

Abstract
IL-32 is a multi-faceted cytokine with a role in infections, autoimmune diseases, and cancer, and it exerts diverse functions, including aggravation of inflammation and inhibition of virus propagation. We previously identified IL-32 as a critical regulator of endothelial cell (EC) functions, and now reveal that IL-32 also possesses angiogenic properties. The hyperproliferative EC of human pulmonary arterial hypertension (PAH) and glioblastoma multiforme exhibited a markedly increased abundance of IL-32, and, significantly, the cytokine colocalized with integrin αVβ3. VEGF receptor blockade, which resulted in EC hyperproliferation, increased IL-32 threefold. siRNA-mediated silencing of IL-32 negated the 58% proliferation of EC that occurred within 24h in scrambled-transfected controls. Reduction of IL-32 neither affected apoptosis (insignificant changes in Bak-1, Bcl-2, Bcl-XL, LDH, annexin V, and propidium iodide) nor VEGF or TGF-β levels, but siIL-32-transfected adult and neonatal EC produced up to 61% less NO, IL-8, and MMP-9, and up to 3-fold more activin A and endostatin. In co-culture-based angiogenesis assays, IL-32γ dose-dependently increased tube formation up to 3-fold; an αVβ3 inhibitor prevented this activity, and reduced IL-32γ-induced IL-8 by 85%. In matrigel plugs loaded with IL-32γ, VEGF, or vehicle, and injected into live mice, we observed the anticipated VEGF-induced increase in neocapillarization (8-fold vs vehicle), but unexpectedly, IL-32γ was equally angiogenic. A second signal such as IFNγ was required to render cells responsive to exogenous IL-32γ; importantly, this was confirmed using a completely synthetic preparation of IL-32γ. In summary, we add angiogenic properties that are mediated by integrin αVβ3 but VEGF-independent, to the portfolio of IL-32, implicating a role for this versatile cytokine in PAH and neoplastic diseases.
Introduction
Since its designation as a cytokine by Kim and colleagues in 2005 (1), considerable progress has been made with elucidating the properties of the unusual cytokine IL-32. Structurally, IL-32 does not share similarities with known cytokine families (1). Seven isoforms, IL-32α to ζ (1, 2) and one additional isoform (3) have been described and alternative splicing appears to have biological relevance. For example, in endothelial cells (EC)3, an isoform switch from α/γ to β/ε occurs upon stimulation with IL-1β or thrombin (4), and a protective function for this splicing event has been suggested (5). Moreover, an isoform switch from IL-32γ to IL-32β in tissues from patients with rheumatoid arthritis is associated with an attenuation of inflammation (6). A receptor for IL-32 is currently unknown, although ligand-affinity column assays have shown that IL-32 can bind to neutrophil proteinase-3 (7), and that subsequent processing alters the biological activity of IL-32α and IL-32γ (8).
The earlier studies on IL-32 focused mainly on its pro-inflammatory properties, for example the induction of other cytokines and chemokines such as IL-1β, IL-6, and TNF as well as Th1 and Th17-associated cytokines in various cells, via activation of the p38 mitogen-activated protein kinase, NF-κB, and AP-1 signal transduction pathways (1, 9). IL-32 is present in increased abundance in a variety of diseases, including chronic obstructive pulmonary disease (10), inflammatory bowel disease and psoriasis (11), allergic rhinitis (12), and myasthenia gravis (13), and its levels are directly related to disease severity in rheumatoid arthritis (14, 15).
We and others have shown that IL-32 possesses anti-viral properties. For instance, silencing of IL-32 by small interfering (si)RNA4 (siIL-32)5 resulted in increased production of human immunodeficiency virus (HIV)-1 (9) as well as higher viral loads of vesicular stomatitis virus (VSV) and herpes simplex virus (HSV)-2 (16). In each of these models, the abundance of IFNs was dependent on the levels of IL-32, but the anti-viral activity of IL-32 was only in part via type I IFNs. IL-32 has also been implicated in the immune response to influenza A (17), hepatitis B (18) and C (19), papillomavirus (20), and the Venezuelan equine encephalitis virus (21).
With regard to neoplastic diseases, IL-32 has been demonstrated to modulate apoptosis in myelodysplastic syndromes and chronic myeloid leukemia (22). IL-32 also exhibited anti-apoptotic properties in pancreatic cancer cells (23) and was associated with a more malignant phenotype in tumors of the lung (24). Conversely, IL-32γ overexpression by transgene or cell transfer inhibited the growth of melanomas and colon tumors (25).
In EC of various origin, IL-32 is a crucial mediator of pro-inflammatory stimuli such as IL-1β, thrombin, LPS, and platelets: We found that the abundance of IL-32 was increased by treatment with these triggers of EC-inflammation, and silencing by siIL-32 resulted in decreased production of the pro-inflammatory IL-1α, IL-6, IL-8, and ICAM-1, as well as increased expression of thrombomodulin/CD141 (4). Furthermore, IL-32 has been shown to mediate giant cell arteritis (26), to interact with integrins (27), and to play an important role at multiple levels in atherosclerosis (5).
A dysregulation of the functions of EC plays a major role in pulmonary arterial hypertension (PAH)6. Several forms of PAH have been classified, but many of them are characterized by complex pulmonary vascular lesions. These lesions are multicellular and demonstrate hyperproliferative EC that grow in an uncontrolled fashion, to the point of obliteration of the vascular lumen (28). Mechanisms likely involved in this pulmonary microvessel disease have recently been reviewed (29, 30). Importantly, the proliferating EC are apoptosis-resistant, form multiple lumina often with the appearance of glomeruloid structures, or structures which are reminiscent of the vascular coils observed in the tumors of glioblastome multiforme (GBM)7, a tumor which originates from EC precursors (31). Increasingly a role for inflammatory cytokines and immune cells in PAH is being recognized (32, 33); for example, IL-1 and IL-6 plasma levels are increased in patients with severe PAH (32). Although a consensus is building that inflammation and immune mechanisms play a role in the pathobiology of severe forms of PAH, little is known as to whether inflammation causes the formation of the vascular lesions or how inflammatory cells and immune cells contribute to the angio-obliteration (angiogenesis) of these remodeled lung vessels (34).
As IL-32 acts as an important regulator of EC biology (4), and because the prototypical angiogenic mediator vascular endothelial growth factor (VEGF)8 is highly expressed in the plexiform vascular lung lesions (35), as well as in psoriatic angioproliferative skin lesions (36), we asked whether IL-32 could also function as an angiogenic factor.
Materials and Methods
Reagents
Human umbilical vein endothelial cells (HUVEC)9 media and additives were from Lonza Inc., Walkersville, MD or from PromoCell, Heidelberg, Germany (ECGM MV, used in the co-culture experiments). Iscove’s DMEM, RPMI 1640, and M199 were obtained from Gibco/Invitrogen, Carlsbad, CA. Human dermal fibroblasts (HSF) and their medium (FGM2) were from PromoCell. PBS, FCS, and penicillin/streptomycin were purchased from Cellgro, Herndon, VA. Pooled human serum and accutase were acquired from MP Biomedicals, Solon, OH. LPS (O55:B5) was from Sigma Aldrich, St. Louis, MO. The Nucleofector II electroporation device and reagents as well as primocin were from Amaxa, Cologne, Germany. siIL-32 was purchased from Thermo Fisher Scientific, Lafayette, CO and comprised 4 ON-TARGETplus duplexes (I–IV) with the antisense sequences I, 5′-uaa uaa gcc gcc acu guc uuu-3′; II, 5′-ccg uaa ucc auc ucu uuc uuu-3′; III, 5′-uca uca gag agg acc uuc guu-3′; IV, 5′-caa gua gag gag uga gcu cuu-3′. One sequence comprised a quarter of the total siRNA concentration; i.e., 25 nM of each sequence were pooled for a total siRNA concentration of 100 nM used in the transfections. Scrambled siRNA (Silencer Negative Control #1) was purchased from Ambion, Austin, TX. Recombinant human IL-32γ and IL-1β, and recombinant murine VEGF were from R&D Systems, Minneapolis, MN. As per the manufacturer, the endotoxin level in rhIL-32γ was <1 EU per μg by the limulus amoebocyte lysate method. All other recombinant cytokines were bought from Peprotech, Rocky Hill, NJ. We generated the affinity-purified anti-human IL-32 antibody and have utilized it previously (1, 9). FITC-labeled Sambuccus nigra lectin was from Vector Laboratories, Burlingame, CA. The αVβ3 inhibitor cyclo [Arg-Gly-Asp-D-Phe-Val] was obtained from Enzo Life Sciences, Farmingdale, NY.
Generation of synthetic IL-32γ
The chemical synthesis of IL-32γ (103 residues) was performed following a 4-fragment strategy of native chemical ligation. Fragments used to assemble the protein were i) the N-terminal fragment, residues 1-35-Cα-COS-R (F1); ii) the intermediate fragment, residues 36-56-Cα-COS-R with an N-terminal Cys introduced as thiazolidine (F2a); iii) another intermediate fragment, residues 57-59-Cα-COS-R with an N-terminal Cys introduced as thiazolidine (F2b); and the C-terminal fragment, residues 60–103 (F3). F1, 2a and 2b were assembled by the solid phase method on a MPAG (β-mercapto-propionic acid-glycine) resin to generate C-terminal thioesters with R = β-mercapto-propionic acid-glycine, following Boc protocols with in situ neutralization and using related chemicals (37). F3 was assembled by standard Fmoc chemistry on a Wang resin to afford a C-terminal free polypeptide. After cleavage, polypeptides were purified to homogeneity by RP-HPLC and characterized by LC-MS to assess purity and identity. Native chemical ligation and thiazolidine opening reactions were performed as reported elsewhere (37). The intermediate ligated polypeptides IL-32 (57–103), IL-32 (36–103) and the final full length 103 residue products were also purified by RP-HPLC and characterized by LCMS to assess purity and identity: F1, expected Mr 4011.6, found 4011.4; purity 90%; F2a, expected Mr 2760.4, found 2760.0; purity 95%; F2b, expected Mr 560.1, found 560.0; purity 97%; F3, expected Mr 5042.5, found 5043.0; purity 95%; full length IL-32γ, expected Mr 11570.4, found 11570.3; purity >95%. The single letter code sequence of the full-length IL-32γ is as follows, with the ligation sites in bold and underlined: MCFPKVLSDD MKKLKARMVM LLPTSAQGLG AWVSACDTKD TVGHPGPWRD KDPALWCQLC LSSQHQAIER FYDKMQNAES GRGQVMSSLA ELEDDFKEGY LET.
