

Abstract
Yeast cell biologists use a variety of fluorescent protein tags for determining protein localization and for measuring protein dynamics using fluorescence recovery after photobleaching (FRAP). Although many modern fluorescent proteins, such as those with photoactivatable and photoconvertible characteristics, have been developed, none has been exploited for studies in budding yeast. We describe here the construction of yeast-tagging vectors containing photoactivatable green fluorescent protein (PA–GFP) for analysis of protein behaviour. We tagged two yeast proteins, Erg6p and Num1p, with PA–GFP and demonstrated specific photoactivation of the fusion proteins in live cells. Fluorescence intensity measurements showed that a short 5 s exposure to 413 nm light is sufficient to produce the maximum level of activated GFP fluorescence. Local photoactivation of cortical Num1p-PA–GFP showed movement of the marked proteins, providing new insights into the behaviour of Num1p at the cell cortex. Since photoactivation can be achieved using standard mercury arc illumination, the PA–GFP tag represents a convenient and economical way to determine protein dynamics in the cell. Thus, the tagging modules should facilitate protein-tracking studies in a wide variety of cell biological processes in yeast.
Keywords: photoactivatable GFP, PA–GFP, protein-tracking
Introduction
Fluorescent protein tags have been widely used in budding yeast to determine the sub-cellular localization of various proteins of interest. Many commonly used fluorescent protein tags, such as green fluorescent protein (GFP), are available in convenient modules for one-step PCR and oligonucleotide-mediated insertion at the 3′-end of the chromosomal gene locus. Expression of the tagged gene readily allows the visualization of the protein’s localization. While some proteins are targeted to multiple sites in the cell, others may be targeted to different sites at different stages of the cell cycle. Time-lapse imaging has provided insights into the steady state distribution of the protein over time. However, in order to track protein movement within the cell, more complex imaging experiments, such as fluorescence recovery after photobleaching (FRAP), are required. FRAP is traditionally performed by selectively photobleaching the fusion proteins at a site with high-intensity light (e.g. laser) and monitoring the movement of fusion proteins into the bleached region. These measurements can provide insights into how a protein interacts with a cellular site (e.g. an actin patch at the cell cortex or chromatin in the nucleus), revealing its role in the assembly, regulation and maintenance of that site.
The recent advancement in the diversity of fluorescent protein tags for mammalian systems has provided new tools for biological imaging (Chudakov et al., 2007; Shaner et al., 2005; Wiedenmann et al., 2004). In particular, the photoactivatable GFP (PA–GFP) has been utilized for monitoring protein movement within living mammalian cells (Lippincott-Schwartz and Patterson, 2008; Patterson and Lippincott-Schwartz, 2004). PA–GFP emits fluorescence only after photoactivation by a brief exposure to UV light (413 nm) (Patterson and Lippincott-Schwartz, 2002). Thus, similar to photobleaching, photoactivation can selectively mark a pool of fusion proteins for in vivo protein-tracking studies. For example, in mammalian cells expressing α-tubulin tagged with PA–GFP, photoactivation of local segments of microtubules allows monitoring of the movement of tubulin subunits in the microtubule polymer (Tulu et al., 2003). Additionally, experiments involving photoactivation of PA–GFP have been reported to yield better signal-to-noise characteristics compared to experiments involving photobleaching of GFP (Tulu et al., 2003). Thus, PA–GFP is an excellent tool for monitoring the dynamic subcellular localization of fusion proteins.
We have constructed new plasmids for tagging yeast genes with PA–GFP. We generated plasmids with different selectable markers, including kanMX6, TRP1 and His3MX6, in order to increase the utility of these plasmids by the yeast community (Figure 1). We demonstrated for the first time specific photoactivation of two proteins (Erg6p and Num1p) tagged with PA–GFP in living yeast cells. Using arc lamp illumination, we locally photoactivated a subpopulation of cortical Num1p-PA–GFP and observed its movement along the cell cortex. Since laser light is not required for photoactivation experiments, these PA–GFP modules represent an economical way to determine protein dynamics in the cell. Thus, they should facilitate the rapid analysis of various proteins in a wide variety of biological processes in budding yeast.
