

Abstract
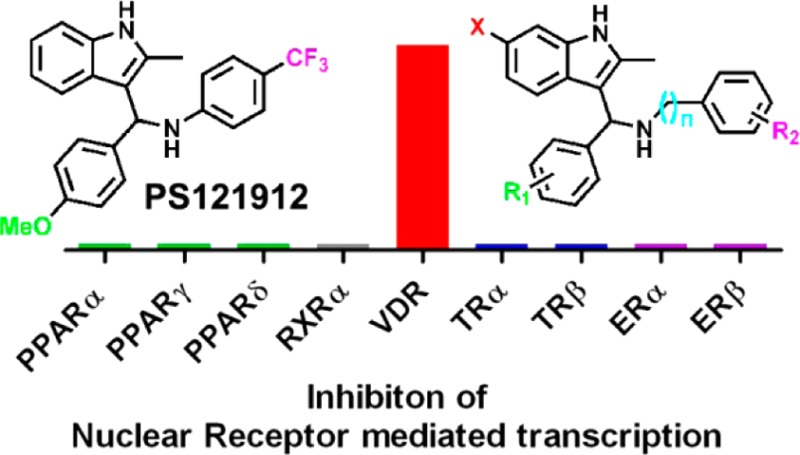
Nuclear receptor coregulators are master regulators of transcription and selectively interact with the vitamin D receptor (VDR) to modulate cell differentiation, cell proliferation, and calcium homeostasis. Herein, we report the syntheses and evaluation of highly potent and selective VDR–coactivator inhibitors based on a recently identified 3-indolylmethanamine scaffold. The most active compound, PS121912, selectively inhibited VDR-mediated transcription among eight other nuclear receptors tested. PS121912 is also selectively disrupting the binding between VDR and the third nuclear receptor interaction domain of the coactivator SRC2. Genetic studies revealed that PS121912 behaves like a VDR antagonist by repressing 1,25-(OH)2D3 activated gene transcription. In addition, PS121912 induced apoptosis in HL-60.
Keywords: Vitamin D receptor, VDR, steroid receptor coactivator, fluorescence polarization, high throughput screening, 3-indolyl-methanamines, CYP24A1, CAMP, apoptosis, HL60
The vitamin D receptor (VDR) is a ligand-inducible transcription factor that belongs to the superfamily of nuclear receptors (NR).1 Like most NRs, VDR has a modular structure consisting of a DNA binding domain and a ligand binding domain (LBD) connected by a flexible hinge region. 1,25-Dihydroxyvitamin D3 (1,25-(OH)2D3), a hormonally active form of vitamin D, is the endogenous high affinity ligand for VDR.2 Traditionally, 1,25-(OH)2D3 is known for its role in cell differentiation and calcium homeostasis, but lately it has been reported to be involved in inflammation, immune response, and cell proliferation.3
Human clinical studies with 1,25(OH)2D3 are dose-limited because of the reported induction of hypercalcemia and hypercalciurea.4,5 This dysregulation can cause psychosis, bone pain, calcification of soft tissue, coronary artery disease, and, in severe cases, coma and cardiac arrest. During the last decades, hundreds of VDR agonists have been synthesized to develop VDR ligands with lower calcemic activity. However, two synthetic VDR ligands that were developed to systemically treat cancer were not active or induced hypercalcemia in clinical trials.6,7 In addition, a smaller number of VDR antagonist were developed, which include the irreversible antagonist TEI-96478 and those bearing bulky side chains such as 25-carboxylic esters (ZK168218 and ZK159222),9 26-adamantly substituted antagonists (ADTT and analogues),10 and 22-butyl-branched compounds that ultimately destabilized the active form of VDR.11 Interestingly, none of these antagonists have been further developed as therapeutics.