Cell Culture
HUVEC were isolated from human umbilical cords after informed consent was obtained from the parents. All experiments were approved by the Colorado Multiple Institutional Review Board. Cords were cleaned by several high-volume rinses with Hank’s balanced Salt Solution. Thereafter, one end of the cord was clamped and a catheter was inserted from the open end, through which the umbilical vein was filled with 0.3% trypsin (Gibco). The catheter was removed, the open end was clamped before the cord was incubated for 30 min at 37°C. Thereafter, the cord was massaged, the clamp was removed, and the cord was squeezed lengthwise to force the content into a collection tube. Two washes with HBSS were used to detach residual cells from the vein. The collection tube was centrifuged at 300 × g for 10 min, the pellet was resuspended in HUVEC growth medium (39% Iscove’s DMEM, 38.6% RPMI 1640, 20% FCS, 0.4% endothelial mitogen (Biomedical Technologies), 17.6 U/ml heparin, 1% penicillin/streptomycin) and transferred into cell culture flasks coated with 1% gelatin. For stimulation, the medium was changed to endothelial cell stimulation medium comprising M199 (Gibco) with 2% FCS, 10 ng/ml human acidic FGF (Peprotech), 1% penicillin/streptomycin, and 17.6 U/ml heparin.
Aortic macrovascular EC as well as coronary and pulmonary microvascular EC were obtained from Lonza (Walkersville, MD). Coronary and pulmonary EC were cultured in EGM-2MV (Lonza) with a final concentration of 5% FCS. Aortic EC were grown in EGM-2 with a final concentration of 2% FCS.
PBMC were isolated and cultured as described previously (38).
For the co-culture experiments, HSF were cultivated in a 48-well plate (5×104 cells/well) and grown to confluence for 3 days in FGM2 plus Supplement Mix (PromoCell). At day 3, HUVEC (5×104 cells/well) were carefully added to the confluent HSF. After 4h of incubation, HUVEC were attached and the incubation media was exchanged with a 1:1 mixture of ECGM MV and FGM2 (plus the respective Supplement Mix) with or without IL-32 (10, 25, 100 ng/ml) and with or without cyclo [Arg-Gly-Asp-D-Phe-Val] (10 μM). Treatment with recombinant human VEGF-165 (40 ng/ml) was used as internal assay control. The cells were co-cultured for 7 days with two media changes at 37°C and 5% CO2. Both HUVEC and HSF were used in passages 2–8 and passaged using the DetachKit (PromoCell).
Transfections
HUVEC were detached, counted, centrifuged at 200 × g for 12 min, and subjected to electroporation using the Amaxa HUVEC Nucleofector Kit and program U001. One cuvette contained 0.8 × 106 cells in 100 μl Nucleofector solution and 25 to 333 nM of either siIL-32 or scrambled. Immediately after electroporation, the cells were incubated in 300 μl prewarmed M199 for 5 min. Thereafter, 0.2 × 106 cells were transferred into gelatinized 6-well plates (Greiner, Ocala, FL) to a final volume of 1 ml growth medium and allowed to recover overnight. On the next day, the medium was replaced with stimulation medium and the cells were stimulated.
Immunohistochemistry
Briefly, after paraffin-embedded sections were cut at 5 μm sections, the slides were deparaffinized and transferred through progressive ethanol gradients for rehydration. High temperature antigen retrieval was performed in 10 mM/l citrate buffer, pH 6.0 for 30 minutes. Endogenous peroxidase was quenched with 3% hydrogen peroxide for 15 minutes twice. All immunostains were performed on the Dako using the Dako Universal LSAB™ + Kit/HRP peroxidase-based visualization kit. Primary antibody incubation was performed at 1:1500 dilution for 30 minutes. Chromagen development was performed with 3,3′-diaminobenzidine and counterstained with hematoxylin. Normal blocking serum without primary antibody was used for the negative control.
Immunofluorescence and confocal microscopy
Paraffin-embedded human lung tissue, obtained from patients with idiopathic PAH and control tissue from patients without pulmonary vascular disease was used for double immunofluorescence. First, slides were steamed for 20 minutes in 0.01 M citrate buffer pH 6.0 and blocked with 1% normal swine serum for 15 minutes. Primary antibody no. 1, IL-32 (1:50, Abcam, Cambridge, MA) was applied overnight at 4°C in 1% normal swine serum. Secondary antibody no. 1, anti-rabbit Alexa Fluor 594 (1:100, Invitrogen, Carlsbad, CA) was incubated for 4 hours at room temperature in PBS. Sections were incubated with primary antibody no. 2, anti-mouse von Willebrand factor (vWF)10 (1:20, LifeSpan Biosciences Inc., Seattle, WA) overnight at 4°C in PBS, then secondary antibody no. 2, anti-mouse Alexa Fluor 488 (1:100, Invitrogen) was applied for 4 hours at room temperature in PBS. Slides were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) at 1:20,000 and mounted with SlowFade Antifade (both Invitrogen). Negative controls with unspecific IgG were run in parallel. Optical sections were acquired by laser- scanning confocal microscopy with a Leica TCS-SP2 confocal microscope and images were analyzed and arranged with ImageJ. Microscopy was performed at the VCU Department of Anatomy and Neurobiology Microscopy Facility, supported, in part, with funding from NIH-NINDS Center core grant (5P30NS047463-02).
Electrochemiluminescence (ECL) assays and ELISAs
Human IL-6 and IL-32 were measured using specific antibody pairs and an Origen Analyzer (Wellstat Diagnostics, Gaithersburg, MD) as described previously (9); antibody pairs for all cytokines were obtained from R&D Systems. The IL-6 ELISA was obtained from Elisakit.com (Scoresby, VIC, Australia). TGF-β1 was determined by ELISA (R&D Systems) according to the manufacturer’s instructions. Recombinant cytokines for ECL or ELISA standards were obtained from R&D Systems or Peprotech (Rocky Hill, NJ).
Real-time PCR analysis
RNA was extracted from HUVECs using the RNA Mini Kit (Bioline, Alexandra, NSW, Australia), then quantified with the NanoDrop (ND-1000) Spectrophotometer (Thermo Scientific, Wilmington, DE). RNA was reversely transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Melbourne, VIC, Australia) as per the manufacturer’s instructions. SYBR Green qRT-PCRs were run on the Applied Biosystems 7900HT Fast RT-PCR system. Oligonucleotide primers were as follows: 18S, ccc ctc gat gct ctt agc tg & ctt tcg ctc tgg tcc gtc tt; activin A, gtt tgc cga gtc agg aac ag & ccc ttt aag ccc act tcc tc; angiogenin, gcc gag gag cct gtg tt & gcg ctt gtt gcc atg aat; angiopoietin 2, agc taa gga ccc cac tgt tg & tga agg gtt acc aaa tcc cac t; Bak-1, cca cca gcc tgt ttg aga gt & aaa ctg gcc caa cag aac ca; Bcl-2, cgc gac tcc tga ttc att gg & cag tct act tcc tct gtg atg ttg t; Bcl-XL, act ctt ccg gga tgg ggt aa & aca aaa gta tcc cag ccg cc; endoglin, gcc cca caa gtc ttg cag aa & gct tgg atg cct gga gag tc; IL-8, act cca aac ctt tcc acc cc & gcg gaa gat gac ctt ctc ct; and u-plasminogen activator, gtc gtg agc gac tcc aaa gg & gac tta tct att tca cag tgc tgc c. No template controls were included in parallel for each gene master mix. The cycling conditions were as follows: denaturation at 95°C for 10mins, followed by 40 cycles of denaturation at 95°C for 15s, annealing and elongation at 60°C for 1 min. These cycles were followed by a melt-curve analysis, 95°C for 15s, 60°C for 15s followed by 95°C for 15s. Relative expression quantification was calculated using Pffafl’s method. All fold-changes in gene expression were normalized to 18S mRNA.
MTS assay
The CellTiter cell proliferation assay was purchased from Promega (Madison, WI) and executed according to the manufacturer’s instructions. Briefly, cells were transfected and 2000 cells in 100 μl stimulation medium were seeded into a 96-well plate. Cells were grown for 3d, then 20 μl of MTS reagent were added to each well, followed by a further 2h incubation. Then the absorbance at 490 nm was recorded in a plate reader. The quantity of the formazan product as measured by the absorbance is proportional to the number of living cells in the cultures.
Determination of nitrate/nitrite
Supernatants from EC cultures were collected and assayed for total nitrate/nitrite concentration using a kit provided by Cayman Chemical (Ann Arbor, MI).
In vitro angiogenesis model
On day 7 of co-culture, cells were washed with PBS, fixed with 2% paraformaldehyde (pH 7.4) for 10 min at RT, treated with 0.1% Triton X-100 and blocked with 2% BSA in PBS. Thereafter, the co-culture was incubated with sambuccus nigra lectin-FITC (20 μg/ml) for 1h at RT in the dark. Cell nuclei were labeled using DAPI. Cells were washed thoroughly, covered with PBS and examined microscopically on an Axiovert, Carl Zeiss, Germany. Five fields of view per condition were randomly chosen and photographed. The number of visible branches was counted using ImageJ.
Matrigel-related procedures in vivo
All animal experiments were approved by the Colorado Multiple Institutional Review Board. Vehicle (NaCl 0.9%), VEGF, or recombinant IL-32γ (both R&D Systems) was mixed with growth factor-reduced high concentration matrigel (BD, San Diego, CA) at 4°C. Thereafter, each mouse (male ICR mice) received a subcutaneous injection with two 200 μl aliquots of matrigel with non-identical contents on the right and left sides of the abdominal wall. The mice were allowed free access to food and water for 14 days, followed by harvest of the plugs as described below.