Figure 1.

PCR plasmid templates for PA–GFP tagging. Shaded boxes represent PA–GFP and the selectable marker as indicated. Arrows inside the boxes indicate directions of transcription, while arrows outside the boxes indicate forward (F) and reverse (R) PCR primers (see Table 1). (A, B) Plasmid templates for single PA–GFP gene tagging. (C) Plasmid template for tandem PA–GFP gene tagging. Sequences to which F2, F5 and F4 primers anneal are shown for each plasmid template. Restriction sites in the sequences are indicated by italics
Materials and methods
Yeast strains and transformation
The wild-type yeast strain used in this study was YWL37, which is identical to YJC2296 (Lee et al., 2003). We transformed yeast strains using the lithium acetate method as previously described (Knop et al., 1999). Stable transformants were selected on YPD plates containing G418 sulphate (EMD Chemicals Inc., NJ, USA) and clonally purified by streaking individual colonies onto selective plates. Proper targeting of the PA–GFP tag was confirmed by PCR. Yeast media were obtained from Sunrise Science Products (San Diego, CA, USA).
Construction of PA–GFP tagging vectors
To generate a PA–GFP yeast-tagging vector, we amplified a fragment containing the PA–GFP open reading frame, using F1 and R1 primers (Table 1) from a mammalian expression vector, pPA–GFP–C1 (a kind gift from Patricia Wadsworth, University of Massachusetts Amherst, USA). The amplified PA–GFP fragment was digested with BamHI and BglII, and ligated into a similarly digested pDH3 plasmid (Yeast Resource Center, University of Washington, USA), replacing the cyan fluorescent protein (CFP) sequence in pDH3. We verified the orientation and sequence of the ligated PA–GFP insert by DNA sequencing (Genewiz Inc., NJ, USA). The resulting plasmid, pDH3–PA–GFP, contains PA–GFP and the Escherichia coli kanr gene as the selectable marker.
Table 1.
Primers used in this study
| Primer | Sequence (5′ → 3′) |
|---|---|
| F1 | CCAGGGATCC ATG GTG AGC AAG GGC GAG GAG1 |
| R1 | CCAGAGATCT CTA CTT GTA CAG CTC GTC CAT1 |
| F2 | (Gene-specific sequence)-ggt gct ggt ATG GTG AGC AAG GGC GAG GA2,3 |
| R2 | (Gene-specific sequence)-GAATTCGAGCTCGTTTAAAC2 |
| F3 | CCAGGTCGAC ATG GTG AGC AAG GGC GAG G1 |
| R3 | CCAG gtc gac agc acc CTT GTA CAG CTC GTC CAT G1,2 |
| F4 | (Gene-specific sequence)-AAG CTT CGT ACG CTG CAG GTC2 |
| F5 | (Gene-specific sequence)-CGC GCC AGA TCC ATG GTG AGC2 |
| R5 | (Gene-specific sequence)-GAATTCGAGCTCGTTTAAAC2 |
The locations of the F2, R2, F4, F5 and R5 primers relative to the plasmid templates are indicated in Figure 1. Reading frame is indicated by spacing in the sequences. Restriction enzyme sites are indicated by italics.
CCAG was included at the 5′ end of the primer to aid digestion of PCR products by the restriction enzyme.
We used 60 nucleotides of gene-specific sequence for homologous integration. Shorter gene-specific sequence may also be used (Longtine et al., 1998).
Lower case indicates sequence encoding the GlyAlaGly linker between the last codon of the tagged gene and the first codon of the PA–GFP tag.
R3 primer is the reverse primer used in the construction of the tandem PA–GFP plasmid template (Figure 1C). Lower case indicates part of the linking sequence between the two PA–GFP open reading frames. The linking sequence connecting the two PA–GFP tags is 5′-GGT GCT GTC GAC GGA TCC-3′, which encodes for GlyAlaValAspGlySer.
