonstrated by gelatin or casein zymography in the rat brain cortical tissue protein extracts from different time periods such as 6 hr, 24 hr, 24 hrR and 48 hr of post-exposure of blast with 123 kPa peak overpressure. Values are mean ±SEM; n=4. Statistically significant, **p<0.01 ***p<0.001 versus control in C. Scale bar = 5 μm in all panels of A.
Leakiness of cerebral vasculature due to MMPs activation is often associated with the disruption of water-channel protein. Thus, we examined the changes in aquaporin-4 (AQP-4, water channel protein) and the development of edema around the vasculature. In deed, enhanced expression of AQP-4 was observed around the perivascular region and within cortical brain tissue of mTBI animals (Figure 8A). We then established the cell phenotypes that are associated with the brain edema formation around the perivascular unit. To achieve this we colocalized AQP-4 with astrocyte marker glial fibrillary acidic protein (GFAP) or with microglia marker ionized calcium binding adaptor molecule 1 (Iba1) in brain tissue cross-sections (8 μm thick), and detected by immunofluorescent staining using specific antibody to respective protein markers. Our data showed that AQP-4 water channel activation in mTBI was associated with astrocyte, most likely towards the astrocyte end-feet surrounding the perivascular region (Figure 8B). We failed to observe any significant colocalization of AQP-4 and microglia marker Iba1 in the brain region (data not shown). This result suggests that cerebrovascular edema formation by AQP-4 activation may promote vascular fluid cavitation and neuroinflamation in blast-induced mild traumatic brain injury.
Figure 8. mTBI activates aquaporin-4.
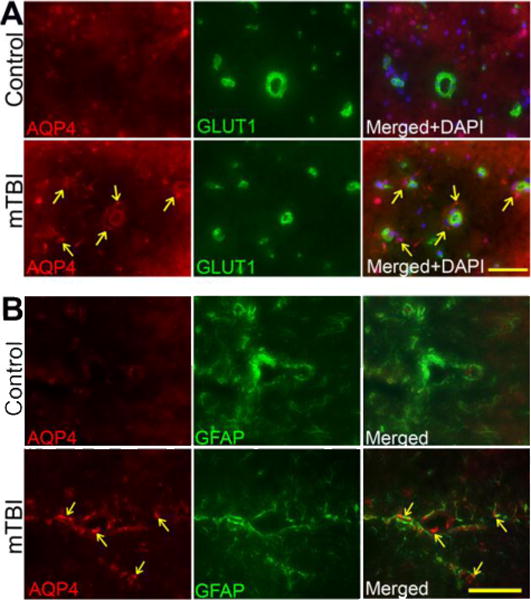
(A) Immunofluorescent staining of aquaporin-4 (AQP-4) (red) and endothelial marker, GLUT1 (green) in brain tissue sections contain microvessels of exposed to primary blast (123 kPa peak overpressure). Cell nuclei were counterstained with DAPI (blue). The arrow indicates the AQP-4 staining surrounding the microvessels. Scale bar = 40 μm in all panels.
(B) Immunofluorescent staining of aquaporin-4 (AQP-4) (red) co-localized with GFAP (astrocyte marker-green) in brain tissue sections contain microvessels of exposed to primary blast (123 kPa peak overpressure). The arrow indicates the AQP-4 staining surrounding the microvessels. Scale bar = 40 μm in all panels.