Histopathological analysis and scoring of matrigel plugs
This analysis was performed by Premier Labs (Boulder, CO). Each of the scientists at Premier Labs was blinded to the content of the matrigel plugs. Matrigel plugs and the surrounding tissue, skin, and muscle were removed at necropsy and placed into 10% neutral buffered formalin. The plugs were bisected, processed, and embedded in paraffin. Multiple sections were cut, one was stained with hematoxylin and eosin and another was immunohistochemically stained for CD31 as follows: Slides were de-waxed in xylene and hydrated to water through a series of alcohol gradients. Prior to staining, the tissue sections were pre-treated with a TRIS/EDTA pH 9.0 target retrieval solution (Dako, K8004) and incubated for 2 hours at 70°C. A 3.0% hydrogen peroxide solution was used for 5 minutes at room temperature to quench any endogenous peroxidase activity within the tissue. Serum-free Protein Block (Dako, X0909) was used to neutralize any charged molecules that may cause non-specific antibody binding. The rat anti-mouse CD31/PECAM-1 antibody (Dianova, Hamburg, Germany) was applied to the tissue sections at a working concentration of 5.0 μg/ml (1:40) from the stock solution for 60 minutes at room temperature. The primary antibody was then conjugated with a biotinylated rabbit anti-rat immunoglobulin (Dako, E0468) at a concentration of 2.125 μg/ml (1:400) for 30 minutes at room temperature. The biotinylated secondary antibody was then amplified with a goat anti-rabbit-labeled polymer (Dako, K4003) for 30 minutes at room temperature. Thereafter, staining was developed with liquid DAB+ for 5 minutes at room temperature (Dako K3468). Counterstaining was developed with automated hematoxylin (Dako, S3301) for 10 minutes. The negative control solution for this immunohistochemical stain was an un-conjugated rat IgG2a isotype antibody (Serotec MCA 1124) that was used at the same working concentration as the primary antibody.
The H&E-stained slides were reviewed by a board certified veterinary pathologist. The evaluation included the degree of fibrosis in the subcutis adjacent to each implant as well as the inflammatory response in the subcutis, and the overall cellularity within each implant. For scoring the metrics identified in the Figures, the most advanced changes within an implant were used for scoring. Neocapillaries were defined by the presence of oval-circular structures containing red blood cells, and associated with concentrically arranged mesenchymal nuclei (fibroblasts and endothelial cells). A subjective, semi-quantitative scoring system was utilized: 0, no significant change; 1, minimal change; 2, mild change; 3, moderate change; and 4, marked change.
The CD31-stained slides were scanned on an Aperio ScanScope XT, the area of matrigel implant was traced and then analyzed via Aperio’s microvessel algorithm that calculated the number of vessels and microvessel density along with other parameters.
Statistical analysis
Datasets (raw data) were first tested for normality by the Kolmogorov-Smirnov method and equal variance (p value to reject 0.05) using the SigmaStat software (Systat Software Inc., San Jose, CA). Thereafter, these data were analyzed by the appropriate statistical test, including Student’s t-test, One-way ANOVA, and One-way ANOVA on ranks.
Results
IL-32 in tissue sections from patients with PAH and in an in vitro model of pulmonary angioproliferation induced by VEGF receptor blockade
We reported that IL-32 is important in EC biology (4). Therefore, a next reasonable step was to investigate the role of this cytokine in diseases that involve EC. We stained tissue sections from patients with pulmonary hypertension (PAH) using two different antibodies against IL-32 (Figure 1A–D and E–H). In a normal human lung arteriole, IL-32 was detected in the vascular smooth muscle cells of the medial layer (shown in red in Figure 1A), but not in the endothelial monolayer, as highlighted by von Willebrand factor (vWF)-staining in green. On the other hand, the EC in lung specimen from patients with idiopathic PAH did express IL-32. Figure 1B shows a patent arteriole, with the arrowheads in the insets pointing at the IL-32-positive EC. When we examined the characteristic plexiform lesions, which are diagnostic for idiopathic PAH (28, 31), we found a yet higher abundance of IL-32 within the hyperproliferative EC that grow to obstruct the blood vessels. In the specimen in Figure 1C, which was obtained from the same patient displayed in panel B, an arteriolar vessel is almost completely obliterated by the IL-32-expressing, hyperproliferative EC. Some of these EC, particularly in the center of the lesion, have very large nuclei (stained by DAPI in blue) and are activated, phenotypically switched cells that have lost the vWF marker. Panel 1D and the insets show more detail at a higher magnification and the colocalization of vWF and IL-32 (arrows). Panels E–H demonstrate a similar pattern of IL-32 expression when we used classical immunohistochemistry. The red arrowheads point at the expression of IL-32 in the proliferating EC. We also found IL-32 in the bronchial epithelium (golden arrows), which was expected from an earlier study (10). For negative control, we performed the staining in the absence of the primary antibodies, and observed no staining on any specimen (data not shown).
FIGURE 1.

IL-32 in PAH. (A–D) Triple-label immunofluorescence images of human lung vessels. IL-32 is stained in red, vWF in green, and nuclei in blue (DAPI). Scale bar: 50 μm. (A) Staining of a normal pulmonary arteriole (arrow) from a patient not suffering from PAH. Image is representative for 3 similar results. (B–D) Specimen from one out of a total of 3 similar patients with idiopathic PAH. (B) An affected, but not obliterated arteriole with activated EC is depicted. Dotted lines indicate areas that are enlarged in insets on right. The arrowheads point to EC expression of IL-32. (C, D) A plexiform lesion is shown, in which the lumen of the blood vessel is obliterated by hyperproliferative EC. Dotted line indicates area that is enlarged in (D), and yet more detail is provided in insets on right. The arrowheads point to the hyperproliferative EC that stain positive for IL-32 and vWF. (E–H) Classical immunohistochemistry of IL-32 in lung specimen of PAH patients. After incubation with the primary antibody to IL-32, slides were stained with diaminobenzidine and hematoxylin. Diseased, hyperproliferative pulmonary blood vessels with a markedly thickened endothelium are shown in overview in panel E (100x magnification) and in detail in panels F–H (200x). The innermost endothelial layer contains a considerable amount of IL-32 protein (red arrows). Staining is also seen in alveolar epithelial cells (golden arrows). Inages are representative for n = 3 patients. (I) HUVEC were plated and grown for 24h. Thereafter, the medium was changed to stimulation medium (2% FCS, see Methods) and either semaxanib (10 μM) or vehicle was added. Cells were harvested after the indicated periods of time and lysates were assayed for IL-32 protein and total protein. IL-32 abundance was normalized to total protein, fold-increases in normalized IL-32 abundance were calculated (IL-32 in semaxanib-treated cells divided by IL-32 in vehicle-treated cells), and IL-32 abundance in vehicle-treated cells was set at 1. Each line indicates fold-change in semaxanib-treated cells over vehicle-treated cells in one time course experiment. n = 4; *, p < 0.05 for semaxanib vs vehicle.
We previously reported that blockade of the VEGF receptor by the small molecule inhibitor semaxanib (Su5416) initially causes apoptosis. However, EC surviving this initial phase become hyperproliferative and grow in an uncontrolled fashion which can result in obstruction of the lumen of artificial capillaries (39), resembling the pathology of PAH (panels A–H). Hence, treatment of EC with semaxanib can be regarded as a model of this disease, which we employed to confirm the immunohistochemistry results. As depicted in Fig. 1I, IL-32 protein levels increased up to 3.4-fold as HUVEC underwent the transformation into the hyperproliferative state. In 3 of the 4 experiments, IL-32 expression peaked on day 3. However, in the EC from the fourth cord which, unlike the other HUVEC, contained considerably less IL-32 in semaxanib-treated cells than did vehicle-treated controls 24h after the initial stimulation, the maximal increase in IL-32 protein expression occurred on day 7 (brown line and points in Fig. 1I).
IL-32 in glioblastoma multiforme (GBM)
Based on the findings in the lung, we hypothesized that expression of IL-32 may be associated with augmented proliferation of EC. To examine this hypothesis, we stained tissue sections from GBM, which is a malignant brain tumor where EC growth (angiogenesis) is of critical importance for tumor growth. Again, we observed only occasional staining of IL-32 in unaffected areas of the brain. The tumor cells, however, showed massive expression of IL-32 protein (red arrows in Figure 2). Negative control experiments were performed as described in the previous section and did not exhibit any color changes (data not shown).
FIGURE 2.
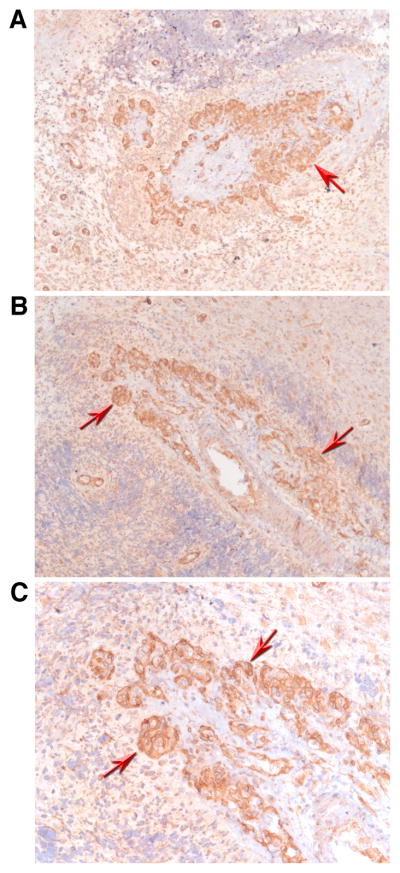
IL-32 in glioblastoma multiforme (GBM). Immunohistochemistry with an antibody against IL-32 in sections of brains affected by GBM was performed and is depicted at an original magnification of 100x (A, B) or x 200 (C). Strong staining for IL-32 protein is observed in areas affected by the tumor (red arrows). Images are representative for those obtained from a total of three GBM patients.
siIL-32 reduces the proliferation of EC, but changes in IL-32 protein abundance do not affect EC apoptosis
Nearly all of the experiments in this study in which siIL-32 was used were performed together with those we have previously published (4). In fact, the same supernatants and lysates were used in most cases; therefore, Figs. 3A and B, which show the concentration-dependent knockdown of IL-32 protein by siIL-32 compared to scrambled siRNA without (panel A) or with IL-1β stimulation (panel B), are recycled from (4).
FIGURE 3.