We subcloned the PA–GFP open reading frame as a BamHI–BglII fragment from pDH3–PA–GFP into the BglII site of pFA6a–kanMX6, pFA6a–TRP1 and pFA6a–His3MX6 (Longtine et al., 1998). Orientation of the PA–GFP insert was verified by restriction enzyme digestion. The resulting plasmids, pFA6a–PA–GFP–kanMX6, pFA6a–PA–GFP–TRP1 and pFA6a–PA–GFP–His3MX6, contain the PA–GFP and the kanr, the Saccharomyces cerevisiae TRP1 gene, or the Schizosaccharomyces pombe his5+ gene as the selectable marker, respectively.
To generate a tandem PA–GFP yeast-tagging vector, we amplified a fragment containing the PA–GFP open reading frame without the stop codon using F3 and R3 primers (Table 1) from the mammalian expression vector pPA–GFP–C1. The amplified fragment was digested with SalI and ligated into a SalI-linearized pDH3–PA–GFP vector, resulting in an in-frame insertion of a second PA–GFP 5′ to the first PA–GFP. We verified the orientation and sequence of the second PA–GFP by DNA sequencing. The resulting plasmid is pDH3–tandemPA–GFP.
Construction of PA–GFP-tagged strains
To tag ERG6 with PA–GFP, we amplified from pDH3–PA–GFP using F2 and R2 primers (Table 1). We chose 60 nucleotides immediately upstream of the stop codon of ERG6 as the gene-specific sequence in the forward F2 primer for homologous integration. The reading frame of the PA–GFP tag was preserved and a linker sequence encoding GlyAlaGly was engineered in the F2 primer, separating the last codon of ERG6 from the first codon of PA–GFP. We chose the reverse complement of 60 nucleotides immediately downstream of the stop codon of ERG6 as the gene-specific sequence in the reverse R2 primer for homologous integration. The amplified product was gel-purified (QIAEX II kit, Qiagen, CA, USA) before transformation into yeast. To tag NUM1 with PA–GFP, we used a similar strategy, except that NUM1 gene-specific sequences were used in F2 and R2 primers for homologous integration. Additionally, we used F5 and R5 primers containing NUM1 gene-specific sequences to tag NUM1 with PA–GFP constructed in pFA6a–PA–GFP–kanMX6, pFA6a–PA–GFP–TRP1 and pFA6a–PA–GFP–His3MX6 plasmids. A linker sequence was not included in the F5 primer, since vector sequences upstream of the PA–GFP start codon in the pFA6a-series plasmids encode a linker of four amino acids (ArgAlaArgSer). Tagging of NUM1 with either F2/R2 or F5/R5 primer pairs resulted in Num1p-PA–GFP that showed similar photoactivated fluorescence (data not shown). To tag NUM1 with tandemPA–GFP, F4 and R2 primers containing NUM1 gene-specific sequences were used. To introduce PGAL1-controlled expression of NUM1-PA–GFP, we replaced 50 bp upstream of the start codon of the chromosomal gene with the GAL1 promoter, using a TRP1 selectable marker as previously described (Longtine et al., 1998). The resulting strain was YWL508.
Photoactivation and image acquisition
Yeast cultures were grown to mid-log phase at 30 °C and analysed on an agarose pad containing non-fluorescent synthetic defined medium (Sunrise Science Products, CA, USA). Analysis of Erg6p–PA–GFP cells was performed on a laser-scanning confocal microscope (Nikon C1). Analysis of Num1p–PA–GFP cells was performed on a Nikon Eclipse TE 300 microscope equipped with a spinning disk confocal scan head (Perkin-Elmer-Cetus, MA, USA) and an ORCA-ER camera (Hamamatsu, NJ, USA), as previously described (Tulu et al., 2003). We used either a ×100 1.40 NA objective or a ×100 1.45 NA objective for acquiring images. We chose to use Num1p-PA–GFP-expressing cells to quantify the GFP fluorescence after photoactivation because the punctate appearance of Num1p cortical patches enables simple and accurate measurement of fluorescence intensities. Briefly, the cells were exposed to 413 nm light for varying lengths of time, using a shuttered 100 W mercury arc epiilluminator (X-Cite 120, EXFO America, TX, USA) and a D405/20 filter cube (Chroma Technology, VT, USA). An image of GFP fluorescence was acquired before and after the 413 nm light exposure using the same camera settings. Using ImageJ (NIH), we measured the specific fluorescence due to photoactivation as a function of the duration of 413 nm irradiation by subtracting the total fluorescence of a patch before photoactivation (= area of patch × mean pixel value of the area) from the total fluorescence after photoactivation. To perform local photoactivation of cortical Num1p-PA–GFP, the area of photoactivation was restricted using a slit (Lennox Laser, MD, USA) mounted in a Ludl filter wheel placed in a conjugate image plane in the epiillumination light path. To photoactivate the entire field of view, an open position in the filter wheel was selected.
