

Abstract
Pneumococcal infections remain a leading cause of death in persons ≥ 65 years of age. Recent reports have illustrated detrimental changes in the ER stress response or unfolded protein response (UPR) in aging and age-related diseases; however the relationship between aging, the UPR, and innate immune responses to Streptococcus pneumoniae has not been fully elucidated. Our results illustrate that STING mediated production of IFNβ during S. pneumoniae infection is decreased in aged hosts. Enhanced ER stress in response to S. pne umoniae augmented IRE1/XBP1 mediated production of Atg9a. Knockdown of Atg9a or treatment with gemcitabine HCL resulted in enhanced STING mediated production of IFNβ by aged macrophages. Consecutive treatments with gemcitabine during in vivo S. pneumoniae infection decreased morbidity and mortality in aged hosts which was associated with decreased Atg9a expression, increased IFNβ production, and improved bacterial clearance from lung tissue. Taken together, data presented in this study provide new evidence as to why older persons are more susceptible to S. pneumoniae and provide a possible mechanism to enhance these responses, thereby decreasing morbidity and mortality in this population.
Introduction
Pneumococcal infections are a leading cause of death in the United States with greater than 90% of deaths occurring in persons greater than 65 years of age (1, 2). It has been well established that aging affects various components of the immune response, which can lead to impaired host defense to pulmonary infections and defective vaccine responses, resulting in a significantly higher risk of elderly persons (≥65 years of age) developing bacterial infections (3-6). The ill effects of primary and secondary pulmonary bacterial infections are increasingly being felt in older populations, with Streptococcus pneumoniae (S. pne) being the most causative organism in both immunocompromised and non-immunocompromised populations (7). With the increased emergence of antibiotic strains of S. pne and increased incidence of co-infections, it is imperative to elucidate the mechanisms that underlie age associated impairments in innate immunity and devise therapeutic treatment strategies to augment these responses (8-11).
Initiation of type I IFN responses to S. pne is mediated by a cytosolic DNA-sensing pathway that involves the intracellular recognition of bacterial DNA by the adaptor molecule stimulator of interferon genes (STING) and phosphorylation of transcription factor IFN regulatory factor 3 (IRF3) (12-14). Function and responsiveness of STING is critical for responses to cytosolic DNA and unique bacterial nucleic acids (cyclic dinucleotides) (15). In resting cells, dimerized STING localizes to the endoplasmic reticulum (ER) or to the mitochondria-associated membrane (MEM), a compartment that connects the ER to the mitochondria (12, 16). Recent work has illustrated that in response to DNA or cyclic dinucleotides, TANK binding kinase 1 (TBK1) and IRF3 are recruited to the carboxy-terminal tail (CTT) of STING, leading to IRF3 activation, nuclear translocation, and subsequent transcription of IFNβ (17, 18). In addition, activated STING has also been shown to localize together with several autophagy-associated proteins, including autophagy related gene 9 (Atg9a) (19). Recent work has illustrated that a loss of Atg9a results in enhanced assembly of STING/TBK1 complexes in response to dsDNA, leading to heightened innate immune responses (19).
The unfolded protein response (UPR) is initiated by increased protein load, misfolding of proteins, and calcium gradient deregulation that disrupt normal ER function and consists of inositol-requiring protein 1 (IRE1), the activating transcription factor 6 (ATF6), and the protein kinase RNA (PKR) like ER kinase (PERK). Recent studies have shown that IRE1 and X-box binding-1 protein (XBP1), which act immediately downstream of IRE1, control the acetylation and activation of Atg9a (20).
While recent evidence has implicated detrimental changes in the ER stress response in aging and age-related diseases, the relationship between aging, the UPR, and the innate immune responses to S. pne has not been fully elucidated. In the present study, we investigated the effect of aging on STING mediated activation of IFNβ in a murine model of S. pne infection. Our results demonstrate that aged hosts have decreased STING mediated production of IFNβ which was associated with increased bacterial titers in lung as well as increased morbidity and mortality in aged mice. Enhanced ER stress in aged macrophages and lung during S. pne infection was associated with increased caspase-12 and caspase-3 mediated induction of apoptosis. Further, increased IRE/XBP1 mediated acetylation of Atg9a in response to S. pne induced ER stress resulted in decreased STING mediated IFNβ production by aged macrophages. Knockdown of Atg9a or treatment with gemcitabine HCL resulted in enhanced STING mediated production of IFNβ by aged macrophages. Consecutive treatments with gemcitabine during in vivo S. pne infection decreased morbidity and mortality in aged hosts which was associated with increased IFNβ production and improved bacterial clearance from lung tissue. Taken together, our findings illustrate a potential mechanism by which an age associated enhancement in ER stress during S. pne infection results in increased Atg9a mediated inhibition of STING dependent IFNβ responses.
Experimental Procedures
Mice
Young (2 months) and aged (19 months) male and female BALB/c mice were purchased from the NIA rodent facility (Charles River Laboratories). Upon receipt, mice were handled under identical husbandry conditions and fed certified commercial feed. Body weights were measured daily and mice were humanely euthanized if they lost more than 15% of their starting body weight. The IACUC at Lovelace Respiratory Research Institute approved the use of animals in this study. No animals were used in the study if they had evidence of skin lesions, weight loss, or lymphadenopathy.