Assessment of cerebral vascular (the BBB) leakage
Disruption of cerebral vascular barrier integrity as a result of TJ protein damage and perivascular cavitation was assessed by leaking in and leaking out of biomarkers across the BBB. Infiltration of immune cells into the brain was assayed by adhesion and migration of Fluo-3 labeled macrophage and permeability of Na-Fl/EB tracers across the BBB assessed the tightness of the vasculature. Leaking out of brain matters into the blood circulation after shock wave exposure was analyzed by detecting S100β and neuronal-specific enolase (NSE) in blood samples that are commonly used in traumatic brain injury event (26). We observed that adhesion/infiltration of Fluo-3 labeled cells was significantly enhanced in the microvessel of mTBI shock wave exposed animal compared with controls (Figure 9A). Adhesion and infiltration of these immune cells appeared to occur at the vascular injury sites. Similarly, mTBI shock wave exposure greatly increased the permeability of small molecular weight NaFl (MW=376) and large molecular weight tracer EB (MW=961) across the BBB compared with respective controls (Figure 9B), which correlated with the enhanced immune cell adhesion and infiltration into the brain. Intriguingly, mTBI shock wave exposure animals also showed significantly higher levels of S100β and NSE in the blood samples compared with controls (Figure 9C). The maximum level of S100β was found at 6 hr where as leaking of NSE across the damaged BBB into the blood stream continued to increase even at 24 hr after the primary blasts. The elevation of S100β and NSE in the plasma of mTBI animals clearly indicated that degenerated/injured glial/neuronal cell body contents were leaked out of the brain and into the circulation. These data also supported the findings that activation of caspase-3 and TUNEL positive cells observed in the brain could well be the degenerated neurons and glial cells.
Figure 9. mTBI causes BBB leakage.
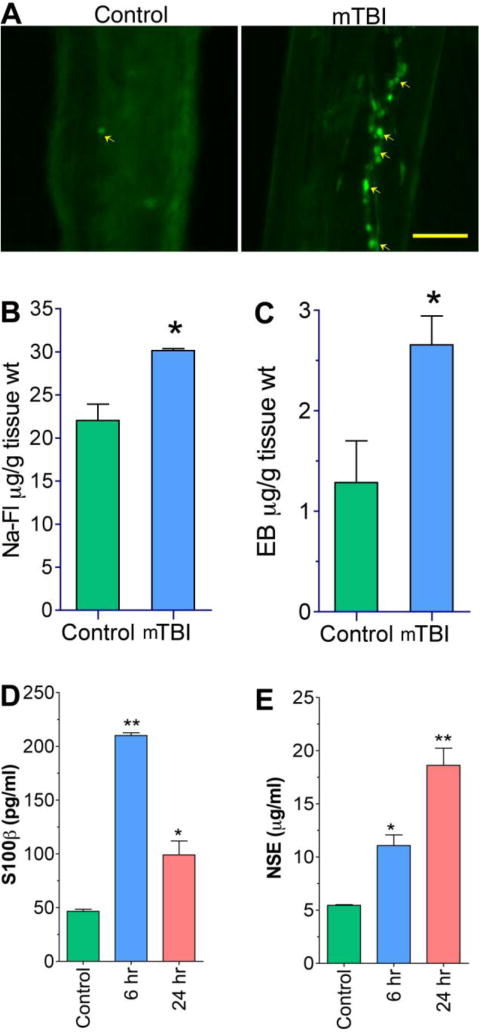
(A) Fluo-3 labeled macrophage adhesion/migration in brain capillary following infusion of cells into the common carotid artery of mTBI exposed rats after 24 hr of blast and compared with controls. (B,C) Graphical representation of In vivo permeability to show the leakage of Evans Blue (EB, 5 μM)(B) and Sodium fluorescein (Na-Fl, 5 μM) (C) in mTBI exposed rats. The carotid artery of rats was exposed and infused EB-NaFl mixture and collected the brain after one hour of infusion and processed for BBB permeability assay as given in materials and methods. (D, E) ELISA shows the levels of S100β (D) and NSE (E) in the blood serum of mTBI exposed rats. The blood serum was collected after 24 hr of blast exposure. Values are mean ±SEM, n=3 in B, C and n=5 in D,E. *p<0.05; **p<0.01 are statistically significant in B–E. Scale bar =40 μm in both panels of A.
Vascular injury and inflammation lead to neurovascular cell apoptosis
To correlate oxidative injury and inflammation with possible cell death around the cerebral vasculature, we then examined the activation of caspase-3 intrinsic apoptotic pathway in the brain tissue sections of control and shock wave experimental condition. We also used glucose transporter 1 (GLUT1) as positive marker for brain endothelium (the BBB). It was obvious that brain microvessel and brain tissue from 123 kPa shock wave exposure showed much higher expression of caspase-3 protein than that of controls (Figure 10A). Western blot analyses further substantiated the significant increase in caspase-3 protein levels in rat brain tissue homogenates of either single or repeated exposure of blast wave compared with controls (Figure 10B). As expected, repeated blast resulted in an increase caspase-3 expression that was higher than the single exposure.