Cell lysis and immunoblotting
Cell pellets from 3 ml cultures were resuspended in 0.5 ml ice cold lysis buffer [20 mM Tris, pH8, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM PMSF, plus protease inhibitor cocktail tablet (Roche Applied Science)] and lysed by bead beating at 4 °C in a Bullet Blender (Next Advance Inc.) five times for 1 min each, with 2 min on ice between each beating. Protein concentration was measured using Bradford Protein Assay (Bio-Rad). Crude lysate was separated by a 6.25% linear SDS–PAGE and electroblotted to nitrocellulose in a semi-dry Transblot (BioRad). Rabbit anti-GFP polyclonal antibody (Invitrogen) and HRP-conjugated goat anti-rabbit antibody (Jackson ImmunoResearch Laboratories) were used at 1 : 1000 and 1 : 10 000 dilutions, respectively. Chemiluminescence signal was acquired and imaged using a G : BOX Chemi HR16 (Syngene) equipped with a sensitive 16-bit CCD camera (Sony ICX285AL; pixel size 6.45 × 6.45 μm). The length of exposures that were used (30 s to 5 min) did not saturate the camera’s pixels. Using ImageJ, we measured the intensity of Num1 bands as the area of the band multiplied by the mean pixel value of the area.
Plasmid requests
Plasmids generated in this study have been deposited at Addgene for distribution to academic researchers (http://www.addgene.org). Find our plasmids and sequence information in the Addgene database by searching with keyword ‘PA–GFP’. Follow the instructions at Addgene’s website to request for plasmids.
Results and discussion
Construction of PA–GFP tagging cassettes
First, we subcloned a DNA fragment containing the open reading frame of PA–GFP (Patterson and Lippincott-Schwartz, 2002; Tulu et al., 2003) from a mammalian expression plasmid into a yeast-tagging plasmid containing the kanr selection marker, generating PA–GFP::kanr cassette (Figure 1A). This PA–GFP has been used in living mammalian cells to tag various proteins for photo-marking experiments, including α-tubulin for tracking microtubule dynamics (Ferenz and Wadsworth, 2007; Tulu et al., 2003). We verified photoactivatability of the PA–GFP in living yeast cells as described below. Next, in order to broaden the utility of the PA–GFP construct, we subcloned the PA–GFP into pFA6a-series plasmids containing kanr, TRP1 and his5+ selectable markers (Long-tine et al., 1998) (Figure 1B). We also subcloned a second PA–GFP upstream to the first PA–GFP in the PA–GFP::kanr cassette (Figure 1C). An intervening GlyAlaValAspGlySer linker separates the tandem PA–GFP coding sequences. The resulting cassette, termed tandemPA–GFP::kanr, was confirmed by DNA sequencing and subsequently tested in vivo as described below.
Demonstration of photoactivation of PA–GFP fusion proteins in yeast
Prior to this study, photoactivatable GFP has not been expressed in yeast. Therefore, it was not clear whether the fluorescent protein would be photoactivatable when fused to endogenous proteins in living yeast cells. In order to address this question, we set out to test the PA–GFP that we constructed in Figure 1A. Using the PCR-mediated integration technique, we inserted the PA–GFP coding sequence at the 3′ end of the chromosomal ERG6 gene, which encodes for an abundant enzyme functioning in the ergosterol biosynthetic pathway (Gaber et al., 1989; Lees et al., 1995; Welihinda et al., 1994). Erg6p is an excellent test protein, not only because it is highly abundant (53 800 molecules/cell; Ghaemmaghami et al., 2003), but also because it localizes to discrete foci of intracellular lipid particles (Gaber et al., 1989; Huh et al., 2003; Lees et al., 1995; Parks et al., 1995), rather than diffusely in the cytoplasm. Additionally, Erg6p localizes to the mitochondrial outer membrane (Zahedi et al., 2006).