Bacterial culture
Streptococcus pneumoniae (ATCC 6303, ATCC, Manassas, VA) was grown on 10% sheep blood agar plates (BD Biosciences, San Jose, CA) overnight or for 4-24 hours in brain heart infusion (BHI) broth (BD Biosciences). Colony forming units (CFU) were assessed by dilution of samples in BHI and titers were determined by colony counts × dilution.
Primary bone marrow isolation and cell culture
Bone marrow cells (BMCs) were prepared from the femurs of mice as previously described (21, 22). Bone marrow derived macrophages were cultured in media alone or media containing S. pne (50 CFU), ciAMP or ciGMP (50-100μM), 1μg/μl ISD/lyovec, 3′-3′-cGAMP or 2′-2′-cGAMP (10μg/ml) (Invivogen, San Diego, CA), tauroursodeoxycholic acid (100μM) (Enzo Life Sciences, Farmingdale, NY), sunitinib malate (200nM) (Enzo Life Sciences), or gemcitabine (10mM) (Selleck Chemicals, Houston, TX) for 4 to 24 hours. In additional experiments, cells were treated with 1μg/μl of isotype (mouse 1gG1) or purified anti-mouse IFNAR-1 (Biolegend, San Diego, CA) 24 hours prior to infection.
Phagocytosis Assay
Phagocytosis assays were performed using the pHrodo Phagocytosis Labeling Kit for Flow Cytometry (Invitrogen) per manufacturer's instructions and analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
Transfection with siRNA
Missense, Atg9a siRNA, and STING siRNA (Flexitube GeneSolution siRNA, Qiagen) (15 nmol/sequence) was complexed with GenMute siRNA transfection reagent (SignaGen Laboratories, Ijamsville, MD) for 15 minutes before addition to macrophage cultures. siRNA transfected cells were cultured for 24 hours prior to S. pne infection. Atg9a and STING silencing was confirmed by real time PCR and western blot analysis.
Flow cytometry
Cells were collected from digested lung samples [collagenase D (1 mg/ml) and DNase I (0.1 mg/ml) (Roche, Indianapolis, IN)] prior to staining with CD45.2-PE, CD11b-PERCP Cy5.5, CD11c-Alexa Fluor 647, CD64-PE-Cy7, F4/80-APC, and/or GR1-PE (eBioscience, San Diego, CA) in staining solution (1% FBS supplemented 1× PBS). Alveolar macrophage populations were analyzed using previously published methods (23). Briefly, CD45+ cell populations were initially selected and CD11c+CD11b-CD64+ cells were quantified as alveolar macrophages. Non-determined macrophage populations were identified as F4/80+CD11b+GR1- cells. Examination of apoptosis was performed using Annexin V/7-AAD staining per manufacturer's instructions (eBioscience, San Diego, CA). All flow cytometry was analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
Co-immunoprecipitation
Co-immunoprecipiation of Atg9a was performed using the Pierce Co-Immunoprecipitation kit (Thermo Scientific, Rockford, IL) according to the manufacturer's instructions using rabbit αAtg9a (Abcam). Briefly, 500μg of in vivo murine lung homogenates were incubated with 1μg of αAtg9a.
RNA purification and real time PCR
RNA samples were extracted and real time PCR was performed using previously published methods (24). QuantiTect Primer Assays and RT2 Profiler™ Arrays (mouse antibacterial response) were used to assess gene expression (Qiagen, Valencia, CA). All reactions were performed in triplicate. Relative levels of messenger RNA (mRNA) was calculated by the comparative cycle threshold method and either βActin or β2M mRNA levels were used as the invariant control for each sample.
ELISA
Culture supernatants, lung homogenates, or serum were analyzed for IFN-β production using ELISA kits purchased from Biolegend (San Diego, CA). BIP/GRP78 and PDI protein expression was assessed by ELISA kits purchased from Enzo Life Sciences (Farmingdale, NY). STING/TMEM173 protein expression was confirmed by ELISA purchased from MyBioSource Inc. (San Diego, CA). Phosphorylated IRF3 levels were quantified using the TransAM IRF3 kit purchased from Active Motif (Carlsbad, CA). Protein levels were assessed per each kits manufacturer's instructions.
Western blotting
Equal amounts of protein (10-45 μg/lane) were loaded onto a 4-12% BIS-TRIS Bolt gel (Invitrogen) and run at 165V for 35 minutes. Protein was transferred to nitrocellulose membrane using the iBlot Western blotting system (Invitrogen). Immunodetection was performed using primary antibodies against STING, phosphorylated IRF3, TBK1, β-Actin (Abcam, Cambridge, MA), CRT, BIP/GRP78, PDI, caspase-3 and 12, ATF4, CHOP, BCL2, IRE1, XBP1, Atg9a, and acetylated lysine (Cell Signaling Technologies, Cambridge, MA) (all 1:1000 dilution) and the ECL Western Blotting Analysis System (Santa Cruz Biologicals, Santa Cruz, CA). Images were acquired using Multi-Gauge software (Fujifilm, Greenwood, SC).