Recently, coregulator proteins have been identified as transcriptional master regulators.12 In its unligated state, VDR is bound with corepressors. Upon binding with 1,25-(OH)2D3, VDR undergoes a conformational change that disrupts corepressor binding and enables the interaction between VDR and coactivators. The activated form of VDR interacts with retinoid X receptor (RxR) to form a heterodimer that binds to vitamin D response elements on the DNA leading to transcription of associated genes. VDR is known to interact with 2700 human DNA sites and effects the expression of 229 gene products.13
A new approach to modulate VDR function without causing hypercalcemia but to induce cell proliferation and differentiation is the design of small molecules that target the interactions between VDR and coregulators. In general, the approach of inhibiting protein–protein interactions is challenging in terms of potency and specificity. Further adding to the complexity is the fact that the family of more than 300 coregulators interact with different NR.14 Thus, selectivity with respect to different NRs and inhibition of specific NR–coregulator interactions is essential for novel promising NR–coregulator inhibitors.
Various small molecule NR–coactivator inhibitors have been discovered for thyroid receptor (TR), pregane X receptor (PxR), estrogen receptor (ER), and the androgen receptor (AR).15 Recently, the first reversible VDR–coactivator inhibitors were developed to disrupt the VDR-mediated gene transcription, but these compounds lack the important selectivity among other NRs.16,17 However, drawing from a pool of hundreds of thousands of molecules, high throughput screening (HTS) is a superior method to identify ideal candidates that not only possess selectivity for VDR but also for the various VDR–coregulator interactions.
The first HTS method to quantify the interaction between VDR and coregulator peptides was introduced by Arai et al. using fluorescence intensity.18 Six compounds were investigated in this study. Recently, we described a HTS campaign in collaboration with NIH investigating 390 000 molecules that led to the identification of GW0742 as a competitive VDR antagonist.19 Furthermore, we reported a second HTS campaign with 275 000 molecules that resulted in the development of an irreversible VDR–coactivator binding inhibitor.20 Herein, we report our recent successes to improve the potency and selectivity of these VDR–coactivator binding inhibitor. Importantly, we demonstrated that these unique compounds modulated gene transcription and cell proliferation.
A diversified library of 3-indolylmethanamines was generated by using one-pot modified microwave-based Aza–Friedel–Craft reaction (Scheme 1).
Scheme 1. Improved Synthesis of 3-Indolylmethanamines.
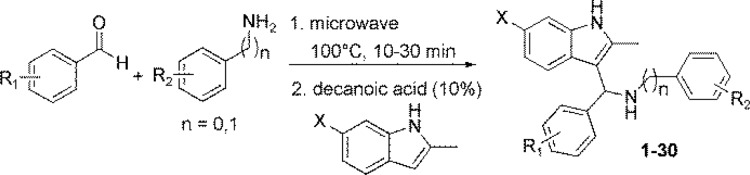
The first step involves the combination of an amine and an aromatic aldehyde to form an imine intermediate at 100 °C within 10 to 30 min using microwave irradiation. To that solution, indole and a catalytic amount of decanoic acid was added to afford the desired products at room temperature, which are summarized in Figure 1. The purified compounds were analyzed using a battery of biophysical and biochemical assays (Table 1).
Figure 1.

Structures of synthesized 3-indolylmethanamines.
Table 1. Summary of Biophysical and Biochemical Properties of 3-Indolylmethanamines.