To test for photoactivation of PA–GFP, we used a laser scanning confocal microscope (Nikon C1) to illuminate a small quadrant within a field of live cells with a pulse of 405 nm light. We found that cells expressing Erg6p fused to PA–GFP became fluorescent upon 488 nm excitation only after intense irradiation by 405 nm laser light. Prior to photoactivation, little fluorescence was detected upon 488 nm excitation (Figure 2B), consistent with published data on PA–GFP in mammalian systems (Ferenz and Wadsworth, 2007; Lippincott-Schwartz and Patterson, 2008; Patterson and Lippincott-Schwartz, 2002; Tulu et al., 2003). As reported (Huh et al., 2003), the localization of Erg6p-PA–GFP was observed as foci within the cytoplasm (Figure 2B, C). A second pulse irradiating a different quadrant in the same field resulted in photoactivation of Erg6p-PA–GFP cells in that new quadrant (box 2 in Figure 2B; Figure 2C). Notably, the fluorescence of activated Erg6p-PA–GFP proteins in the first quadrant appeared stable and unchanged (Figure 2C).
Figure 2.
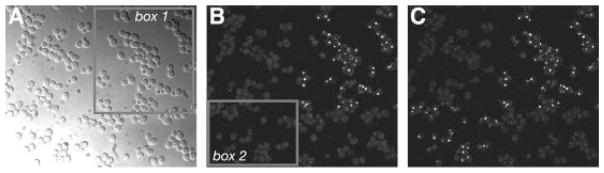
Photoactivation of Erg6p-PA–GFP. Live cells expressing Erg6p tagged at the chromosomal locus with single PA–GFP were examined using a Nikon C1 laser scanning confocal microscope. (A) DIC image. Cells within box 1 were selectively irradiated with a 405 nm laser for 20 s. (B) GFP fluorescence image after irradiation of box 1. Cells within box 2 were subsequently irradiated as in (A). (C) GFP fluorescence image after irradiation of box 2. Note that cells that were photoactivated by the first irradiation remained fluorescent after the second irradiation. Strain: YWL492, Erg6p-PA–GFP
Next, using the PCR-mediated integration technique, we inserted PA–GFP at the 3′ end of the chromosomal NUM1 gene, which encodes for a cortical protein functioning in mitochondrial distribution (Cerveny et al., 2007) and nuclear migration (Farkasovsky and Kuntzel, 2001; Heil-Chapdelaine et al., 2000). We chose Num1p as a test protein because it localizes as stationary cortical patches (Figure 3A) (Farkasovsky and Kuntzel, 1995; Heil-Chapdelaine et al., 2000), yet it remains unclear how these patches are assembled. Furthermore, once Num1p patches are assembled, it is unknown whether Num1p molecules remain stably associated within a patch or are subsequently capable of moving to a secondary site along the cortex.
Figure 3.