In vivo procedures
S. pne infection: All mice were anesthetized with isoflurane (5% for induction and 2% for maintenance) prior to intranasal instillation with 1×103 CFU of S. pne (ATCC 6303) (50μL volume in PBS). As previously described, the following clinical scores were assigned: 0=normal, 1=slightly ruffled, 2=ruffled fur, 3= ruffled fur and inactive, 4=hunched/moribund, and 5= dead (25). Gemcitabine HCl administration: Mice received a 100μL volume of 0.03 mg/mL dose of gemcitabine (Selleck Chemicals) (1× PBS as vehicle) intraperitoneally starting at 4 hours post infection post S. pne infection and repeated every 24 hours post infection thereafter.
Statistical Analysis
Survival analysis between groups was calculated using the Mantel Cox test. Comparison of groups was performed using a two-tailed t-test, one-way or two-way ANOVA, when appropriate. Samples obtained were normally or approximately normally distributed. All samples were independent and contained the same sample size for analysis. Variances of the populations were equal. All data were analyzed using GraphPad prism software (San Diego, CA). Statistical significance was considered by a p value < 0.05.
Results
Increased Morbidity in Aged Mice in Response to S. pneumoniae Infection
It is well established that the elderly have increased morbidity and mortality to Streptococcus pneumoniae (S. pne) infections (26, 27). We infected young (2 months of age) and aged (19 months of age) male and female BALB/c mice with ATCC 6303, a highly virulent type 3 strain of S. pne commonly associated with an increased relative risk of death in older persons (28). During S. pne infection, aged mice had increased morbidity, as illustrated by enhanced weight loss at early time points post infection (Figure 1A: two-way ANOVA, p<0.0001) as well as increased bacterial titers in lung (Figure 1B: two-way ANOVA, p<0.0001) and enhanced gene expression of several key bacterial lytic genes (Figure 1C: two-way ANOVA, p=0.0018). We next examined cellular recruitment to lung in young and aged lung at early time points post S. pne infection. At 24 hours post infection there were similar levels of cellular infiltration in both young and aged lung, with cell numbers remaining elevated in aged lung at 48 hours post infection (Figure 1D; t-test, p=0.0026). As macrophages play a key role in S. pne infection, we next examined the number and phenotype of macrophages present in young and aged lung at early time points post infection. Despite similar numbers of alveolar macrophages (E; two-way ANOVA, NS), when compared to young, there was an increase in the number of F4/80+CD11b+GR1- macrophages present in aged lung (F; two-way ANOVA, p=0.0470).
Figure 1. Increased Morbidity in Aged Mice in Response to S. pneumoniae Infection.
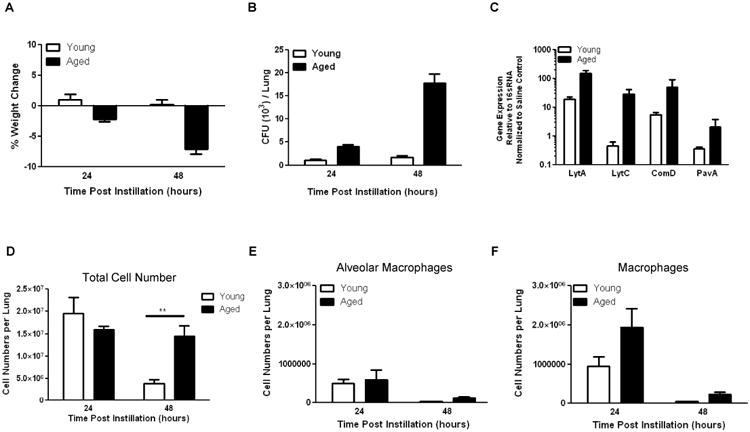
Young (2 months) and aged (19 months) male and female BALB/c mice received 1×103 CFU of S. pne (ATCC 6303) or saline via intranasal instillation. (A) Weights were collected at 24 and 48 hours post infection (two-way ANOVA, p<0.0001). (B) Lungs were harvested from S. pne infected mice and homogenates were diluted and plated on sheep blood agar plates overnight prior to calculation of CFU titers (two-way ANOVA, p<0.0001). (C) RNA was harvested at 24 hours post infection and bacterial lytic gene expression in young and aged lung was assessed by real time PCR (two-way ANOVA, p=0.0018). (D-F) Lung tissue was digested with collagenase D and the number of total leukocytes (D: t-test (48 hours), p=0.0026), alveolar macrophage (E: two-way ANOVA, NS), and macrophage populations (F: two-way ANOVA, p=0.0470) are shown. Similar results were obtained from at least three independent experiments with greater than N=5 per group and results are shown as the mean ± SEM.