| compd | solubility (μM)a | permeabilityb log (Pe) (cm/s) | VDR-SRC2–3 IC50 (μM)c | VDR-mediated transcription IC50 (μM)d | toxicity LC50 (μM)e |
|---|---|---|---|---|---|
| 1 | 3.6 | 7.36 ± 0.10 | 12.8 ± 0.8 | n.o. | 36.4 ± 7.4 |
| 2 | 57.9 | 7.03 ± 0.07 | 7.2 ± 0.4 | 3.0 ± 0.7 | >50 |
| 3 | 96.5 | 6.26 ± 0.02 | 22.8 ± 2.2 | 2.2 ± 0.5 | 15.3 ± 1.7 |
| 4 | 91.5 | 6.64 ± 0.02 | 21.8 ± 1.7 | 4.4 ± 2.1 | 14.1 ± 1.6 |
| 5 | 94.0 | 6.16 ± 0.02 | 11.3 ± 0.5 | 3.2 ± 0.7 | 41.7 ± 13.3 |
| 6 | 48.1 | 5.86 ± 0.03 | n.o. | 9.3 ± 3.1 | >100 |
| 7 | 25.2 | 6.26 ± 0.01 | 17.2 ± 4.8 | 5.0 ± 3.4 | >75 |
| 8 | 8.5 | 7.42 ± 0.52 | 15.6 ± 1.4 | 5.2 ± 2.7 | >50 |
| 9 | 19.8 | 7.68 ± 0.31 | 63.1 ± 13.6 | 7.3 ± 2.9 | >100 |
| 10 | 29.4 | n.d. | n.o. | 5.8 ± 3.1 | >75 |
| PS121912 | 68.9 | 6.36 ± 0.02 | 12.4 ± 0.7 (8.1 ± 2.3)f | 0.59 ± 0.1 | 27.3 ± 2.7 |
| 12 | 60.4 | 6.21 ± 0.01 | 7.2 ± 0.4 | 5.4 ± 3.5 | 42.7 ± 2.7 |
| 13 | 45.5 | 5.60 ± 0.19 | 59.9 ± 4.5 | 5.8 ± 2.1 | >75 |
| 14 | 18.2 | 6.67 ± 0.04 | 35.2 ± 12.5 | 9.1 ± 2.2 | 53.7 ± 16.8 |
| 15 | 20.1 | 6.41 ± 0.01 | 9.5 ± 0.4 | 3.4 ± 0.6 | 10.8 ± 1.6 |
| 16 | 123.3 | 6.47 ± 0.11 | >75 | 3.2 ± 1.4 | >100 |
| 17 | 128.3 | 6.50 ± 0.14 | 51.5 ± 10.4 | 3.7 ± 1.8 | >100 |
| 18 | 52.8 | 7.57 ± 0.32 | 14.2 ± 1.4 | 14.1 ± 6.6 | >100 |
| 19 | 3.6 | n.d. | 66.8 ± 10.3 | n.o. | >100 |
| 20 | 176.7 | 6.71 ± 0.01 | 16.7 ± 0.8 | 6.1 ± 2.5 | >75 |
| 21 | 97.9 | 6.14 ± 0.13 | 11.9 ± 0.7 | 3.8 ± 2.1 | >75 |
| 22 | 99.3 | 6.92 ± 0.16 | n.o. | n.o. | >100 |
| 23 | 114.4 | n.d. | >75 | n.o. | >75 |
| 24 | 3.0 | n.d. | n.o. | >25 | >100 |
| 25 | 95.3 | 6.68 ± 0.11 | 15.8 ± 1.2 | >25 | >75 |
| 26 | 130.1 | 5.87 ± 0.01 | 101.4 ± 15.2 | 12.2 ± 3.3 | >75 |
| 27 | 120.3 | 6.09 ± 0.01 | 29.3 ± 5.7 | 6.7 ± 2.5 | >100 |
| 28 | 101.8 | 6.14 ± 0.04 | 17.3 ± 0.7 | 8.5 ± 2.7 | >50 |
| 29 | 150.2 | 5.78 ± 0.01 | 32.3 ± 6.4 | 5.4 ± 2.1 | 18.6 ± 2.5 |
| 30 | 144.8 | 5.88 ± 0.02 | n.o. | n.d. | >75 |
Solubility was determined in phosphate-buffered saline at pH 7.4.
Permeability was measured using the parallel artificial membrane permeation assay (PAMPA) at neutral pH (pH = 7.4).