Photoactivation of Num1p-PA–GFP. (A) Bright-field and fluorescence images of cells expressing endogenous levels of Num1p-GFP (top row) compared to cells expressing PGAL1-Num1p-PA–GFP (bottom row). The PGAL1-controlled strain was grown overnight in a synthetic defined medium containing 2% galactose instead of 2% glucose. The overnight culture was diluted 1 : 50 into fresh medium with 2% galactose and 2% glucose, grown to log phase for 5 h at 30 °C and then imaged for GFP fluorescence after 20 s irradiation of 413 nm light, using a mercury arc lamp. (B) Immunoblot analysis of Num1p-PA–GFP protein levels. Equal amount of total cell lysate were separated by a 6.25% SDS–PAGE gel, transferred to nitrocellulose, and immunoblotted with anti-GFP antibody. (Left) Total cell lysate of PGAL1 –Num1p-PA–GFP strain after overnight growth (15–18 h) in 2% galactose medium. (Right) Total cell lysate of Num1p–GFP (expressed from the endogenous promoter) and PGAL1 –Num1p-PA–GFP strains, both of which were grown to log phase in 2% galactose and 2% glucose for 5 h at 30 °C after 1 : 50 dilution from an overnight culture in 2% galactose medium. Arrows indicate the Num1p fusion protein bands. (C) Specific fluorescence of Num1p-PA–GFP plotted as a function of the duration of 413 nm light irradiation. Cells were imaged before and after irradiation. The total fluorescence of a patch was measured as the area of the patch multiplied by its mean pixel value. The specific fluorescence due to photoactivation was calculated as the difference between the total fluorescence before and after photoactivation. Error bars represent standard error (n = 60 patches measured for each duration). Strains: YWL537, Num1p–GFP; YWL508, PGAL1 –Num1p-PA–GFP
Previous reports have suggested that there are <50 Num1p molecules/cell (Ghaemmaghami et al., 2003). Therefore, before we could begin to track the dynamics of Num1p within cortical patches, it was necessary to determine whether Num1p-PA–GFP could be detected at the cell cortex. Using a Nikon Eclipse TE300 microscope equipped with an ORCA-ER camera and a 413 nm mercury arc illumination lamp for photoactivation (see Materials and methods), we found that Num1p-PA–GFP was below our detection limit. In addition, Num1p tagged with tandemPA–GFP was not detected upon 488 nm excitation after photoactivation (data not shown). In a second effort to intensify Num1p-PA–GFP fluorescence, we replaced the endogenous promoter with a GAL1 promoter at the 5′ end of the chromosomal NUM1-PA–GFP gene. Immunoblot analysis showed that cells expressing PGAL1-controlled Num1p-PA–GFP expressed a high level of Num1p-PA–GFP after an overnight growth (15–18 h) in the presence of 2% galactose (Figure 3B, left). However, we found that if the overnight cultures were subsequently diluted and cultured to log phase for 5 h in the presence of 2% galactose and 2% glucose, the level of Num1p-PA–GFP decreased to a level that is only 1.2-fold higher than Num1p-GFP expressed from the endogenous promoter (Figure 3B, right). Under this condition, cells expressing mildly elevated levels of Num1p-PA–GFP showed (upon photoactivation) cortical Num1p localization that is comparable to a control strain expressing endogenous levels of Num1p tagged with a conventional GFP (Figure 3A) (Heil-Chapdelaine et al., 2000). We conclude that Num1p-GFP fluorescence is brighter than photoactivated Num1p-PA–GFP fluorescence, given that the protein levels of Num1p-PA–GFP must be elevated by ~20% for detection under our imaging conditions.
In order to determine the maximum fluorescence intensity that can be generated by photoactivation, we quantified the total fluorescence intensity as a function of the duration of 413 nm irradiation. Cells expressing mildly elevated levels of PGAL1-controlled Num1p-PA–GFP (cultured as described above) were photoactivated for different lengths of time (Figure 3C). The total fluorescence due to photoactivation was measured for individual Num1p-PA–GFP cortical patches. We found that a 5 s irradiation is sufficient to elicit maximal level of fluorescence after photoactivation (Figure 3C). Furthermore, increasing the duration of irradiation resulted in a lower level of fluorescence after photoactivation. This indicates that prolonged photoactivation may reduce the PA–GFP signal due to photobleaching.
PA–GFP enables photo-marking of proteins in yeast
Next, we wanted to selectively mark and track the movement of Num1p-PA–GFP molecules over time. We restricted photoactivation to a small region of the cortex followed by time-lapse imaging with 488-nm excitation (Figure 4). We observed a subpopulation of photoactivated Num1p-PA–GFP molecules moving away from the photoactivated area to another region along the cortex (arrows, Figure 4). This result suggests that Num1p molecules may have more than a structural role in patch assembly. Because Num1p tagged with conventional GFP is functional (Heil-Chapdelaine et al., 2000), we expect that Num1p-PA–GFP is also functional.