Decreased IFNβ Production in Aged Mice during S. pneumoniae Infection
Production of type I interferon by macrophages, such as IFNβ, during S. pne infection plays an important role in the initiation of key innate immune signaling cascades that mediate defense against bacterial pathogenesis (13, 29). We examined IFNβ production in young and aged lung homogenates (Figure 2A) and serum (Figure 2B) collected at 24 hours post S. pne infection. While overall IFNβ production was increased in both age groups, when compared to young, IFNβ levels were significantly lower in aged lung (Figure 2A; two-way ANOVA, p<0.0001) and serum (Figure 2B; two-way ANOVA, p=0.0015) at 24 hours post infection. Examination of additional cytokines, illustrated that age associated alterations in innate cytokine production also occurred in response to S. pne (Supplemental Figure 1A). We next examined IFNβ production by young and aged bone-marrow derived macrophages in response to S. pne infection. Despite similar levels of phagocytosis (Supplemental Figure 1B) when compared to young, aged macrophages produced significantly less IFNβ (Figure 2C; two-way ANOVA, p=0.0006) as well as had higher bacterial titers and enhanced bacterial lytic gene expression (Supplemental Figure 1C-E) in response to S. pne infection. To confirm these results, we treated young and aged macrophages with either isotype and αIFNAR antibodies or missense and STING specific siRNA and examined the impact on IFNβ production in response to S. pne. There was decreased production of IFNβ and enhanced S. pne growth in both young and aged macrophages in response to αIFNAR or STING siRNA treatment (Supplemental Figure 1C-E). We next examined IFNβ production by young and aged macrophages in response to cyclic dinucleotides, interferon stimulatory DNA (ISD), and cyclic guanosine monophosphate-adenosine monophosphate (cGAMP). While IFNβ production was increased in both age groups, when compared to young, aged macrophages produced significantly less IFNβ in response to c-di-AMP, c-di-GMP, and ISD (Figure 2D; two-way ANOVA, p=0.0022). Similarly, IFNβ production in response 3′-3′ and 2′-2′ cGAMP sequences, was significantly decreased in aged macrophages (Figure 2E; two-way ANOVA, p<0.0001).
Figure 2. Decreased IFNβ Production in Aged Mice during S. pneumoniae Infection.
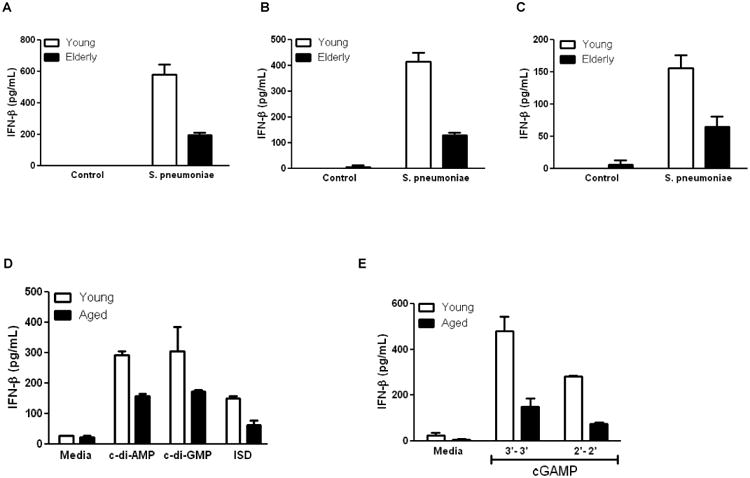
Young (2 months) and aged (19 months) male and female BALB/c mice received 1×103 CFU of S. pne (ATCC 6303) or saline via intranasal instillation. IFNβ production in (A) lung homogenates (two-way ANOVA, p<0.0001) and (B) serum (two-way ANOVA, p=0.0015) were assessed by ELISA. (C) Macrophages were cultured with media alone or with S. pne (50 CFU) for 24 hours. Cell culture supernatants were collected and IFNβ production (two-way ANOVA, p=0.0006) was assessed by ELISA. (D-E) In additional experiments, IFNβ production by macrophages cultured with c-di-AMP, c-di-GMP, and ISD (D: two-way ANOVA, p=0.0022), or 3′-3′ and 2′-2′ cGAMP (F: two-way ANOVA, p<0.0001) was assessed by ELISA. Similar results were obtained from three or more independent experiments. For in vitro experiments, the values represent N=6 or greater per experiment and are expressed as the mean ± SEM. For in vivo experiments, the values are representative of five or more mice per group and are expressed as the mean ± SEM.
Initiation of type I IFN responses to S. pne is mediated by a STING, TBK1, and IRF3 mediated pathway that results in the production of IFNβ (13, 14). We next examined the expression of STING, TBK1, and IRF3 in young and aged macrophages in response to in vitro S. pne infection. Expression of STING, TBK1, IRF3, and IFNβ was increased in both young and aged macrophages (Figure 3A-E), however, when compared to young, gene and protein expression was significantly lower in aged macrophages (Figure 3A; two-way ANOVA, p<0.0001, 3B; two-way ANOVA, p<0.0001, E; two-way ANOVA, p=0.0071). To confirm these results, we next examined STING mediated gene expression in young and aged macrophages in response to cGAMP. When compared to young, there was decreased STING, TBK1, IRF3, and IFNβ mRNA expression in response to 3′-3′ and 2′-2′ cGAMPs (Supplemental Figure 1F-G). We next examined STING, TBK1, and IRF3 expression in young and aged lung 24 hours post S. pne infection. As illustrated in Figure 3F-I, when compared to young, there was significantly less STING, IRF3, and TBK1 mRNA expression in aged lung post S. pne infection (Figure 3F; two-way ANOVA, p<0.0001). In addition, protein expression of STING and IRF3 activation was significantly decreased in aged lung in response to S. pne (Figure 3G-I; 3H; two-way ANOVA, p=0.0224, 3I, two-way ANOVA, p<0.0001).