Figure 10. mTBI causes cell apoptosis.
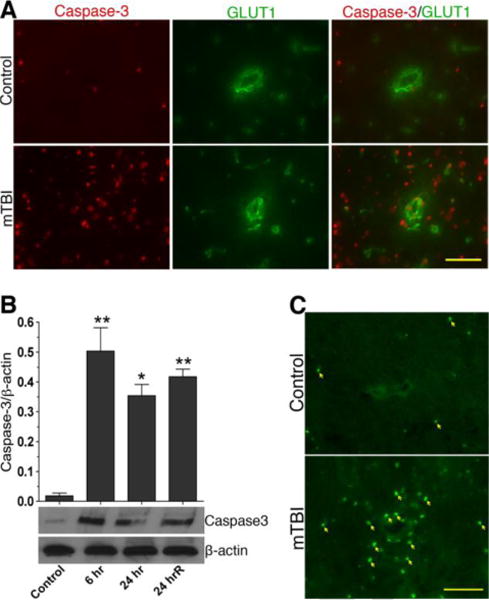
(A) Changes in the expression of active caspase-3 (red) in brain microvessels and cortical tissue sections of control and mTBI blast (123 kPa) exposed animals. (B) Immunoblot analysis to show the alterations in active caspase-3 protein levels in brain cortical tissue and microvessel homogenates proteins. Bar graphs show the results, which are expressed as ratio of caspase-3 (active) to that of β-actin bands. Values are mean ±SEM; n=4 to 5. *p<0.05; **p<0.01 are statistically significant. (C) TUNEL staining in rat brain cortical tissue section. The arrow indicates the TUNEL-positive cells. Scale bar = 20 μm in all panels of A and B.
To validate the activation of caspase-3, we evaluated cell apoptosis by TUNEL, and we confirmed that TUNEL positive cells were present much higher in the brain tissue of blast wave exposure than the control, similar to the data of caspase-3 activation (Figure 10C). The activation of caspase-3 and TUNEL positive cells were not only confined to the cerebral vascular but appeared to spread throughout the brain parenchyma in mTBI. Taken together, these results clearly indicate that oxidative injury of the BBB by mTBI shock wave pressure causes vascular edema, BBB leakage, and neurovascular inflammation and degeneration.
DISCUSSION
To the best of our knowledge, the present findings are the first to describe the mechanisms of cerebral vascular damage by shock wave in mTBI exposure. We established that low frequency of shock wave pressure causes biochemical cerebral vascular/brain injury, which occurs within a window of 6 – 24 hours after the exposure. The nature of vascular injury is oxidative damage and inflammation, which is initiated by oxidative/nitrosative reactive stress via the induction of NOX1 and iNOS by blast wave. The signature of oxidative damage (4HNE) and nitrated protein (3NT) in the microvessel correlated well with the induction of NOX1/iNOS and its kinetic profile. Based on these findings, it is apparent that any neurovascular inflammation and neuronal degeneration are expected to occur within this time frame, following single shock wave exposure in animal model. Thus, in a single mild shock wave exposure, one can miss the cerebral vascular injury and neuropathological signature outside this time frame, since oxidative injury and inflammatory response are gradually diminishing after 24 hours. A constant counteracting injury repair process (wound healing process) operating in a dynamic self-organizing system of the tissue organ may account for the disappearance of the vascular injury after 24 hr. This brings to a vital issue of the experimental design that single low shock wave intensity in animal model may not be ideal for the investigation of long-term neurocognitive deficits, behavioral changes, and neurological disorders such as PTSD. A repeated exposure to mild primary blasts is suggested for such type of neurocognitive and behavioral studies, which is demonstrated here by a prolonged vascular damage in repeated primary blast exposures.