Figure 4.
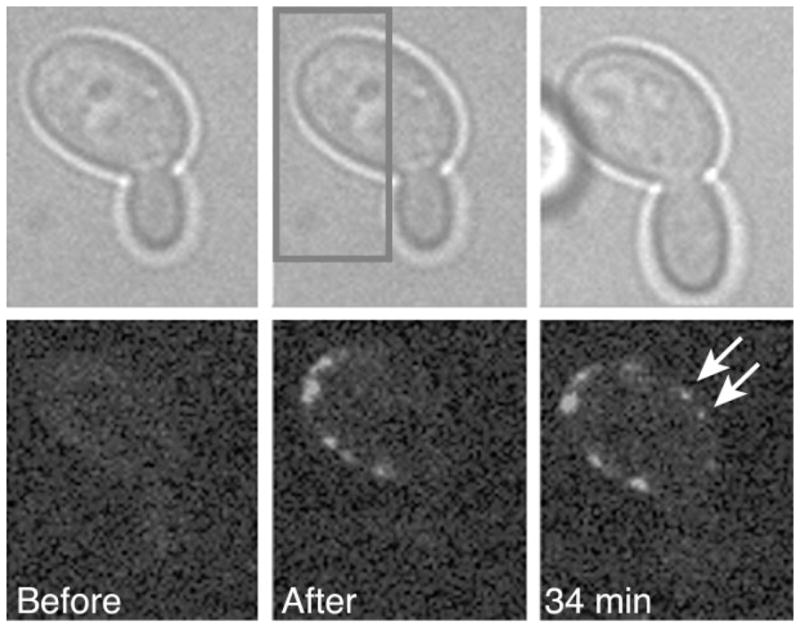
Local photoactivation of Num1p-PA–GFP. Cells expressing PGAL1-controlled Num1p-PA–GFP tagged at the chromosomal locus were grown overnight in synthetic defined medium containing 2% galactose instead of 2% glucose. The overnight culture was diluted 1 : 50 into medium with 2% galactose and 2% glucose and grown to log phase for 5 h at 30 °C. Bright-field (top row) and GFP fluorescence (bottom row) images were acquired before photoactivation (left column), immediately after (middle column) and 34 min after photoactivation (right column). Photoactivation was targeted locally to a selected region of the cortex. Num1p-PA–GFP proteins marked by photoactivation moved from the boxed region to a new region of the cell cortex (arrows). Strain: YWL508, PGAL1 –Num1p-PA–GFP
We have demonstrated that PA–GFP is a useful tool that has a broad application for protein-tracking studies in yeast. The PA–GFP-tagged proteins were photoactivated by an exposure to filtered light from a shuttered mercury arc epiilluminator, hence an expensive laser was not necessary. Although the microscope that we used to examine Num1p-PA–GFP is equipped with a spinning disc confocal scan head, we envision that time-lapse imaging of photoactivated proteins can also be performed with a standard wide-field fluorescence microscope. We restricted the area of photoactivation using slits mounted in the excitation light path. For studies requiring a smaller area of photoactivation, the area can be selected by reducing the field diaphragm or by using pinholes. Photo-marking a selective pool of proteins allows us to track the movement of proteins as the cell progresses through mitosis. Unlike GFP, this technique will help distinguish photo-marked proteins from newly synthesized proteins. Thus, we think that the plasmids developed in this study will be broadly useful to yeast researchers studying the spatial and temporal targeting of proteins.
Acknowledgments
We thank Xianying Tang for critically reading the manuscript. We are very grateful to Dr Patricia Wadsworth and Dr Magdalena Bezanilla for letting us use their confocal microscope for photoactivation experiments. This work was supported by an HHMI Academic Research Internship and a University of Massachusetts Amherst Biology Department Junior Fellowship to C.V., and an NIH/NIGMS grant (No. 1R01GM076094) to W.-L.L.
Footnotes
This article is a U.S. Government work and is in the public domain in the U.S.A.