Proliferation and NO abundance after silencing of IL-32 in HUVEC. All statistical comparisons are *, p < 0.05; **, p < 0.01; and ***, p < 0.001 for siIL-32 vs scrambled siRNA. (A, B) These panels are reproduced from (4), since the results in this Figure were obtained from the same lysates and supernatants as those in Figs. 4 and 5 in (4). Transfection of HUVEC with either the indicated concentrations of siIL-32 or scrambled siRNA was followed by stimulation with 10 ng/ml IL-1β (B) or vehicle (A) for 20h. Mean ± SEM of IL-32 abundance in cell lysates normalized to total protein (t.p.) is depicted; n = 10. (C) HUVEC were gently detached from the plates and viable cells were counted by hand using the trypan blue exclusion method. The number of cells that was seeded after transfection was set as baseline. Mean percent change in siIL-32-transfected cells compared to scr-transfected cells ± SEM is shown; n = 4. (D) After transfection with the indicated concentration of siIL-32 or the same concentration of scrambled siRNA, cells were plated, IL-1β (10 ng/ml) or vehicle was added, and the cultures were incubated for 3d. Thereafter, MTS assays were performed. The graph shows means of percent changes ± SEM in the number of live cells, comparing 25 or 100 nM siIL-32 to the appropriate concentration of scrambled siRNA, which is set at 100%; n = 6. (E) Supernatants from HUVEC transfected with 100 nM of siIL-32 or scrambled siRNA were collected after 48h and assayed for total nitrate/nitrite concentration. Mean concentration of total NO normalized to total protein (t.p.) ± SEM is depicted; n = 6.
Since the abundance of IL-32 was increased in hyperproliferative EC, we reasoned that silencing of this cytokine should result in a reduction of proliferation of HUVEC. When we knocked down IL-32 with siRNA, we observed that a reduction of IL-32 protein had moderate effects on cell numbers in vehicle-treated HUVEC (left pair of bars in Fig. 3C). However, upon stimulation with IL-1β, a reduction of IL-32 by 76% resulted in a decrease in proliferation from +58% in scrambled siRNA-transfected HUVEC to −15% (right pair of bars in Fig. 3C). To gather additional evidence, we used an MTS assay in which the production of dye is proportional to the number of live cells. As shown in Fig. 3D, the proliferation of IL-1β-stimulated HUVEC was reduced by more than 50% upon silencing of IL-32 compared to scrambled siRNA-transfected cells. Similar to Fig. 3C, the effect was less pronounced in unstimulated HUVEC.
Next, we performed experiments to answer the important question whether the reduction in cell numbers conferred by siIL-32 was due to an increase in apoptosis or truly due to a decrease in proliferation. Using flow cytometric quantification of annexin V to detect the presence of ongoing apoptosis and propidium iodide (PI) to identify cells during late apoptosis and necrosis, we found that siIL-32 conferred a moderate (statistically not significant) increase of annexin V and PI in HUVEC (up to 1.4-fold ± 11% more annexin V and up to 1.2-fold ± 9% more PI compared to scrambled siRNA, n = 6, p > 0.2). In pulmonary microvascular EC, this trend was slightly more pronounced but still not significant with 1.7-fold ± 0.32 more annexin V and 1.6-fold ± 41% more PI with siIL-32, n = 8, p > 0.1. Furthermore, we performed measurements of LDH in the culture supernatants, which also showed that siIL-32 did not result in an increase in cell death (no difference between scrambled and siRNA to IL-32). To be as certain as possible about this crucial aspect, we next treated HUVEC with exogenous IL-32γ in the presence or absence of IFNγ pre-treatment (described below) and measured the mRNA abundance of bak1, bcl2, and bclxl by real-time PCR. None of the three mediators of apoptosis exhibited a significant change in mRNA abundance in IL-32γ-stimulated cells compared to controls (bak1 up to 19% ± 7% increased, bcl2 up to 19% ± 6% decreased, and bclxl up to 17% ± 7% increased, n = 4, p > 0.3). These findings are highly unlikely to be an artifact as the data on IL-8 (which show a pronounced regulation, see Fig. 7G) were obtained from the same samples. Taken together, these data show that the angiogenic effect of IL-32 is not due to a change in apoptotic programing.
FIGURE 7.

Colocalization of integrin αVβ3 with IL-32 and functional relevance of the interaction. (A–F) Representative optical sections acquired by confocal microscopy of double immunofluorescence staining for IL-32 (red) and αVβ3 (green). Nuclei are counterstained with DAPI (blue). (A) In lung tissue from a representative control patient (without pulmonary vascular disease) single αVβ3+ cells are seen in the endothelial layer or perivascular region of a pulmonary artery, indicated by double-headed arrow. One αVβ3+/IL-32+ cell (single-headed arrow) was found in the perivascular tissue (cytoplasmic staining). (B–F) In lung tissue from a representative patient, αVβ3 colocalized with IL-32 in the plexiform lesions. This colocalization occurred in the nucleus, as indicated by arrowheads, as well as in the cytoplasm and the cell membrane, pointed at by arrows. Cells positive for IL-32 alone are indicated with open arrows. (C and E) depict the regions enclosed by the dotted line in (B and D, respectively) at higher magnification. Original magnification was 400x. Scale bar: 50 μm. n = 3 controls and 3 PAH patients. (G) HUVEC were pretreated with IFNγ (10 ng/ml) or vehicle for 24h, followed by addition of recombinant IL-32γ at the indicated concentrations (ng/ml) and/or the αVβ3 inhibitor cyclo [Arg-Gly-Asp-D-Phe-Val] at 10 μM. Six hours thereafter, cells were lysed and subjected to real-time PCR analysis. Fold-changes in abundance of IL-8 mRNA normalized to 18S over control (which is set at 1) ± SEM are depicted. n = 3; *, p < 0.05 and ***, p < 0.001 for IFNγ alone vs IL-32γ plus IFNγ; #, p < 0.05 and ##, p < 0.01 for IL-32γ plus IFNγ vs IL-32γ plus IFNγ plus cyclo [Arg-Gly-Asp-D-Phe-Val].
siIL-32 in EC: TGF-β1, nitric oxide (NO), and VEGF
We further explored the actions of IL-32 using EC in which IL-32 was silenced by siRNA and measured the concentrations of TGF-β1 and NO, two mediators known to play a role in PAH (40). As demonstrated in Fig. 3E, we found that the concentration of NO decreased by 61% from 13 to 5 pg/mg total protein in unstimulated HUVEC in which IL-32 was reduced. By contrast, the abundance of TGF-β1 did not change significantly with siIL-32 (data not shown). Unexpectedly, this was the case for VEGF as well; in fact, the abundance of VEGF was slightly though non-significantly higher in EC transfected with siIL-32 (vehicle: 400 ± 72 vs 508 ± 64 pg/mg total protein in scrambled-transfected vs siIL-32-transfected cultures, respectively, p = 0.09; IL-1β-stimulated HUVEC: 810 ± 87 vs 1082 ± 195 pg/mg total protein in scr vs siIL-32, p = 0.54; n = 9).
Pre-treatment with IFNγ sensitizes EC to exogenous IL-32γ
Next, we attempted to confirm the findings obtained by silencing IL-32 by incubating the HUVEC with exogenous recombinant IL-32γ. We expected to observe inverse effects, but instead exogenous IL-32γ did not affect any of the parameters described previously at concentrations between 1 and 500 ng/ml. However, when HUVEC were pretreated with IFNγ for 24 h, stimulation with exogenous IL-32γ resulted in the expected findings; in IFNγ-pretreated cells, the concentrations of IL-32γ needed to elicit IL-6 production were as low as 10 ng/ml. As demonstrated in Fig. 4A, IFNγ alone conferred a moderate, up to 39%, reduction in the production of IL-6. Treatment with IL-32γ alone had only marginal effects, but together with IFNγ, the abundance of IL-6 increased up to 2.3-fold over control and 3.8-fold over IFNγ alone. This increase was dependent on the concentration of both IL-32γ and IFNγ.
FIGURE 4.

IFNγ sensitizes cells to exogenous IL-32γ. (A) After the medium was changed from growth- to stimulation medium, HUVEC were treated with the indicated concentrations of IFNγ (in ng/ml) or vehicle for 24h. Thereafter, recombinant IL-32γ or vehicle was added at the concentrations indicated (ng/ml). IL-6 was measured in the culture supernatants after another 24h. The graph shows means of fold-changes in normalized IL-6 protein abundance comparing vehicle (control, set as 1) with stimulated conditions ± SEM; n = 7; *, p < 0.05 and **, p < 0.01 for IFNγ alone vs IL-32γ plus IFNγ. (B, C) PBMC were incubated with 10 ng/ml of IFNγ or vehicle for 24h, followed by addition of synthetic (s)IL-32γ at the indicated final concentrations. Supernatants were harvested 24h later and assayed for IL-6 (B) and TNF (C). Absolute concentrations of both cytokines ± SEM are depicted, n = 4 healthy donors. Grey bar (very left), medium alone; black bar (second from left), IFNγ alone; open bars, sIL-32γ alone; striped bars, sIL-32γ plus IFNγ. *, p < 0.05 and **, p < 0.01 for sIL-32γ alone vs sIL-32γ plus IFNγ. n.d., not detected.
Interestingly, the sensitization effect was specific to IFNγ. We also pre-treated the HUVEC cultures with IL-1β (1 and 10 ng/ml), LPS (10 and 100 ng/ml), muramyl dipeptide (0.2 and 1 μg/ml), thrombin (0.1 and 0.5 U/ml), or VEGF at 1 and 10 ng/ml, none of which increased the responsiveness of the cells to exogenous IL-32γ.
To further characterize this unexpected effect, we tested whether IFNγ pre-treatment increased the response to a different preparation of exogenous IL-32γ in a different cell type. We generated a completely synthetic IL-32γ peptide and added this protein to PBMC cultures that had been pretreated with IFNγ or vehicle. Fig. 4B and C show that IFNγ indeed sensitized PBMC: The abundance of IL-6 was up to 8-fold higher and that of TNF was up to 24-fold increased compared to exogenous synthetic IL-32γ alone, whereas the effects of IFNγ alone were negligible.
In vivo angiogenesis assays (matrigel)
To obtain in vivo evidence for the angiogenic properties of IL-32, we employed the matrigel method (41, 42). Growth factor-reduced matrigel was loaded with two different concentrations of recombinant IL-32γ (25 and 100 ng/ml), VEGF (25 ng/ml) for positive control, or vehicle for negative control, and injected s.c. The plugs and the surrounding tissue were harvested 14 days later. After cutting, the sections were stained for the endothelial marker CD31 and then analyzed for the formation of capillaries using both a semi-quantitative scoring system, in which 0 represented no change, 4 stood for maximum change, and 1, 2, and 3 for intermediate levels of change, as well as an automated algorithm that was based on CD31+ cells. Functionality of the new vessels was ascertained by confirming the presence of red blood cells within them. In addition, we assessed the infiltration of the plugs by other cells, which were identified by morphology and subjected to the semi-quantitative scoring system described above.