Figure 3. STING Mediated Production of IFNβ is Decreased in Aged Hosts during S. pneumoniae infection.
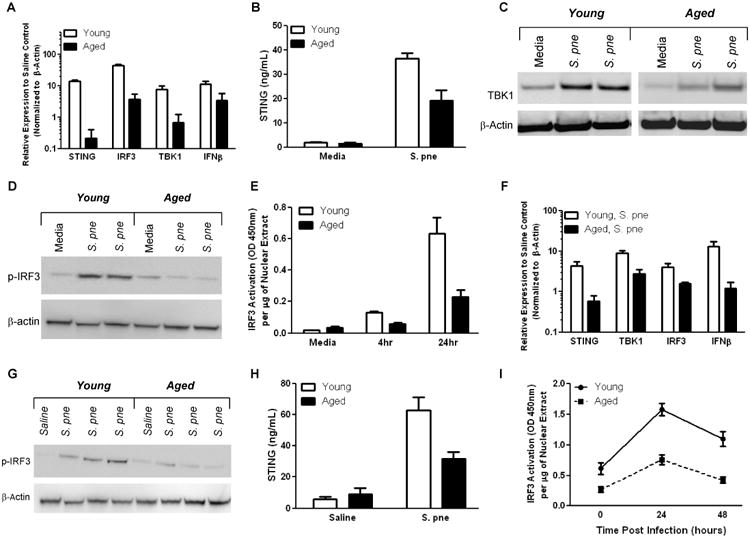
(A-C) Macrophages were cultured with media alone or with S. pne (50 CFU) for 24 hours prior to RNA or protein isolation. (A) Gene specific expression of STING, TBK1, IRF3, and IFNβ, relative to media control, was assessed by real time PCR (two-way ANOVA, p<0.0001). (B) STING protein levels were assessed by ELISA (two-way ANOVA, p<0.0001). (C-D) Protein was isolated from cultured macrophages and TBK1 and phosphorylated IRF3 protein expression was examined by western blotting. (E) IRF3 activation was assessed using TransAM IRF3 ELISA (two-way ANOVA, p=0.0071). (F-I) Young (2 months) and aged (19 months) male and female BALB/c mice received 1×103 CFU of S. pne (ATCC 6303) or saline via intranasal instillation. RNA and protein was collected from lung tissue samples at 24 hours post infection. (F) Gene specific expression of STING, TBK1, IRF3, and IFNβ, relative to saline control, was assessed by real time PCR (two-way ANOVA, p<0.0001). (G) Protein was isolated from lung tissue and phosphorylated IRF3 protein expression was examined by western blotting. (H) STING protein levels were assessed by ELISA (two-way ANOVA, p=0.0224). (I) IRF3 activation was assessed using TransAM IRF3 ELISA (two-way ANOVA, p<0.0001). Similar results were obtained from three or more independent experiments. For in vitro experiments, the values represent N=4 or greater per experiment and are expressed as the mean ± SEM. For in vivo experiments, the values are representative of five or more mice per group and are expressed as the mean ± SEM.
Enhanced ER Stress in Aged Lung during S. pneumoniae Infection
The UPR is initiated by disruptions to normal ER function, such as increased protein load, protein misfolding, and calcium gradient deregulation (30). We evaluated ER function and activation of the UPR in young and aged macrophages in response to in vitro infection with S. pne. To this extent, we assessed the expression of binding immunoglobulin protein (BIP/GRP78), an essential component of the translocation machinery induced under conditions where there is an accumulation of unfolded polypeptides in the ER (31). BIP/GRP78 expression was increased in response to S. pne, with significantly higher levels detected in aged macrophages (Figure 4A; two-way ANOVA, p<0.0001). To confirm these results, we next evaluated BIP/GRP78 expression in young and aged S. pne infected macrophages post treatment with ER stress reducing agent, tauroursodeoxycholic acid (TUDCA), or ER stress inducing agent, sunitinib malate (SM) (32, 33). In response to TUDCA treatment, there was a significant reduction in S. pne mediated BIP/GRP78 expression, while levels were significantly enhanced in response to SM treatment (Figure 4A; two-way ANOVA, p<0.0001). In response to elevated ER stress, an additional protein associated with the UPR, protein disulfide isomerase (PDI), promotes disulfide reduction, rearrangement, and re-oxidation until the correct protein conformation is achieved (31). Similar to BIP/GRP78, PDI expression was increased post treatment with S. pne, with significantly higher levels present in aged macrophages (Figure 4B; two-way ANOVA, p=0.0063). To confirm these results, we next evaluated changes in BIP/GRP78 and PDI expression in young and aged lung at 24 hours post in vivo S. pne infection. In agreement with our in vitro findings, we detected increased BIP/GRP78 (Figure 4C; two-way ANOVA, p=0.0006) and PDI (data not shown) expression in response to S. pne infection, with significantly higher levels expressed in aged lung.