A fluorescence polarization competition assay was carried out using VDR-LBD (1 μM), Alexa Fluor-labeled peptide SRC2–3 (7 nM), VDR-agonist LG190178 (5 μM), and serial diluted small molecules. IC50 values were obtained by fitting data obtained after 2 h to the following equation: Y = bottom + (top – bottom)/(1 + 10((log IC50 – X)hillslope)) using three independent experiments in quadruplet.
Transcription assay: HEK293T cells were transfected with CMV-VDR and a CYP24A1 promoter driven luciferase expression vector in the presence of 1,25(OH)2D3.
Toxicity was determined under the conditions of the transcription assay using CellTiter-Glo.
Two-hydrid assay: HEK293T cells were transfected with a VP16-VDR-LBD, SRC1-GAL4, and luciferase reporter plasmid vector in the presence of 1,25(OH)2D3.21 n.d. = not determined; n.o. = not observed.
The biophysical properties determined include small molecule solubility and permeability. The solubility of synthesized 3-indolylmethanamines in PBS buffer (pH 7.4) with 5% DMSO ranged between 150 and 3 μM. The compounds substituted with polar heterocyclic side chains showed excellent solubility (>100 μM). The small molecule permeability was determined using a parallel artificial membrane permeation assay (PAMPA) employing a hexadecane membrane. In comparison to the used standards (ranitidine = −8.02 ± 0.074 cm/s (low permeability), carbamazepine = −6.81 ± 0.0011 cm/s (medium permeability), and verapamil = −5.93 ± 0.015 cm/s (high permeability), the majority of 3-indolylmethanamines exhibited medium to high permeability (Table 1).
A fluorescence polarization (FP) assay was used to determine the ability of synthesized molecules to inhibit the interactions between VDR-LBD and Alexa Fluor 647 labeled coactivator peptide SRC2–3. The compounds were analyzed in a dose-dependent manner, and potencies are reported as IC50 values. In order to assess the ability of 3-indolylmethanamines to inhibit the VDR–coactivator interaction in cells, a VDR two-hybrid assay and a VDR-mediated transcription assay was used. The toxicity of compounds under the conditions of the transcription assay was determined with CellTiter-Glo (Promega).
All 3-indolylmethanamines in group A (Table 1, compounds 1–10 and PS121912) exhibited cellular activities in the low micromolar to nanomolar range. The compound activities measured with the biochemical FP assay are generally higher probably due to compound off-targets effects. The compound toxicities are ranging between 14.1 and >100 μM. The compound PS121912 exhibited the highest activity in the VDR-mediated transcription assay (IC50 = 590 ± 100 nM) and largest therapeutic index.
For compounds in group B, bearing benzylamine substitutents, low micromolar activities were determined for the transcriptional inhibition of VDR. The activities for the FP assay ranged between 7.2 to 59.9 μM. Importantly, 3-indolylmethanamines are irreversible inhibitors acting through the formation of an azafulvenium salt that react with nucleophilies like mercaptoethanol (see Supporting Information). Thus the activity of these inhibitors depends on the incubation time, the environment, and the electronic substituent effects.20 Compound 15 was the most toxic compound within the library of 3-indolylmethanamines with a LD50 value of 10.8 ± 1.6 μM.
For compounds in group C, various heterocyclic substituents were introduced. Interestingly, the majority of these 3-indolylmethanamine were not toxic but very active inhibitors of VDR-mediated transcription. Compound 16 exhibited the largest therapeutic index of more than 31 in group C, but it was still inferior to compound PS121912 with a therapeutic index of 46. The substitution of the secondary nitrogen by oxygen or carbon prevented the generation of a reactive electrophilic compound and thus resulted in inactive compounds 22 and 23.
The NR-selectivity of the most potent compound, PS121912, was determined by measuring the inhibition of transcription for a panel of nine different NRs. These include the peroxisome proliferator-activated receptors α, γ, and δ, the retinoic acid receptor α, the thyroid receptors α and β, and the estrogen receptors α and β. The results are summarized in Table 2.