It may be noted here that there are dramatic changes at 24 hours in a single 123kPa blast and a delayed onset effects of repeated blast exposure on the outcome of oxidative markers. This effect is also manifested by reversibility of the effect in a single primary blast wave and a prolonged effect of the repeated blasts that are not easily reversible. These acute dramatic changes and a delayed onset effects may impact the pathological signature of the single and repeated blast wave brain injury and the outcome of cognitive behavior changes in long-term exposure to primary blasts. We are currently investigating the significance of these acute dramatic and delayed onset effects of single and repeated exposure to blast wave in relation to the outcome of cognitive behavior changes.
It is evident from this study that mild shock wave causes cerebral vascular injury, thus, moderate to severe shock wave primary blast intensity is expected to exacerbate cerebral vascular injury. Here we demonstrate the pathophysiological evidence that disruption of the BBB and perivascular components by primary blast wave pressure contributes to neuroinflammation and neurotrauma. The association of cerebral vascular oxidative injury with a concomitant reduction of the BBB TJ proteins and the perivascular units with subsequent enhanced immune cell infiltration justified the notion that vascular barrier break down precedes neuroinflammation. Loosening of this barrier interface by shock wave exposure is mediated by oxidative stress-induced MMPs and fluid channel aquaporin-4 activation in the perivascular units. Pericyte is an integral unit of the neurovascular components, which contributes to the integrity of the BBB, and destruction of this barrier leads to neurodegenerative disease (27, 28). Our findings suggest that degradation of the TJ proteins and perivascular units by MMPs promotes BBB leakiness, vascular fluid cavitation, edema formation and neuroinflammation.
In the context of these findings, we can emphasize that cerebral vascular oxidative injury and inflammation may precede the development of blast-induced neurotrauma. This claim is substantiated by the fact that there was a strong association between vascular BBB damage and neuronal specific protein leakage into the blood stream. Leakage of brain matters from the cerebrospinal fluid into the blood circulation is possible only if the BBB function is impaired and brain cells (neurons, astrocyte) in the proximity of the perivascular area are either injured or dead. This distance close proximity to BBB may also explain the trend as why the levels of S100β in plasma samples elevated much faster (at 6 hr) than that of NSE (at 24 hr) following mTBI exposure as demonstrated in the present studies. In deed, our findings validated that caspase-3 activation and cell apoptosis (TUNEL positive cells) were observed mostly around the perivascular region of the brain. Our concept of cerebral vascular injury occurring prior to neurotrauma and neurological disease in single or repeated multiple shock wave exposure is strongly supported by the recent findings of Goldstein et al. (29). In this neurotrauma mouse model of primary blast, the authors demonstrated an impressive neurodegeneration in the form of phosphorylated tau protein clearly localized around the perivascular region. This distinctive localization of tauopathy surrounding the perivascular region lends inevitable evidence that vascular BBB oxidative injury/inflammation paves the path for neuroinflammation and neuronal degeneration within the neurovascular layers. This interconnected event is functionally demonstrated in the present studies by validating the detection of biochemical and pathological biomarkers with evidence of enhanced BBB permeability (Na-Fl/EB tracers), neuroinflammation (increase in infiltration of immune cells), and neuro-astroglial degeneration as evident by the elevated leakage of NSE/S100β into the blood stream. We conclude that induction of oxidative stress and subsequent MMPs activation by shock wave pressure leads to cerebral vascular BBB leakage, vascular edema formation, neuroinflammation and neurodegeneration.
Highlights.
In mTBI, oxidative injury causes the development of blast-induced neurotrauma.
Oxidative stress activates MMPs that causes the loosening of the vascular unit.
BBB damage is characterized by the reduction of the tight junction proteins in mTBI.
mTBI leads to elevation of level of NSE/S100β into the circulation.
mTBI leads to cerebral vascular leakage, neuroinflammation and neurodegeneration.