Figure 4. Enhanced ER Stress in Aged Hosts during S. pneumoniae Infection.
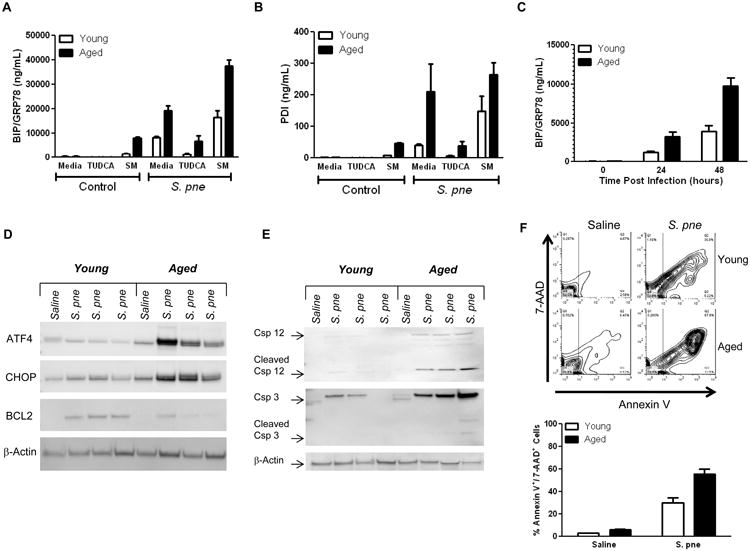
(A-B) Macrophages were cultured with media alone, media containing tauroursodeoxycholic acid (TUDCA), sunitinib malate (SM), and/or S. pne overnight. Protein was isolated and expression of BIP/GRP78 and PDI were assessed by ELISA (A: two-way ANOVA, p<0.0001; B: two-way ANOVA, p=0.0063). (C-F) Young (2 months) and aged (19 months) male and female BALB/c mice received 1×103 CFU of S. pne or saline via intranasal instillation. Protein was collected from lung tissue samples at 24 hours post infection and expression of BIP/GRP78 was assessed by ELISA (two-way ANOVA, p=0.0006). (D) Expression of ATF4, CHOP, and BCL2 were assessed by western blotting. (E) Expression of caspase-12 and -3 in young and aged lung homogenates collected at 24 hours post infection was assessed by western blotting. (F) Apoptosis of lung cells was assessed by flow cytometric staining with Annexin V and 7-AAD (two-way ANOVA, p=0.0007). Similar results were obtained from three or more independent experiments. For in vitro experiments, the values represent N=5 or greater per experiment and are expressed as the mean ± SEM. For in vivo experiments, the values are representative of five or more mice per group and are expressed as the mean ± SEM.
Increased ER Stress Induced Apoptosis in Aged Lung in Response to S. pneumoniae Infection
It is well established that an inability of the UPR response to resolve prolonged ER stress results in apoptosis (30). Dissociation of BIP/GRP78 from its ER stress receptor results in the upregulation of activating transcription factor 4 (ATF4) and subsequent transcription of genes required to restore ER homeostasis (34). In response to S. pne infection, there was an increase in ATF4 expression with higher levels detected in aged lung (Figure 4D). ATF4 mediated transcription of C/EBP homologous protein (CHOP), signifies the commitment of a highly ER stressed cell to switch from pro-survival to apoptotic cell signaling cascades (35-38). We next examined the expression of CHOP in young and aged lung at early time points post S. pne infection. While CHOP expression was increased in lung in response to S. pne infection, there were higher levels of CHOP present in aged lung homogenates (Figure 4D). Previous work has shown that CHOP represses B-cell lymphoma 2 (BCL2), an important regulator of apoptosis, which increases the proportion of pro-apoptotic proteins in the cell, leading to apoptosis induction (35). We next evaluated BCL2 expression in young and aged lung post S. pne infection. In accordance with CHOP expression, BCL2 expression was detectable in both age groups; however there was a lower level in aged lung when compared to young S. pne infected lung (Figure 4D). Further, when compared to young, there was increased expression and cleavage of caspase-12 and -3 in aged lung (Figure 4E), resulting in increased ER mediated apoptosis and cell death in this cohort (Figure 4F; two way ANOVA, p=0.007). Taken together, our current results illustrate that there is an age associated increase in ER stress induced apoptosis in lung during the early time points of S. pne infection.
Increased Atg9a Mediated Inhibition of STING in Aged Lung Contributes to Decreased IFNβ Production during S. pneumoniae Infection
Subsequently following BIP/GRP78 dissociation, inositol-requiring enzyme 1 (IRE1), a serine-threonine kinase containing an endoribonuclease domain, becomes activated (39). When compared to young, IRE1 expression was upregulated in aged macrophages (Figure 5A, Supplemental Figure 2A) and lung (Figure 5B) in response to S. pne infection. Activation of IRE1 results in cleavage and splicing of X-box binding protein 1 (XBP1), a transcription factor that controls the transcription of chaperones that aid in restoring ER function, into its activated form (20, 40-42). Recent work has illustrated an immunoregulatory role for XBP1 in mediating IFNβ induction upon TLR stimulation (30, 43, 44). We detected an increase in XBP1 expression in S. pne infected macrophages, with higher expression levels in aged macrophages (Figure 5A, Supplemental Figure 2A). XBP1 levels were also elevated in aged lung during S. pne infection (Figure 5B).