Table 2. Inhibition of NR-Mediated Transcription in the Presence of Compound PS121912b.
| compd | nuclear receptor | IC50 (μM)a |
|---|---|---|
| 1 | VDR | 0.59 ± 0.1b |
| 2 | PPAR-α | 20.8 ± 1.1c |
| 3 | PPAR-γ | >25d |
| 4 | PPAR-δ | >30e |
| 5 | RxR-α | >25f |
| 6 | TR-α | >30g |
| 7 | TR-β | 24.1 ± 1.1g |
| 8 | ER-α | 26.2 ± 3.3h |
| 9 | ER-β | >25h |
Three independent experiments were conducted in quadruplicate, and data was analyzed using nonlinear regression with variable slope (GraphPrism).
1,25(OH)2D3 (10 nM).
GW7647 (30 nM).
Rosiglitazone (300 nM).
GW0742 (50 nM).
Bexarotene (200 nM).
T3 (10 nM).
Estradiol (10 nM).
The 3-indolylmethanamine PS121912 selectively inhibited the transcription of VDR at nanomolar concentrations, whereas for all other NRs we determined IC50 values ranging between 20 and 26 μM. It is important to point out that the LD50 value of PS121912 in HEK293T cells was 27.3 μM; thus, the reduced luciferase activity measured at concentrations above 20 μM is probably due to cell death and not caused by inhibition of NR-mediated transcription. Therefore, the NR selectivity of PS121912 is at least 35-fold.
The selectivity of PS121912 to inhibit the interaction between VDR and coactivator SRC1,22 SRC2,23 SRC3,24 or DRIP20525 was assessed using FP assays employing different NR interaction domains (NIDs) of these coactivators (Figure 2).
Figure 2.

Selectivity studies for VDR–coactivator interactions inhibition in the presence of PS121912 using FP assay. VDR-LBD (1–10 μM), VDR agonist LG190178 (5 μM), and different coactivator peptides (7 nM) were incubated for 3 h in the presence of different concentrations of compound PS121912.
The quantification of interactions between VDR and different coactivator peptides was reported recently.26 Among the interactions tested, only the interactions between VDR and coactivator peptide SRC2–3 was fully inhibited by compound PS121912 with an IC50 value of 12.4 ± 0.7 μM (Table 1). The NIDs of SRC1, SRC3, and DRIP2, which exhibit a similarly strong interaction with VDR, were not inhibited by PS121912. A very weak partial inhibition for the VDR–SRC2–2 interaction was observed in the presence of PS121912 leaving 88% of the VDR–coactivator binding intact.
Furthermore, the expression levels of VDR target genes CYP24A127 and CAMP28 were determined in HL-60 cells treated with 7.5 μM of compound PS121912 in the presence and absence of 20 nM 1,25-(OH)2D3 for 18 h. The results are summarized in Figure 3.
Figure 3.

Gene regulation by PS121912 (7.5 μM) in HL-60 cells after 18 h in the presence and absence of 1,25-(OH)2D3 (20 nM). Standard errors of mean were calculated from two biological independent experiments performed in triplicate. Stars represent P < 0.001 (***) (Student’s t test).
A strong induction of CYP24A1 and CAMP by 1,25-(OH)2D3 was observed. Cells treated 1,25-(OH)2D3 and compound PS121912 exhibited a loss of induction of transcription, whereas cells treated with only PS121912 showed comparable expression to that of vehicle treated HL-60 cells. The transcriptional inhibition of the CYP24A1 gene product 24-hydroxylase by PS121912 is important because it has been shown that deactivation of 24-hydroxylase, using vitamin D analogues29 or general P450 enzyme inhibitors30 promotes antiproliferation and differentiation of cancer cells.
Therefore, viable, apoptotic, and dead HL-60 cells were determined in the presence of different concentrations of PS121912 as depicted in Figure 4.
Figure 4.