Acknowledgments
This work was supported by NIH/NIAAA grants R21 AA020370-01A1 and RO1 AA017398 to JH and US Army Research Office project “Army-UNL Center of Trauma Mechanics” (Contract No. W911NF- 08-10483 to NC) to NC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA. Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N Engl J Med. 2008;358:453–463. doi: 10.1056/NEJMoa072972. [DOI] [PubMed] [Google Scholar]
- 2.Vanderploeg RD, Belanger HG, Horner RD, Spehar AM, Powell-Cope G, Luther SL, Scott SG. Health Outcomes Associated With Military Deployment: Mild Traumatic Brain Injury, Blast, Trauma, and Combat Associations in the Florida National Guard. Arch Phys Med Rehabil. 2012 doi: 10.1016/j.apmr.2012.05.024. [DOI] [PubMed] [Google Scholar]
- 3.Vasterling JJ, Verfaellie M, Sullivan KD. Mild traumatic brain injury and posttraumatic stress disorder in returning veterans: perspectives from cognitive neuroscience. Clin Psychol Rev. 2009;29:674–684. doi: 10.1016/j.cpr.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Trudeau DL, Anderson J, Hansen LM, Shagalov DN, Schmoller J, Nugent S, Barton S. Findings of mild traumatic brain injury in combat veterans with PTSD and a history of blast concussion. J Neuropsychiatry Clin Neurosci. 1998;10:308–313. doi: 10.1176/jnp.10.3.308. [DOI] [PubMed] [Google Scholar]
- 5.Santiago PN, Wilk JE, Milliken CS, Castro CA, Engel CC, Hoge CW. Screening for alcohol misuse and alcohol-related behaviors among combat veterans. Psychiatr Serv. 2010;61:575–581. doi: 10.1176/ps.2010.61.6.575. [DOI] [PubMed] [Google Scholar]
- 6.Wilk JE, Bliese PD, Kim PY, Thomas JL, McGurk D, Hoge CW. Relationship of combat experiences to alcohol misuse among U.S. soldiers returning from the Iraq war. Drug Alcohol Depend. 2010;108:115–121. doi: 10.1016/j.drugalcdep.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 7.Otis JD, McGlinchey R, Vasterling JJ, Kerns RD. Complicating factors associated with mild traumatic brain injury: impact on pain and posttraumatic stress disorder treatment. J Clin Psychol Med Settings. 2011;18:145–154. doi: 10.1007/s10880-011-9239-2. [DOI] [PubMed] [Google Scholar]
- 8.Kamnaksh A, Kovesdi E, Kwon SK, Wingo D, Ahmed F, Grunberg NE, Long J, Agoston DV. Factors affecting blast traumatic brain injury. J Neurotrauma. 2011;28:2145–2153. doi: 10.1089/neu.2011.1983. [DOI] [PubMed] [Google Scholar]
- 9.Vandevord PJ, Bolander R, Sajja VS, Hay K, Bir CA. Mild neurotrauma indicates a range-specific pressure response to low level shock wave exposure. Ann Biomed Eng. 2012;40:227–236. doi: 10.1007/s10439-011-0420-4. [DOI] [PubMed] [Google Scholar]
- 10.Higashida T, Kreipke CW, Rafols JA, Peng C, Schafer S, Schafer P, Ding JY, Dornbos D, 3rd, Li X, Guthikonda M, Rossi NF, Ding Y. The role of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J Neurosurg. 2011;114:92–101. doi: 10.3171/2010.6.JNS10207. [DOI] [PubMed] [Google Scholar]
- 11.Haorah J, Floreani NA, Knipe B, Persidsky Y. Stabilization of superoxide dismutase by acetyl-l-carnitine in human brain endothelium during alcohol exposure: novel protective approach. Free Radic Biol Med. 2011;51:1601–1609. doi: 10.1016/j.freeradbiomed.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rump TJ, Abdul Muneer PM, Szlachetka AM, Lamb A, Haorei C, Alikunju S, Xiong H, Keblesh J, Liu J, Zimmerman MC, Jones J, Donohue TM, Jr, Persidsky Y, Haorah J. Acetyl-L-carnitine protects neuronal function from alcohol-induced oxidative damage in the brain. Free Radic Biol Med. 2010;49:1494–1504. doi: 10.1016/j.freeradbiomed.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alikunju S, Abdul Muneer PM, Zhang Y, Szlachetka AM, Haorah J. The inflammatory footprints of alcohol-induced oxidative damage in neurovascular components. Brain Behav Immun. 