Figure 5. Increased Atg9A Mediated Inhibition of STING in Aged Lung during S. pneumoniae Infection.
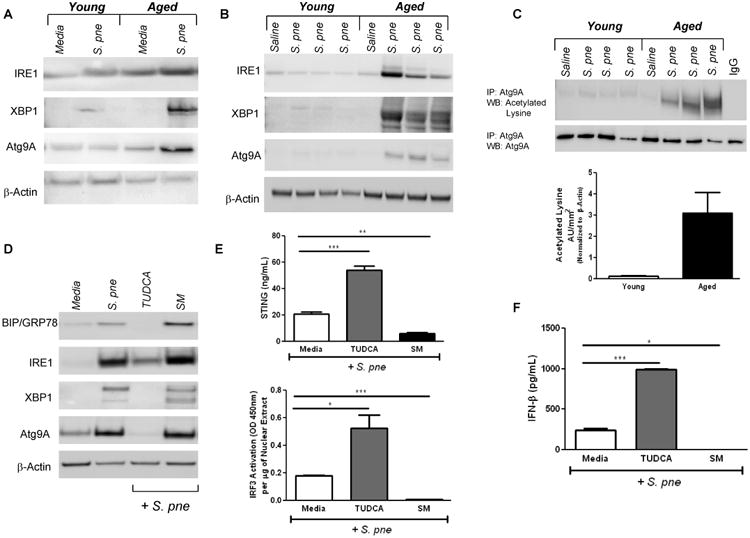
(A-B) Macrophages were cultured with media alone or with S. pne for 24 hours. Protein was isolated and expression of IRE1, XBP1, and Atg9a were assessed by western blotting. (B) Young (2 months) and aged (19 months) male and female BALB/c mice received 1×103 CFU of S. pne or saline via intranasal instillation. Protein was collected from lung tissue samples at 24 hours post infection and expression of IRE1, XBP1, and Atg9a were assessed by western blotting. (C) Co-immunoprecipitation using α-Atg9a followed by western blotting for acetylated lysine (one-way ANOVA, p=0.0391). (D-F) Aged macrophages were cultured with media alone, media containing tauroursodeoxycholic acid (TUDCA), sunitinib malate (SM), and/or S. pne overnight. (D) Protein was isolated and expression of BIP/GRP78, IRE1, XBP1, and Atg9a in aged macrophages was assessed by western blot. (E) STING protein and IRF3 activation were assessed by ELISA (t-test comparisons of media and compound treatment, p<0.05). IFNβ production was assessed by ELISA (t-test comparisons between media and compound treatment, p<0.05). Similar results were obtained from three or more independent experiments. For in vitro experiments, the values represent N=5 or greater per experiment and are expressed as the mean ± SEM. For in vivo experiments, the values are representative of five or more mice per group and are expressed as the mean ± SEM.
Recent reports have established a role for IRE1/XBP1 in controlling the induction of Atg9a expression (20). Atg9a, an ER-based protein, is a regulator of STING-mediated innate immune responses, with a loss of Atg9a resulting in enhanced dsDNA-induced assembly of STING/IRF3 dependent innate immune responses (19). When compared to young, there was an increase in Atg9a expression in aged macrophages (Figure 5A and Supplemental Figure 2B) and lung (Figure 5B) during S. pne infection. As the acetylation status of Atg9a serves as a sensor for its initiation, we next examined if an age associated alteration in acetylation of Atg9a may contribute to altered production of IFNβ in response to S. pne infection (20). Protein lysates collected from young and aged lung post infection with S. pne were immunoprecipitated with α-Atg9a followed by western blot analysis of acetylated lysine. As shown in Figure 5C, when compared to young, there was significantly higher levels of acetylated Atg9a in aged lung in response to S. pne (t-test, p=0.0391).
To examine if Atg9a expression in aged macrophages was dependent on levels of ER stress and IRE1 activation, we treated aged macrophages with TUDCA or IRE1 activator, SM. In response to TUDCA, there was decreased BIP/GRP78, IRE1, XBP1, and Atg9A during S. pne infection (Figure 5D), which corresponded with enhanced STING and IRF3 expression (Figure 5E; STING t-test, p=0.0007 and IRF3 t-test, p=0.0108) and increased IFNβ production (Figure 5F; TUDCA, t-test, p=0.0009). We next examined the impact of decreased ER stress on IFNβ production by aged macrophages in response to 3′-3′ and 2′-2′ cGAMPs. In response to TUDCA, there was a significant increase in IFNβ production in aged macrophages in response to cGAMPs (Supplemental Figure 2C). In contrast, in response to treatment with SM, there was enhanced BIP/GRP78, IRE1, XBP1, Atg9A (Figure 5D), which corresponded with decreased STING expression and IRF3 activation (Figure 5E; STING t-test, p=0.0014 and IRF3 t-test, p=0.0008) and diminished production of IFNβ (Figure 5F; SM, t-test, p=0.0067). Similarly, SM treatment decreased IFNβ production by aged macrophages in response to cGAMPs (Supplemental Figure 2C). To confirm these results, we examined the impact of enhanced ER stress on IFNβ production by young macrophages in response to S. pne. In response to SM treatment, there was a significant decrease in STING, IRF3, and IFNβ in response to S. pne infection and cGAMP stimulation (Supplemental Figure 2D-E).