HL-60 viability, toxicity, and apoptosis assay after 18 h in the presence of PS121912.
Three different assays were used: (a) CellTiter-Glo that quantifies the amount of ATP (toxicity), (b) CellTiter-Fluor that quantifies the amount of active cellular proteases (viability), and (c) Caspase-Glo 3/7 that quantifies the amount of active cellular caspase 3 and 7 (apoptosis). As expected, the amount of intact and dead HL-60 cells at the same concentration of PS121912 was inversely proportional with EC50 values of around 20 μM. Importantly, a dose-dependent increase of caspase 3 and 7 activity was observed in the presence of PS121912. The increase of the apoptotic enzymes was proportional to the amount of dead cells, confirming that programmed cell death and not necrosis was the mode of action of PS121912 at higher concentrations.
Acknowledgments
We thank Stephen D. Ayers for providing us with the nuclear receptor plasmids.
Glossary
ABBREVIATIONS
- NR
nuclear receptors
- VDR
vitamin D receptor
- LBD
ligand binding domain
- SRC
steroid receptor coactivator
- DMSO
dimethylsulfoxide
- 1,25-(OH)2D3
1,25-dihydroxyvitamin D3
- RxR
retinoid X receptor
- TR
thyroid receptor
- PxR
pregame X receptor
- ER
estrogen receptor
- AR
androgen receptor
- HTS
high throughput screening
- PAMPA parallel artificial membrane permeation assay
- CYP24A1
cytochrome P450_24A1
- CAMP
cathelicidin antimicrobial peptide gene
Supporting Information Available
Detailed procedures and characterization of all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the University of Wisconsin–Milwaukee [to L.A.A.], the UWM Research Growth Initiative (RGI grant 2012) [to L.A.A.], the NIH R03DA031090 [to L.A.A.], the UWM Research Foundation (Catalyst grant), the Lynde and Harry Bradley Foundation [to L.A.A.], the Richard and Ethel Herzfeld Foundation [to L.A.A.], and RO1 AR050023 [to D.D.B.].
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Brumbaugh P. F.; Haussler M. R. 1 Alpha,25-dihydroxycholecalciferol receptors in intestine. I. Association of 1 alpha,25-dihydroxycholecalciferol with intestinal mucosa chromatin. J. Biol. Chem. 1974, 24941251–7. [PubMed] [Google Scholar]
- Holick M. F.; Schnoes H. K.; DeLuca H. F.; Suda T.; Cousins R. J. Isolation and identification of 1,25-dihydroxycholecalciferol. A metabolite of vitamin D active in intestine. Biochemistry 1971, 10142799–804. [DOI] [PubMed] [Google Scholar]
- Feldman D.; Pike J. W.; Glorieux F. H.. Vitamin D, 2nd ed.; Elsevier: Burlington, MA, 2005; Vol. 1–2. [Google Scholar]
- Gross C.; Stamey T.; Hancock S.; Feldman D. Treatment of early recurrent prostate cancer with 1,25-dihydroxyvitamin D3 (calcitriol). J. Urol. 1998, 15962035–9and discussion 2039–40. [DOI] [PubMed] [Google Scholar]
- Beer T. M.; Myrthue A. Calcitriol in cancer treatment: from the lab to the clinic. Mol. Cancer Ther. 2004, 33373–81. [PubMed] [Google Scholar]
- Gulliford T.; English J.; Colston K. W.; Menday P.; Moller S.; Coombes R. C. A phase I study of the vitamin D analogue EB 1089 in patients with advanced breast and colorectal cancer. Br. J. Cancer 1998, 7816–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R. K.; Trump D. L.; Egorin M. J.; Fernandez M.; Johnson C. S.; Ramanathan R. K. A phase I study of the vitamin D3 analogue ILX23-7553 administered orally to patients with advanced solid tumors. Invest. New Drugs 2011, 2961420–5. [DOI] [PubMed] [Google Scholar]