2011 doi: 10.1016/j.bbi.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haorah J, Ramirez SH, Floreani N, Gorantla S, Morsey B, Persidsky Y. Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic Biol Med. 2008;45:1542–1550. doi: 10.1016/j.freeradbiomed.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abdul Muneer PM, Alikunju S, Szlachetka AM, Haorah J. The mechanisms of cerebral vascular dysfunction and neuroinflammation by MMP-mediated degradation of VEGFR-2 in alcohol ingestion. Arterioscler Thromb Vasc Biol. 2012;32:1167–1177. doi: 10.1161/ATVBAHA.112.247668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DePalma RG, Burris DG, Champion HR, Hodgson MJ. Blast injuries. N Engl J Med. 2005;352:1335–1342. doi: 10.1056/NEJMra042083. [DOI] [PubMed] [Google Scholar]
- 17.Moore DF, Radovitzky RA, Shupenko L, Klinoff; A, Jaffee MS, Rosen JM. Blast physics and central nervous system injury. Future Neurology. 2008;3:243–250. [Google Scholar]
- 18.Chen YC, Smith DH, Meaney DF. In-vitro approaches for studying blast-induced traumatic brain injury. J Neurotrauma. 2009;26:861–876. doi: 10.1089/neu.2008.0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2:81–84. doi: 10.1016/s0140-6736(74)91639-0. [DOI] [PubMed] [Google Scholar]
- 20.Okie S. Traumatic brain injury in the war zone. N Engl J Med. 2005;352:2043–2047. doi: 10.1056/NEJMp058102. [DOI] [PubMed] [Google Scholar]
- 21.Chandra N, Holmberg A, Feng R. U.S. Provisional patent application no. 61542354. Controlling the shape of the shock wave profile in a blast facility. 2011 Oct 3; 2011.
- 22.Ganpule S, Alai A, Plougonven E, Chandra N. Mechanics of blast loading on the head models in the study of traumatic brain injury using experimental and computational approaches. Biomech Model Mechanobiol. 2012 doi: 10.1007/s10237-012-0421-8. [DOI] [PubMed] [Google Scholar]
- 23.Sundaramurthy A, Alai A, Ganpule S, Holmberg A, Plougonven E, Chandra N. Blast-Induced Biomechanical Loading of the Rat: An Experimental and Anatomically Accurate Computational Blast Injury Model. J Neurotrauma. 2012 doi: 10.1089/neu.2012.2413. [DOI] [PubMed] [Google Scholar]
- 24.Abdul Muneer PM, Alikunju S, Szlachetka AM, Murrin LC, Haorah J. Impairment of brain endothelial glucose transporter by methamphetamine causes blood-brain barrier dysfunction. Mol Neurodegener. 2011;6:23. doi: 10.1186/1750-1326-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haorah J, Schall K, Ramirez SH, Persidsky Y. Activation of protein tyrosine kinases and matrix metalloproteinases causes blood-brain barrier injury: Novel mechanism for neurodegeneration associated with alcohol abuse. Glia. 2008;56:78–88. doi: 10.1002/glia.20596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berger RP, Adelson PD, Pierce MC, Dulani T, Cassidy LD, Kochanek PM. Serum neuron-specific enolase, S100B, and myelin basic protein concentrations after inflicted and noninflicted traumatic brain injury in children. J Neurosurg. 2005;103:61–68. doi: 10.3171/ped.2005.103.1.0061. [DOI] [PubMed] [Google Scholar]
- 27.Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14:1398–1405. doi: 10.1038/nn.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, Goletiani CJ, Maglakelidze GM, Casey N, Moncaster JA, Minaeva O, Moir RD, Nowinski CJ, Stern RA, Cantu RC, Geiling J, Blusztajn JK, Wolozin BL, Ikezu T, Stein TD, Budson AE, Kowall NW, Chargin D, Sharon A, Saman S, Hall GF, Moss WC, Cleveland RO, Tanzi RE, Stanton PK, McKee AC. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012;4:134ra160. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]