References
- Cerveny KL, Studer SL, Jensen RE, Sesaki H. Yeast mitochondrial division and distribution require the cortical Num1 protein. Dev Cell. 2007;12:363–375. doi: 10.1016/j.devcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Chudakov DM, Lukyanov S, Lukyanov KA. Tracking intracellular protein movements using photoswitchable fluorescent proteins PS-CFP2 and Dendra2. Nat Protoc. 2007;2:2024–2032. doi: 10.1038/nprot.2007.291. [DOI] [PubMed] [Google Scholar]
- Farkasovsky M, Kuntzel H. Yeast Num1p associates with the mother cell cortex during S/G2 phase and affects microtubular functions. J Cell Biol. 1995;131:1003–1014. doi: 10.1083/jcb.131.4.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkasovsky M, Kuntzel H. Cortical Num1p interacts with the dynein intermediate chain Pac11p and cytoplasmic microtubules in budding yeast. J Cell Biol. 2001;152:251–262. doi: 10.1083/jcb.152.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenz NP, Wadsworth P. Prophase microtubule arrays undergo flux-like behaviour in mammalian cells. Mol Biol Cell. 2007;18:3993–4002. doi: 10.1091/mbc.E07-05-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaber RF, Copple DM, Kennedy BK, et al. The yeast gene ERG6 is required for normal membrane function but is not essential for biosynthesis of the cell cycle-sparking sterol. Mol Cell Biol. 1989;9:3447–3456. doi: 10.1128/mcb.9.8.3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, et al. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Heil-Chapdelaine RA, Oberle JR, Cooper JA. The cortical protein Num1p is essential for dynein-dependent interactions of microtubules with the cortex. J Cell Biol. 2000;151:1337–1344. doi: 10.1083/jcb.151.6.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, et al. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Knop M, Siegers K, Pereira G, et al. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast. 1999;15:963–972. doi: 10.1002/(SICI)1097-0061(199907)15:10B<963::AID-YEA399>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Lee WL, Oberle JR, Cooper JA. The role of the lissencephaly protein Pac1 during nuclear migration in budding yeast. J Cell Biol. 2003;160:355–364. doi: 10.1083/jcb.200209022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees ND, Skaggs B, Kirsch DR, Bard M. Cloning of the late genes in the ergosterol biosynthetic pathway of Saccharomyces cerevisiae. Lipids. 1995;30:221–226. doi: 10.1007/BF02537824. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Patterson GH. Fluorescent proteins for photoactivation experiments. Methods Cell Biol. 2008;85:45–61. doi: 10.1016/S0091-679X(08)85003-0. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, III, Demarini DJ, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Parks LW, Smith SJ, Crowley JH. Biochemical and physiological effects of sterol alterations in yeast. Lipids. 1995;30:227–230. doi: 10.1007/BF02537825. [DOI] [PubMed] [Google Scholar]
- Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- Patterson GH, Lippincott-Schwartz J. Selective photolabeling of proteins using photoactivatable GFP. Methods. 2004;32:445–450. doi: 10.1016/j.ymeth.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- Tulu US, Rusan NM, Wadsworth P. Peripheral, non-centrosome-associated microtubules contribute to spindle formation in centrosome-containing cells. Curr Biol. 2003;13:1894–1899. doi: 10.1016/j.cub.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Welihinda AA, Beavis AD, Trumbly RJ. Mutations in LIS1 (ERG6 ) gene confer increased sodium and lithium uptake in Saccharomyces cerevisiae. Biochim Biophys Acta. 1994;1193:107–117. doi: 10.1016/0005-2736(94)90339-5. [DOI] [PubMed] [Google Scholar]
- Wiedenmann J, Ivanchenko S, Oswald F, et al. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc Natl Acad Sci USA. 2004;101:15905–15910. doi: 10.1073/pnas.0403668101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahedi RP, Sickmann A, Boehm AM, et al. Proteomic analysis of the yeast mitochondrial outer membrane reveals accumulation of a subclass of preproteins. Mol Biol Cell. 2006;17:1436–1450. doi: 10.1091/mbc.E05-08-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]