- Miura D.; Manabe K.; Ozono K.; Saito M.; Gao Q.; Norman A. W.; Ishizuka S. Antagonistic action of novel 1alpha,25-dihydroxyvitamin D3–26, 23-lactone analogs on differentiation of human leukemia cells (HL-60) induced by 1alpha,25-dihydroxyvitamin D3. J. Biol. Chem. 1999, 2742316392–9. [DOI] [PubMed] [Google Scholar]
- Bury Y.; Steinmeyer A.; Carlberg C. Structure activity relationship of carboxylic ester antagonists of the vitamin D(3) receptor. Mol. Pharmacol. 2000, 5851067–74. [DOI] [PubMed] [Google Scholar]
- Igarashi M.; Yoshimoto N.; Yamamoto K.; Shimizu M.; Ishizawa M.; Makishima M.; DeLuca H. F.; Yamada S. Identification of a highly potent vitamin D receptor antagonist: (25S)-26-adamantyl-25-hydroxy-2-methylene-22,23-didehydro-19,27-dinor-20-epi-vita min D3 (ADMI3). Arch. Biochem. Biophys. 2007, 4602240–53. [DOI] [PubMed] [Google Scholar]
- Inaba Y.; Yoshimoto N.; Sakamaki Y.; Nakabayashi M.; Ikura T.; Tamamura H.; Ito N.; Shimizu M.; Yamamoto K. A new class of vitamin D analogues that induce structural rearrangement of the ligand-binding pocket of the receptor. J. Med. Chem. 2009, 5251438–49. [DOI] [PubMed] [Google Scholar]
- McKenna N. J.; Lanz R. B.; O’Malley B. W. Nuclear receptor coregulators: cellular and molecular biology. Endocr. Rev. 1999, 203321–44. [DOI] [PubMed] [Google Scholar]
- Ramagopalan S. V.; Heger A.; Berlanga A. J.; Maugeri N. J.; Lincoln M. R.; Burrell A.; Handunnetthi L.; Handel A. E.; Disanto G.; Orton S. M.; Watson C. T.; Morahan J. M.; Giovannoni G.; Ponting C. P.; Ebers G. C.; Knight J. C. A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res. 2010, 20101352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna N. J.; O’Malley B. W. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 2002, 1084465–74. [DOI] [PubMed] [Google Scholar]
- Caboni L.; Lloyd D. G. Beyond the Ligand-Binding Pocket: Targeting Alternate Sites in Nuclear Receptors. Med. Res. Rev. 2013, 33, 1081–118. [DOI] [PubMed] [Google Scholar]
- Mita Y.; Dodo K.; Noguchi-Yachide T.; Miyachi H.; Makishima M.; Hashimoto Y.; Ishikawa M. LXXLL peptide mimetics as inhibitors of the interaction of vitamin D receptor with coactivators. Bioorg. Med. Chem. Lett. 2010, 2051712–7. [DOI] [PubMed] [Google Scholar]
- Mita Y.; Dodo K.; Noguchi-Yachide T.; Hashimoto Y.; Ishikawa M. Structure–activity relationship of benzodiazepine derivatives as LXXLL peptide mimetics that inhibit the interaction of vitamin D receptor with coactivators. Bioorg. Med. Chem. 2013, 214993–1005. [DOI] [PubMed] [Google Scholar]
- Arai M. A.; Takeyama K.; Ito S.; Kato S.; Chen T. C.; Kittaka A. High-throughput system for analyzing ligand-induced cofactor recruitment by vitamin D receptor. Bioconjugate Chem. 2007, 183614–20. [DOI] [PubMed] [Google Scholar]