Most importantly, we observed that, compared to vehicle, 100 ng/ml of IL-32γ in the matrigel plug induced a marked angiogenic response; in fact, this response was stronger than that of 25 ng/ml VEGF. Fig. 5 shows slides representative of this effect. Whereas in the VEGF-loaded plugs (a representative one shown in Fig. 5B) the mean score for the formation of new capillaries was 1.65, that score was 1.70 in the plugs loaded with 100 ng/ml IL-32γ (exemplary plug at 10x and 20x magnification, Figs. 5E and 5F, respectively). 25 ng/ml of IL-32γ still resulted in a mean score of 0.78 (Figs. 5C, D), with the vehicle-loaded plugs scoring 0.20 (Fig. 5A; a quantitative summary is depicted in Fig. 6A). These results obtained by semi-quantitative scoring were confirmed by subjecting the same slides to automated counting using the capillary algorithm of the Aperio software. As shown in Fig. 6B, this method produced similar increases, namely 3.5-, 7-, and 6-fold for 25 and 100 ng/ml IL-32γ and VEGF, respectively, over control. Due to the greater variation of the data, only the neocapillarization induced by 100 ng/ml IL-32γ was statistically significant when the computer-based analysis was used. The mean vessel area of the neocapillaries induced by IL-32γ was moderately larger than in control and VEGF plugs (49 μm2 in control and VEGF plugs vs 55 and 72 μm2 in 25 and 100 ng/ml of IL-32γ, respectively (Fig. 6C)).
FIGURE 5.

IL-32γ induces angiogenesis in vivo. Growth factor-reduced high concentration matrigel was mixed with vehicle (A), 25 ng/ml VEGF (B), or recombinant IL-32γ at 25 ng/ml (C, D) or at 100 ng/ml (E, F); n = 10 plugs for each condition. Two 200 μl aliquots of matrigel with non-identical contents were then injected on the right and left sides of the abdominal wall of male ICR mice. The plugs were harvested 14d later, followed by cutting and staining for CD31 (chromagens were diaminobenzidine and hematoxylin). The coworkers involved in the analysis of the matrigel experiments were blinded to the reagent with which each individual plug was loaded. Blinding was lifted only after the completion of all aspects of the analysis. Representative plugs loaded with vehicle (A, 10x magnification), VEGF at 25 ng/ml (B, 20x magnification), or IL-32γ at 25 ng/ml (C, D at 10x and 20x magnification, respectively) or 100 ng/ml (E, F at 10x and 20x magnification, respectively) are depicted. The green line highlights the matrigel plugs, differentiating them from the surrounding tissue, which was left in situ.
FIGURE 6.

Quantitative analysis of neocapillarization, lipoidosis, and influx of inflammatory cells into the matrigel plugs. After cutting and CD31- as well as H&E staining, the slides of the matrigel plugs (n = 10 per group) were assessed for the formation of capillaries (A, B) and for the surface area of these capillaries (C), as well as for the presence of adipocytes (D) and white blood cells (E). (A) Neocapillarization was determined by hand-counting and scored semi-quantitatively with 0 representing no increase in neocapillarization, 1 minimal increase, 2 mild increase, 3 moderate increase, and 4 marked increase in neocapillarization. Mean score ± SEM is shown; **, p < 0.01 for IL-32γ or VEGF vs vehicle. (B) The same slides were assessed by automated analysis using Aperio’s microvessel algorithm. The graph depicts means of absolute numbers of microvessels per μm2 × 10−6 ± SEM; *, p < 0.05 for IL-32γ vs vehicle. (C) Computer-based analysis of the surface area of the capillaries in the plugs. Means of the surface area of the microvessels on each slide ± SEM are shown. (D, E) The same semi-quantitative scoring system as in (A) was used to categorize the degree of lipoidosis (D) and inflammation (E). Mean score ± SEM is shown; *, p < 0.05 for IL-32γ vs vehicle.
To ascertain specificity, we also performed the matrigel assay with plugs loaded with 10 ng/ml of LPS. Angiogenesis in these plugs was equal to that in vehicle controls.
With regard to other observations, the overall cellularity within the implants was mainly composed of inflammatory cells, lipid-containing cells, neocapillaries, and fibroblasts. In the tissue adjacent to the matrigel plugs, the scores for the presence of inflammatory cells as well as fibroblasts showed only minor differences. In the implants, however, IL-32γ induced an increase in lipoidosis at both concentrations (Fig. 6D), whereas this effect was mild and not significant with VEGF. As shown in Fig. 6E, a degree of infiltration with inflammatory cells was observed in each of the slides, and there was no difference between the treatment groups. The inflammatory response was mixed, with both polymorphonuclear cells (PMNs) as well as lymphocytes present. Where the inflammation was more advanced, and extended further into surrounding tissues, PMNs predominated. When the inflammatory response was minimal or mild, a scattering of PMNs and/or lymphocytes/macrophages was apparent within or associated with the subcutis and fibrous connective tissue layer.
The integrin αVβ3 colocalizes with IL-32 in PAH and mediates its angiogenic activities
We recently reported the interaction of IL-32γ with the integrin αVβ3 (27). Therefore, we investigated whether this interaction occurred in human PAH. Indeed, confocal microscopy experiments demonstrated that IL-32 colocalized with αVβ3 in the plexiform lesions of PAH (Figure 7B–F), but not in the endothelium of normal pulmonary blood vessels (Figure 7A), in which the expression of αVβ3 was low.
To further explore the mechanisms by which IL-32 exerted its angiogenic properties, we first subjected samples from siIL-32- or scrambled-transfected and vehicle- or IL-1β-stimulated HUVEC to angiogenesis profiling analysis. Consistent with the pro-angiogenic state conferred by IL-32, we found that the anti-angiogenic activin A, endostatin, and angiopoietin 2 were increased 3-, 2-, and 2-fold when IL-32 was reduced, whereas the pro-angiogenic IL-8, u-plasminogen activator, and MMP-9 were up to 55% decreased. On the other hand, there was up to 3.5-fold more angiogenin and endoglin in siIL-32 transfected HUVEC under unstimulated conditions, but no change was detected after treatment with IL-1β. Next, we treated HUVEC cultures with exogenous recombinant IL-32γ in the presence or absence of the αVβ3 inhibitor cyclo [Arg-Gly-Asp-D-Phe-Val] and of IFNγ pre-treatment and performed real-time PCR analysis. As shown in Fig. 7G, exogenous IL-32γ dose-dependently induced IL-8 but only when the cells were pretreated with IFNγ (up to 2.5-fold over IFNγ alone). Importantly, blockade of the integrin αVβ3 reduced the IL-32γ + IFNγ-induced increase in IL-8 mRNA by up to 85%. We also performed PCR analysis of the other mediators of angiogenesis that exhibited a change on the profiler; these experiments showed a reduction by IL-32γ + IFNγ and a blockade of the effect by the αVβ3 inhibitor for activin A, but only small changes for the other mediators. This weaker effect of exogenous IL-32 even with IFNγ pre-treatment in comparison with siIL-32 is consistent with our previous experience with this cytokine (9, 16).
To demonstrate that αVβ3 indeed contributes to the angiogenic effects of IL-32, we next performed an angiogenesis assay in which HUVEC or human aortic EC (HAoEC)11 were co-cultured with human dermal fibroblasts (43). In this assay, exogenous recombinant IL-32γ did not require IFNγ, but without a costimulus markedly increased tube formation in a dose-dependent fashion (up to 3-fold, Fig. 8). Consistent with the regulation of IL-8 described above, the IL-32-induced angiogenesis was dependent on functional αVβ3, as cyclo [Arg-Gly-Asp-D-Phe-Val] potently reduced the angiogenetic activity of IL-32γ (by up to 72%, Fig. 8B, C). In HAoEC, the angiogenetic activity of IL-32γ at 100 ng/ml was comparable to that of 40 ng/ml VEGF (3-fold vs 3.6-fold increase in tube formation), thus resembling our findings in the matrigel experiments. This effect was somewhat weaker, though still significant, in the HUVEC (2-fold vs 3.5-fold increase in tube formation).
FIGURE 8.

IL-32γ-induced in vitro EC tube formation requires functional αVβ3. Human dermal fibroblasts were grown to confluence for 3d, followed by careful addition of HUVEC or HAoEC. Four hours later, treatment with the indicated concentrations of recombinant IL-32γ and/or 10 μM of the αVβ3 inhibitor cyclo [Arg-Gly-Asp-D-Phe-Val] was commenced. Treatment with recombinant human VEGF-165 (40 ng/ml) was used as internal assay control. On day 7 of co-culture, newly formed EC tubes were stained with sambuccus nigra lectin-FITC (green), nuclei were labeled with DAPI (blue), and the cells were then examined microscopically. Five fields of view per condition were randomly chosen and photographed. (A) One represesentative image of five independently performed HAoEC experiments is shown. (B, C) The number of visible branches in the co-cultures containing HAoEC (B) or HUVEC (C) was counted using ImageJ. Graphs illustrate the fold-changes in the number of branches of the newly formed EC tubes over vehicle-stimulated co-cultures (which are set as 1). *, p < 0.05, **, p < 0.01, and ***, p < 0.001 for IL-32γ or VEGF vs control; #, p < 0.05, ##, p < 0.01, and ###, p < 0.001 for IL-32γ vs the αVβ3 inhibitor; and ◆, p < 0.05, ◆◆, p < 0.01, and ◆◆◆, p < 0.001 for IL-32γ vs IL-32γ plus the αVβ3 inhibitor.
Discussion
The major finding of our investigation is the discovery that IL-32 possesses angiogenic properties. In addition, we revealed that these properties, which are at least in part mediated by the integrin αVβ3, are relevant in vivo (exemplified by examination of EC in PAH and GBM), and are not mediated by VEGF. Moreover, we demonstrate that pre-treatment with IFNγ sensitizes EC of neonatal as well as adult pulmonary origin to exogenous IL-32γ, which without IFNγ is inactive when used as a stimulant in these cells. This finding may be clinically relevant, as there is some evidence that IFNγ may play a detrimental role in PAH (44, 45). In coculture of HUVECs or HAoECs with HSF and in vivo in matrigel assays, IFNγ was not required, and exogenous IL-32γ effectively induced angiogenesis when added alone. However, the preparation of recombinant human IL-32γ we used contained a small amount of LPS (see below). This small quantity of LPS, or another factor present in these complex experimental systems, may have substituted for IFNγ in providing a cofactor that renders IL-32 functional. This concept is backed by similar data obtained with a completely synthetic IL-32γ preparation, which also required a cofactor (IFNγ) to exert its biological activity. One possible explanation is that IFNγ may induce expression and/or transport to the cell surface of the yet unknown receptor for IL-32. Since we moreover confirmed the colocalization and functional relationship between IL-32 and the integrinαVβ3, and since integrins are known to be IFNγ-inducible (46, 47), it is possible that αVβ3 may act as a receptor for IL-32 on EC. In more general terms, these data show that a second signal is necessary to render cells responsive to exogenous IL-32.
We have previously reported that the production of IL-32 protein by EC is enhanced by various stimuli, most notably by IL-1β which increases the abundance of IL-32 in EC lysates up to 15-fold, even at low concentrations (4). In that report, we furthermore demonstrated that IL-32 mediates the pro-inflammatory effects of IL-1β in EC, as silencing of IL-32 reduced the IL-1β-triggered production of IL-1α, IL-6, IL-8, and ICAM-1, and simultaneously increased the expression of thrombomodulin/CD141.
Each of these proteins has been implicated in the regulation of angiogenesis directly or indirectly, but IL-1 and IL-8 are particularly well established as potent promoters of angiogenesis (for examples see (42, 48–51)). Therefore, it was a logical next step to explore whether IL-32 itself exerted any effects on the proliferation of EC.
We localized IL-32 to the abnormal endothelial cells which populate the so-called plexiform lesions in the lungs from patients with idiopathic PAH (34). These complex lung vascular abnormalities are characterized by high expression of hypoxia-inducible factor (HIF-)1α, VEGF, and VEGF receptor 2 genes and proteins, thus presenting an angiogenic signature (35). One pathogenetic concept is that EC injury and apoptosis occur at sites of precapillary arterial bifurcations (28) and that the remaining apoptosis-resistant EC undergo a phenotypic switch, become hyperproliferative and eventually obliterate the vascular lumen (31, 35, 52). We have previously characterized this group of angioproliferative lung vascular disorders as quasi-malignant or precancerous (52), but features of immune dysregulation as in autoimmune diseases are also present (53). For example, angioproliferative PAH is associated with the limited form of scleroderma, the so-called CREST syndrome, with systemic lupus erythematosus and Sjögren syndrome (30). In this regard, it is interesting to note that IL-32 appears to regulate the production of NO, which in turn has been implicated in the pathogenesis of PAH (40).
The presence of IL-32 in the angioproliferative plexiform lesions of PAH is also consistent with the expression of this cytokine in psoriatic skin lesions (11, 54), as increased angiogenesis is an important feature of psoriasis. A similar comparison can be made between plexiform lesions and the malignant brain tumor GBM (31), and indeed, we found the abundance of IL-32 protein to be increased in this highly vascularized tumor as well. This is in accord with another study that screened mRNA levels in this neoplastic disease as well as in breast cancer, where mRNA for IL-32 was also elevated (55).
Together with our previous results, these data suggested that production of IL-32 in EC was associated with activation and proliferation of these cells. To determine whether IL-32 acts as a causal nexus in the regulation of angioproliferation, we reduced the abundance of this cytokine by siRNA in neonatal HUVEC and adult pulmonary microvascular EC and HAoEC, and also employed an in vivo and an in vitro angiogenesis assay. Our finding that reduction of IL-32 arrested the proliferation of HUVEC is consistent with data that IL-1β was found to possess angiogenic properties in matrigel assays (42) and in tumors (56). Similar properties have been described for IL-1α (49, 57), although it should be noted that under different circumstances, IL-1α has been shown to inhibit angiogenesis and tumor growth (58).
Whereas our in vitro proliferation assays showed that IL-32 plays an important role in the proliferation of EC, both the matrigel studies as well as the co-culture experiments demonstrated that an IL-32 gradient did not simply attract EC in a disorganized fashion. Rather, IL-32 conferred a marked increase in the formation of new EC tubes in the co-cultures and new capillaries in the matrigel plugs. The capillaries were functional, as they carried red blood cells. In fact, at 100 ng/ml IL-32γ was more efficient at achieving such neocapillarization than 25 ng/ml of the prototypical pro-angiogenic mediator VEGF. These striking data establish IL-32 as a player on the stages of PAH, neoplastic diseases, and wound healing.
The significance of the larger size of the IL-32-induced new blood vessels compared to those induced by VEGF, and of the increase in lipoidosis that was also conferred by IL-32γ, will have to be determined. However, the latter observation is intriguing, as one may infer an involvement of IL-32 in atherogenesis; moreover, it represents the first indication for a possible role of this cytokine in obesity. The absence of a difference in the influx of inflammatory cells between IL-32γ and vehicle-controls indicates that the angiogenic properties are not dependent on intermediate chemokine release.
It is important to note that the manufacturer tests rhIL-32γ for endotoxin by the Limulus amebocyte lysate assay, which does not detect low amounts of LPS. It thus remained unclear whether the reported effects of rhIL-32γ would be identical in the complete absence of microbial products; therefore, we generated a synthetic and completely endotoxin-free IL-32γ in order to eliminate the problems of microbial contamination. The fact that synthetic IL-32γ alone was inactive at 150 and 500 ng/ml supports the pivotal concept that IL-32 requires cofactors such as TLR agonists or IFN-γ for its biological activities. Notwithstanding this fact, because the LPS-loaded matrigel plugs did not differ from those loaded with vehicle, the angiogenic effect was indeed due to IL-32, not the LPS contamination of the recombinant protein preparation.
Importantly, our results not only confirm the recent finding that IL-32 interacts with integrins, but also demonstrate that the IL-32-induced angiogenesis and cytokine production by EC are at least in part mediated by the integrin αVβ3. We demonstrated interaction of IL-32 with the αVβ3 and αVβ6 integrins and their downstream signaling intermediates focal adhesion kinase (FAK) and paxillin (27). Integrins, including αVβ3 (59), and FAK (60, 61) contribute to angiogenesis and inhibitors of αVβ3 inhibit this process (62); therefore, FAK may also contribute to the pro-angiogenic properties of IL-32 and αVβ3. The high expression of the αVβ3 integrin in lung endothelial cells in a rat model of severe PAH has recently been reported (63). Furthermore, our data suggest that IL-32 may utilize IL-8, u-plasminogen activator, and/or MMP-9 while subduing the anti-angiogenic properties of activin A, endostatin, and/or angiopoietin 2 in order to achieve its pro-angiogenic programing of EC.
Of note, although we did observe a mild trend towards an increase in cell death in EC when there was less IL-32, it should be stated that changes in the abundance of IL-32 did not significantly affect the regulation of apoptosis in these cells; for example, Bak-1, Bcl-2, and Bcl-XL, and LDH remained unchanged. We infer that at least in EC, IL-32 has moderate effects on apoptotic programs at best. The effect of this cytokine on cell cycle-related pathways was not part of this study, but appears an interesting topic of further research.
Although not in EC, the role of IL-32 in apoptosis and cancer has been investigated previously; but the results of these studies are not homogenous. On the one hand, IL-32γ-overexpression resulted in reduced growth of melanoma and colon cancer in mice and was associated with reduced expression of anti-apoptotic genes such as bcl2 and iap and an increase in caspase3 and 9 (25). Other studies found that IL-32 contributed to activation-induced cell death in normal T-cells (2), HEK cells, HeLa cells, and Mycobacterium tuberculosis-stimulated THP-1 cells, and that IL-32γ-induced apoptosis was dependent on caspase-3 (5). In contrast, siIL-32 transfected into bone marrow stromal cells conferred reduced apoptosis in malignant cells in chronic myelomonocytic leukemia (22), and was also shown to induce the proliferation of hematopoietic progenitor cells (64). In pancreatic cancer, silencing of IL-32 suppressed apoptosis and mRNA expression of bcl2, bclxl, and mcl1 (23). Importantly, IL-32 was also demonstrated to be associated with a more malignant and invasive phenotype of lung cancer, and IL-32 levels correlated with those of IL-6, IL-8, and VEGF as well as with a higher microvessel density in the tumors and a poor clinical outcome (24). It appears likely that the overall effect of IL-32 on apoptosis and cancer growth in experimental models depends on the cell type in which this cytokine is (over-)expressed or blocked and whether other factors such as immune cells are a part of the respective model or not.
Only few studies have investigated IL-32 in the biology of the endothelium - among them our own (4, 16) and some others (55, 64–67) - but these studies mainly focused on the role of IL-32 in endothelial inflammation. In addition to our own finding that VEGF was unaffected by silencing of IL-32 in EC, others have reported the following data related to VEGF: Treatment of EC with VEGF did not change the abundance of IL-32 protein (4); blockade of IL-32 by siRNA reduced the production of VEGF from a human bone marrow stroma cell line (22); and the abundance of IL-32 correlated with that of VEGF in lung cancer (24). The last two papers as well as the data we present here appear to contrast the report of an increase in VEGF abundance in human bronchial epithelial cells stimulated with cytokines or rhinovirus after silencing of IL-32 (68). Supernatants from these cells conferred enhanced angiogenesis in HUVEC in vitro. This study was conducted in the setting of asthma, and IL-32 was silenced in epithelial, not endothelial cells. Overall, the data obtained in EC in this report and by others favor the conclusion that the angiogenesis driven by IL-32 is not mediated by VEGF.
In conclusion, we further expand the portfolio of properties of IL-32, adding angiogenesis to the functions of this versatile cytokine. Mechanistically, these pro-angiogenetic activities likely utilize regulation of IL-8, MMP-9, activin A, and endostatin, but not VEGF or TGF-β1. A second signal is required to render cells responsive to exogenous IL-32, and IL-32-induced angiogenesis is at least in part dependent on the integrin αVβ3. Therefore, IL-32 emerges as a key nexus in endothelial cell biology at which the pathways of inflammation and angiogenesis converge.
Acknowledgments
We are grateful for outstanding technical assistance by Nana Burns.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft grant No 747/1-1 to MFN, by National Institute of Health grants AI-15614 and CA-04 6934 to CAD, and by the Victorian Government’s Operational Infrastructure Support Program.