- Nandhikonda P.; Yasgar A.; Baranowski A. M.; Sidhu P. S.; McCallum M. M.; Pawlak A. J.; Teske K.; Feleke B.; Yuan N. Y.; Kevin C.; Bikle D. D.; Ayers S. D.; Webb P.; Rai G.; Simeonov A.; Jadhav A.; Maloney D.; Arnold L. A. Peroxisome proliferation-activated receptor delta agonist GW0742 interacts weakly with multiple nuclear receptors, including the vitamin D receptor. Biochemistry 2013, 52244193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandhikonda P.; Lynt W. Z.; McCallum M. M.; Ara T.; Baranowski A. M.; Yuan N. Y.; Pearson D.; Bikle D. D.; Guy R. K.; Arnold L. A. Discovery of the first irreversible small molecule inhibitors of the interaction between the vitamin D receptor and coactivators. J. Med. Chem. 2012, 55104640–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima M.; Lu T. T.; Xie W.; Whitfield G. K.; Domoto H.; Evans R. M.; Haussler M. R.; Mangelsdorf D. J. Vitamin D receptor as an intestinal bile acid sensor. Science 2002, 29655711313–6. [DOI] [PubMed] [Google Scholar]
- Masuyama H.; Brownfield C. M.; St-Arnaud R.; MacDonald P. N. Evidence for ligand-dependent intramolecular folding of the AF-2 domain in vitamin D receptor-activated transcription and coactivator interaction. Mol. Endocrinol. 1997, 11101507–17. [DOI] [PubMed] [Google Scholar]
- Hong H.; Kohli K.; Garabedian M. J.; Stallcup M. R. GRIP1, a transcriptional coactivator for the AF-2 transactivation domain of steroid, thyroid, retinoid, and vitamin D receptors. Mol. Cell. Biol. 1997, 1752735–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Gomes P. J.; Chen J. D. RAC3, a steroid/nuclear receptor-associated coactivator that is related to SRC-1 and TIF2. Proc. Natl. Acad. Sci. U.S.A. 1997, 94168479–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachez C.; Suldan Z.; Ward J.; Chang C. P.; Burakov D.; Erdjument-Bromage H.; Tempst P.; Freedman L. P. A novel protein complex that interacts with the vitamin D3 receptor in a ligand-dependent manner and enhances VDR transactivation in a cell-free system. Genes Dev. 1998, 12121787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichert A.; Arnold L. A.; Otieno S.; Oda Y.; Augustinaite I.; Geistlinger T. R.; Kriwacki R. W.; Guy R. K.; Bikle D. D. Quantification of the vitamin D receptor-coregulator interaction. Biochemistry 2009, 4871454–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K. S.; DeLuca H. F. Cloning of the human 1 alpha,25-dihydroxyvitamin D-3 24-hydroxylase gene promoter and identification of two vitamin D-responsive elements. Biochim. Biophys. Acta 1995, 126311–9. [DOI] [PubMed] [Google Scholar]
- Wang T. T.; Nestel F. P.; Bourdeau V.; Nagai Y.; Wang Q.; Liao J.; Tavera-Mendoza L.; Lin R.; Hanrahan J. W.; Mader S.; White J. H. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J. Immunol. 2004, 17352909–12. [DOI] [PubMed] [Google Scholar]
- Posner G. H.; Crawford K. R.; Yang H. W.; Kahraman M.; Jeon H. B.; Li H.; Lee J. K.; Suh B. C.; Hatcher M. A.; Labonte T.; Usera A.; Dolan P. M.; Kensler T. W.; Peleg S.; Jones G.; Zhang A.; Korczak B.; Saha U.; Chuang S. S. Potent, low-calcemic, selective inhibitors of CYP24 hydroxylase: 24-sulfone analogs of the hormone 1alpha,25-dihydroxyvitamin D3. J. Steroid Biochem. Mol. Biol. 2004, 89–901–55–12. [DOI] [PubMed] [Google Scholar]
- Schuster I.; Egger H.; Astecker N.; Herzig G.; Schussler M.; Vorisek G. Selective inhibitors of CYP24: mechanistic tools to explore vitamin D metabolism in human keratinocytes. Steroids 2001, 663–5451–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.