EC, endothelial cells
siRNA, small interfering RNA
siIL-32, siRNA to IL-32
PAH, pulmonary arterial hypertension
GBM, glioblastoma multiforme,
VEGF, vascular endothelial growth factor
HUVEC, human umbilical vein endothelial cells
vWF, von Willebrand factor
HAoEC, human aortic endothelial cells
References
- 1.Kim SH, Han SY, Azam T, Yoon DY, Dinarello CA. Interleukin-32: a cytokine and inducer of TNFalpha. Immunity. 2005;22:131–142. doi: 10.1016/j.immuni.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Goda C, Kanaji T, Kanaji S, Tanaka G, Arima K, Ohno S, Izuhara K. Involvement of IL-32 in activation-induced cell death in T cells. Int Immunol. 2006;18:233–240. doi: 10.1093/intimm/dxh339. [DOI] [PubMed] [Google Scholar]
- 3.Imaeda H, Andoh A, Aomatsu T, Osaki R, Bamba S, Inatomi O, Shimizu T, Fujiyama Y. A new isoform of interleukin-32 suppresses IL-8 mrna expression in the intestinal epithelial cell line ht-29. Mol Med Report. 2011;4:483–487. doi: 10.3892/mmr.2011.442. [DOI] [PubMed] [Google Scholar]
- 4.Nold-Petry CA, Nold MF, Zepp JA, Kim SH, Voelkel NF, Dinarello CA. IL-32-dependent effects of IL-1{beta} on endothelial cell functions. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0813334106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heinhuis B, Netea MG, van den Berg WB, Dinarello CA, Joosten LA. Interleukin-32: A predominantly intracellular proinflammatory mediator that controls cell activation and cell death. Cytokine. 2012 doi: 10.1016/j.cyto.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 6.Heinhuis B, Koenders MI, van de Loo FA, Netea MG, van den Berg WB, Joosten LA. Inflammation-dependent secretion and splicing of IL-32{gamma} in rheumatoid arthritis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4962–4967. doi: 10.1073/pnas.1016005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Novick D, Rubinstein M, Azam T, Rabinkov A, Dinarello CA, Kim SH. Proteinase 3 is an IL-32 binding protein. Proc Natl Acad Sci U S A. 2006;103:3316–3321. doi: 10.1073/pnas.0511206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim S, Lee S, Her E, Bae S, Choi J, Hong J, Jaekal J, Yoon D, Azam T, Dinarello CA. Proteinase 3-processed form of the recombinant IL-32 separate domain. BMB Rep. 2008;41:814–819. doi: 10.5483/bmbrep.2008.41.11.814. [DOI] [PubMed] [Google Scholar]
- 9.Nold MF, Nold-Petry CA, Pott GB, Zepp JA, Saavedra MT, Kim SH, Dinarello CA. Endogenous IL-32 controls cytokine and HIV-1 production. J Immunol. 2008;181:557–565. doi: 10.4049/jimmunol.181.1.557. [DOI] [PubMed] [Google Scholar]
- 10.Calabrese F, Baraldo S, Bazzan E, Lunardi F, Rea F, Maestrelli P, Turato G, Lokar-Oliani K, Papi A, Zuin R, Sfriso P, Balestro E, Dinarello CA, Saetta M. IL-32, a novel proinflammatory cytokine in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;178:894–901. doi: 10.1164/rccm.200804-646OC. [DOI] [PubMed] [Google Scholar]
- 11.Dinarello CA, Kim SH. IL-32, a novel cytokine with a possible role in disease. Ann Rheum Dis. 2006;65(Suppl 3):iii61–64. doi: 10.1136/ard.2006.058511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeong HJ, Shin SY, Oh HA, Kim MH, Cho JS, Kim HM. IL-32 up-regulation is associated with inflammatory cytokine production in allergic rhinitis. J Pathol. 2011 doi: 10.1002/path.2899. [DOI] [PubMed] [Google Scholar]
- 13.Na SJ, So SH, Lee KO, Choi YC. Elevated serum level of interleukin-32alpha in the patients with myasthenia gravis. J Neurol. 2011 doi: 10.1007/s00415-011-6036-7. [DOI] [PubMed] [Google Scholar]
- 14.Cagnard N, Letourneur F, Essabbani A, Devauchelle V, Mistou S, Rapinat A, Decraene C, Fournier C, Chiocchia G. Interleukin-32, CCL2, PF4F1 and GFD10 are the only cytokine/chemokine genes differentially expressed by in vitro cultured rheumatoid and osteoarthritis fibroblast-like synoviocytes. Eur Cytokine Netw. 2005;16:289–292. [PubMed] [Google Scholar]
- 15.Joosten LA, Netea MG, Kim SH, Yoon DY, Oppers-Walgreen B, Radstake TR, Barrera P, van de Loo FA, Dinarello CA, van den Berg WB. IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2006;103:3298–3303. doi: 10.1073/pnas.0511233103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zepp JA, Nold-Petry CA, Dinarello CA, Nold MF. Protection from RNA and DNA viruses by IL-32. Journal of immunology. 2011;186:4110–4118. doi: 10.4049/jimmunol.1000081. [DOI] [PubMed] [Google Scholar]
- 17.Li W, Sun W, Liu L, Yang F, Li Y, Chen Y, Fang J, Zhang W, Wu J, Zhu Y. IL-32: a host proinflammatory factor against influenza viral replication is upregulated by aberrant epigenetic modifications during influenza A virus infection. J Immunol. 2010;185:5056–5065. doi: 10.4049/jimmunol.0902667. [DOI] [PubMed] [Google Scholar]
- 18.Pan X, Cao H, Lu J, Shu X, Xiong X, Hong X, Xu Q, Zhu H, Li G, Shen G. Interleukin-32 expression induced by hepatitis B virus protein X is mediated through activation of NF-kappaB. Mol Immunol. 2011 doi: 10.1016/j.molimm.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 19.Moschen AR, Fritz T, Clouston AD, Rebhan I, Bauhofer O, Barrie HD, Powell EE, Kim SH, Dinarello CA, Bartenschlager R, Jonsson JR, Tilg H. Interleukin-32: A new proinflammatory cytokine involved in hepatitis C virus-related liver inflammation and fibrosis. Hepatology. 2011 doi: 10.1002/hep.24285. [DOI] [PubMed] [Google Scholar]
- 20.Lee S, Kim JH, Kim H, Kang JW, Kim SH, Yang Y, Kim J, Park J, Park S, Hong J, Yoon DY. Activation of the interleukin-32 pro-inflammatory pathway in response to human papillomavirus infection and over-expression of interleukin-32 controls the expression of the human papillomavirus oncogene. Immunology. 2011;132:410–420. doi: 10.1111/j.1365-2567.2010.03377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishimoto KP, Laust AK, Nelson EL. A human dendritic cell subset receptive to the Venezuelan equine encephalitis virus-derived replicon particle constitutively expresses IL-32. Journal of immunology. 2008;181:4010–4018. doi: 10.4049/jimmunol.181.6.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marcondes AM, Mhyre AJ, Stirewalt DL, Kim SH, Dinarello CA, Deeg HJ. Dysregulation of IL-32 in myelodysplastic syndrome and chronic myelomonocytic leukemia modulates apoptosis and impairs NK function. Proc Natl Acad Sci U S A. 2008;105:2865–2870. doi: 10.1073/pnas.0712391105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishida A, Andoh A, Inatomi O, Fujiyama Y. Interleukin-32 expression in the pancreas. The Journal of biological chemistry. 2009;284:17868–17876. doi: 10.1074/jbc.M900368200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sorrentino C, Di Carlo E. Expression of IL-32 in human lung cancer is related to the histotype and metastatic phenotype. American journal of respiratory and critical care medicine. 2009;180:769–779. doi: 10.1164/rccm.200903-0400OC. [DOI] [PubMed] [Google Scholar]
- 25.Oh JH, Cho MC, Kim JH, Lee SY, Kim HJ, Park ES, Ban JO, Kang JW, Lee DH, Shim JH, Han SB, Moon DC, Park YH, Yu DY, Kim JM, Kim SH, Yoon DY, Hong JT. IL-32gamma inhibits cancer cell growth through inactivation of NF-kappaB and STAT3 signals. Oncogene. 2011 doi: 10.1038/onc.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ciccia F, Alessandro R, Rizzo A, Principe S, Raiata F, Cavazza A, Guggino G, Accardo-Palumbo A, Boiardi L, Ferrante A, Principato A, Giardina A, De Leo G, Salvarani C, Triolo G. Expression of IL-32 in the inflamed arteries of patients with giant cell arteritis. Arthritis Rheum. 2011 doi: 10.1002/art.30374. [DOI] [PubMed] [Google Scholar]
- 27.Heinhuis B, Koenders MI, van den Berg WB, Netea MG, Dinarello CA, Joosten LA. Interleukin 32 (IL-32) contains a typical alpha-helix bundle structure that resembles focal adhesion targeting region of focal adhesion kinase-1. The Journal of biological chemistry. 2012;287:5733–5743. doi: 10.1074/jbc.M111.288290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cool CD, Stewart JS, Werahera P, Miller GJ, Williams RL, Voelkel NF, Tuder RM. Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell-specific markers. Evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am J Pathol. 1999;155:411–419. doi: 10.1016/S0002-9440(10)65137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hassoun PM, Mouthon L, Barbera JA, Eddahibi S, Flores SC, Grimminger F, Jones PL, Maitland ML, Michelakis ED, Morrell NW, Newman JH, Rabinovitch M, Schermuly R, Stenmark KR, Voelkel NF, Yuan JX, Humbert M. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. 2009;54:S10–19. doi: 10.1016/j.jacc.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 30.Nicolls MR, Taraseviciene-Stewart L, Rai PR, Badesch DB, Voelkel NF. Autoimmunity and pulmonary hypertension: a perspective. The European respiratory journal: official journal of the European Society for Clinical Respiratory Physiology. 2005;26:1110–1118. doi: 10.1183/09031936.05.00045705. [DOI] [PubMed] [Google Scholar]
- 31.Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol. 1994;144:275–285. [PMC free article] [PubMed] [Google Scholar]
- 32.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. American journal of respiratory and critical care medicine. 1995;151:1628–1631. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 33.Tuder RM, Voelkel NF. Pulmonary hypertension and inflammation. J Lab Clin Med. 1998;132:16–24. doi: 10.1016/s0022-2143(98)90020-8. [DOI] [PubMed] [Google Scholar]
- 34.Voelkel NF, Gomez-Arroyo JG, Abbate A, Bogaard HJ, Nicolls MR. Pathobiology of pulmonary arterial hypertension and right ventricular failure. The European respiratory journal: official journal of the European Society for Clinical Respiratory Physiology. 2012 doi: 10.1183/09031936.00046612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L, Kasahara Y, Cool CD, Bishop AE, Geraci M, Semenza GL, Yacoub M, Polak JM, Voelkel NF. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol. 2001;195:367–374. doi: 10.1002/path.953. [DOI] [PubMed] [Google Scholar]
- 36.Canavese M, Altruda F, Ruzicka T, Schauber J. Vascular endothelial growth factor (VEGF) in the pathogenesis of psoriasis--a possible target for novel therapies? J Dermatol Sci. 2010;58:171–176. doi: 10.1016/j.jdermsci.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 37.Hackeng TM, Fernandez JA, Dawson PE, Kent SB, Griffin JH. Chemical synthesis and spontaneous folding of a multidomain protein: anticoagulant microprotein S. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:14074–14078. doi: 10.1073/pnas.260239797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nold MF, Nold-Petry CA, Zepp JA, Palmer BE, Bufler P, Dinarello CA. IL-37 is a fundamental inhibitor of innate immunity. Nature immunology. 2010;11:1014–1022. doi: 10.1038/ni.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sakao S, Taraseviciene-Stewart L, Lee JD, Wood K, Cool CD, Voelkel NF. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. FASEB J. 2005;19:1178–1180. doi: 10.1096/fj.04-3261fje. [DOI] [PubMed] [Google Scholar]
- 40.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 41.Voronov E, Shouval DS, Krelin Y, Cagnano E, Benharroch D, Iwakura Y, Dinarello CA, Apte RN. IL-1 is required for tumor invasiveness and angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2645–2650. doi: 10.1073/pnas.0437939100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carmi Y, Voronov E, Dotan S, Lahat N, Rahat MA, Fogel M, Huszar M, White MR, Dinarello CA, Apte RN. The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. Journal of immunology. 2009;183:4705–4714. doi: 10.4049/jimmunol.0901511. [DOI] [PubMed] [Google Scholar]
- 43.Baumer Y, Scholz B, Ivanov S, Schlosshauer B. Telomerase-based immortalization modifies the angiogenic/inflammatory responses of human coronary artery endothelial cells. Exp Biol Med (Maywood) 2011;236:692–700. doi: 10.1258/ebm.2011.010300. [DOI] [PubMed] [Google Scholar]
- 44.Wang W, Wang YL, Chen XY, Li YT, Hao W, Jin YP, Han B. Dexamethasone attenuates development of monocrotaline-induced pulmonary arterial hypertension. Mol Biol Rep. 2011;38:3277–3284. doi: 10.1007/s11033-010-0390-x. [DOI] [PubMed] [Google Scholar]
- 45.Wort SJ, Ito M, Chou PC, Mc Master SK, Badiger R, Jazrawi E, de Souza P, vans TWE, Mitchell JA, Pinhu L, Ito K, Adcock IM. Synergistic induction of endothelin-1 by tumor necrosis factor alpha and interferon gamma is due to enhanced NF-kappaB binding and histone acetylation at specific kappaB sites. The Journal of biological chemistry. 2009;284:24297–24305. doi: 10.1074/jbc.M109.032524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee MS, Sarvetnick N. Induction of vascular addressins and adhesion molecules in the pancreas of IFN-gamma transgenic mice. Journal of immunology. 1994;152:4597–4603. [PubMed] [Google Scholar]
- 47.Westphal JR, Willems HW, Tax WJ, Koene RA, Ruiter DJ, de Waal RM. The proliferative response of human T cells to allogeneic IFN-gamma-treated endothelial cells is mediated via both CD2/LFA-3 and LFA-1/ICAM-1 and -2 adhesion pathways. Transpl Immunol. 1993;1:183–191. doi: 10.1016/0966-3274(93)90045-a. [DOI] [PubMed] [Google Scholar]
- 48.Farkas L, Gauldie J, Voelkel NF, Kolb M. Pulmonary Hypertension and Idiopathic Pulmonary Fibrosis - A Tale of Angiogenesis, Apoptosis and Growth Factors. Am J Respir Cell Mol Biol. 2010 doi: 10.1165/rcmb.2010-0365TR. [DOI] [PubMed] [Google Scholar]
- 49.Matsuo Y, Sawai H, Ochi N, Yasuda A, Takahashi H, Funahashi H, Takeyama H, Guha S. Interleukin-1alpha secreted by pancreatic cancer cells promotes angiogenesis and its therapeutic implications. J Surg Res. 2009;153:274–281. doi: 10.1016/j.jss.2008.04.040. [DOI] [PubMed] [Google Scholar]
- 50.Shi CS, Shi GY, Chang YS, Han HS, Kuo CH, Liu C, Huang HC, Chang YJ, Chen PS, Wu HL. Evidence of human thrombomodulin domain as a novel angiogenic factor. Circulation. 2005;111:1627–1636. doi: 10.1161/01.CIR.0000160364.05405.B5. [DOI] [PubMed] [Google Scholar]
- 51.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 52.Rai PR, Cool CD, King JA, Stevens T, Burns N, Winn RA, Kasper M, Voelkel NF. The cancer paradigm of severe pulmonary arterial hypertension. American journal of respiratory and critical care medicine. 2008;178:558–564. doi: 10.1164/rccm.200709-1369PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, Jennings S, Barker L, Nicklin P, Walker C, Budd DC, Pepke-Zaba J, Morrell NW. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122:920–927. doi: 10.1161/CIRCULATIONAHA.109.933762. [DOI] [PubMed] [Google Scholar]
- 54.Kempuraj D, Conti P, Vasiadi M, Alysandratos KD, Tagen M, Kalogeromitros D, Kourelis T, Gregoriou S, Makris M, Stavrianeas NG, Theoharides TC. IL-32 is increased along with tryptase in lesional psoriatic skin and is up-regulated by substance P in human mast cells. Eur J Dermatol. 2010;20:865–867. doi: 10.1684/ejd.2010.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kobayashi H, Lin PC. Molecular characterization of IL-32 in human endothelial cells. Cytokine. 2009;46:351–358. doi: 10.1016/j.cyto.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shchors K, Shchors E, Rostker F, Lawlor ER, Brown-Swigart L, Evan GI. The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1beta. Genes Dev. 2006;20:2527–2538. doi: 10.1101/gad.1455706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Salven P, Hattori K, Heissig B, Rafii S. Interleukin-1alpha promotes angiogenesis in vivo via VEGFR-2 pathway by inducing inflammatory cell VEGF synthesis and secretion. The FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2002;16:1471–1473. doi: 10.1096/fj.02-0134fje. [DOI] [PubMed] [Google Scholar]
- 58.Nazarenko I, Marhaba R, Reich E, Voronov E, Vitacolonna M, Hildebrand D, Elter E, Rajasagi M, Apte RN, Zoller M. Tumorigenicity of IL-1alpha- and IL-1beta-deficient fibrosarcoma cells. Neoplasia. 2008;10:549–562. doi: 10.1593/neo.08286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ruegg C, Dormond O, Foletti A. Suppression of tumor angiogenesis through the inhibition of integrin function and signaling in endothelial cells: which side to target? Endothelium. 2002;9:151–160. doi: 10.1080/10623320213635. [DOI] [PubMed] [Google Scholar]
- 60.Cabrita M, Jones LM, Quizi JL, Sabourin LA, McKay BC, Addison CL. Focal adhesion kinase inhibitors are potent anti-angiogenic agents. Mol Oncol. 2011;5:517–526. doi: 10.1016/j.molonc.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen TL, Park A, Alcaraz A, Peng X, Jang I, Koni P, Flavell RA, Gu H, Guan J. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J Cell Biol. 2005;169:941–952. doi: 10.1083/jcb.200411155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nisato RE, Tille JC, Jonczyk A, Goodman SL, Pepper MS. alphav beta 3 and alphav beta 5 integrin antagonists inhibit angiogenesis in vitro. Angiogenesis. 2003;6:105–119. doi: 10.1023/B:AGEN.0000011801.98187.f2. [DOI] [PubMed] [Google Scholar]
- 63.Al Husseini A, Bagnato G, Farkas L, Gomez-Arroyo J, Farkas D, Mizuno S, Kraskauskas D, Abbate A, Van Tassel Pharm DB, Voelkel NF, Bogaard HJ. Thyroid hormone is highly permissive in angioproliferative pulmonary hypertension in rats. The European respiratory journal: official journal of the European Society for Clinical Respiratory Physiology. 2012 doi: 10.1183/09031936.00196511. [DOI] [PubMed] [Google Scholar]
- 64.Moldenhauer A, Futschik M, Lu H, Helmig M, Gotze P, Bal G, Zenke M, Han W, Salama A. Interleukin 32 promotes hematopoietic progenitor expansion and attenuates bone marrow cytotoxicity. European journal of immunology. 2011 doi: 10.1002/eji.201040986. [DOI] [PubMed] [Google Scholar]
- 65.Kobayashi H, Huang J, Ye F, Shyr Y, Blackwell TS, Lin PC. Interleukin-32beta propagates vascular inflammation and exacerbates sepsis in a mouse model. PLoS ONE. 2010;5:e9458. doi: 10.1371/journal.pone.0009458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kobayashi H, Yazlovitskaya EM, Lin PC. Interleukin-32 positively regulates radiation-induced vascular inflammation. Int J Radiat Oncol Biol Phys. 2009;74:1573–1579. doi: 10.1016/j.ijrobp.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cho KA, Jun YH, Suh JW, Kang JS, Choi HJ, Woo SY. Orientia tsutsugamushi induced endothelial cell activation via the NOD1-IL-32 pathway. Microb Pathog. 2010;49:95–104. doi: 10.1016/j.micpath.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 68.Meyer N, Christoph J, Makrinioti H, Indermitte P, Rhyner C, Soyka M, Eiwegger T, Chalubinski M, Wanke K, Fujita H, Wawrzyniak P, Burgler S, Zhang S, Akdis M, Menz G, Akdis C. Inhibition of angiogenesis by IL-32: Possible role in asthma. J Allergy Clin Immunol. 2012 doi: 10.1016/j.jaci.2011.12.1002. [DOI] [PubMed] [Google Scholar]