We next examined the impact of enhanced Atg9a on age associated alterations in IFNβ production in response to infection with S. pne. Silencing of Atg9a using gene specific siRNA resulted in enhanced STING, IRF3, and TBK1 mRNA and protein expression (Figures 6A-D; 6A: two-way ANOVA, p<0.0001; 6B: t-test, p=0.0025; and 6D: t-test, p=0.0164) as well as augmented IFNβ production (Figure 6E; t-test, p<0.0001) by aged macrophages in response to S. pne infection. In addition, decreased Atg9a expression resulted in a decrease in the number of S. pne colonies recovered from both young and age macrophages at 24 hours post in vitro infection (Figure 6F, two-way ANOVA, p=0.0004). Taken together, the results of our current studies illustrate that age associated enhancements in Atg9a expression contribute to decreased IFNβ production in response to S. pne.
Figure 6. Knockdown of Atg9a Restores STING Mediated Production of IFNβ Production during S. pneumoniae Infection.
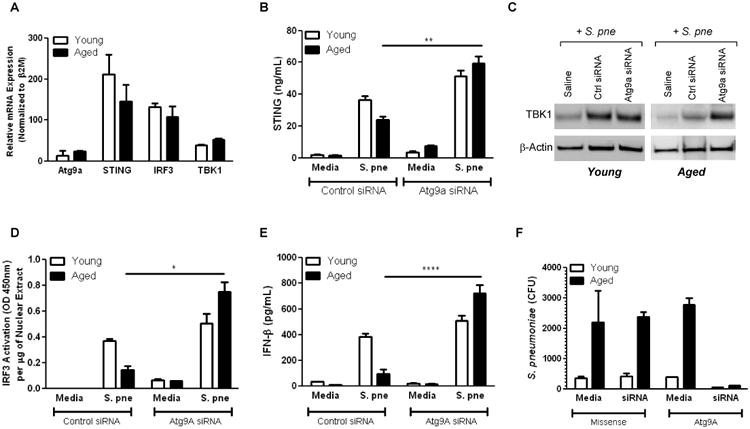
(A-G) Cells were treated with missense or Atg9a specific siRNA for 24 hours prior to S. pne infection. (A) RNA was isolated from cultured macrophages and expression, relative to control was assessed for Atg9A, STING, IRF3, and TBK1 by real time PCR. Gene expression was normalized to β2M (A: two-way ANOVA, p<0.0001). (B-D) Protein was collected and expression of STING, TBK1, and IRF3 was assessed (B: t-test, p=0.0025; D: t-test, p=0.0164). (E) Cell culture supernatants were collected and IFNβ production was assessed by ELISA (t-test, p<0.0001). (G) Bacterial titer was assessed by serial dilutions of cell culture supernatants (two-way ANOVA, p=0.0004). Similar results were obtained from three or more independent experiments. For in vitro experiments, the values represent N=5 or greater per experiment and are expressed as the mean ± SEM. For in vivo experiments, the values are representative of five or more mice per group and are expressed as the mean ± SEM.
Gemcitabine HCl Decreases Atg9A Mediated Inhibition of STING and Enhances IFNβ Production in Aged Lung during S. pneumoniae Infection
Recent studies have illustrated a potential role for FDA/EMEA-approved gemcitabine hydrochloride (GEM), a nucleoside metabolic inhibitor, in stimulating IFNβ production during influenza infection (45). We next examined the impact of GEM treatment on IFNβ production during in vitro and in vivo S. pne infections. GEM treatment, which only partially decreased S. pne replication in vitro (Supplemental Figure 3A), resulted in a subsequent decrease in Atg9A mRNA expression (Supplemental Figure 3B) as well as increased STING, IRF3, and TBK1 expression in aged macrophages during S. pne infection (Supplemental Figure 3C). GEM treatment also resulted in augmented STING expression, IRF3 activation, and IFNβ production in aged macrophages (Supplemental Figure 3D-E) as well as enhanced bacterial clearance (Supplemental Figure 3F). Further, consecutive GEM treatments post S. pne infection improved morbidity (Figure 7A: two-way ANOVA, p<0.0001; Figure 7B: two-way ANOVA, p<0.0001) and mortality (Figure 7C: Mantel Cox, p=0.0001) in aged hosts. While GEM treatment of aged hosts resulted in similar expression of CHOP in lung when compared to S. pne controls (Supplemental Figure 4A), we detected decreased expression of IRE1 and Atg9a as well as acetylated Atg9a, which was associated with an increase in STING mediated production of IFNβ (Figure 7D: t-test 24h, p=0.0063 and t-test 48h, p<0.0001; Figure 7E: t-test 24h, p=0.0482 and 48h t-test, p<0.0001; and Figure 7F: two-way ANOVA, p<0.0001). Consecutive treatments with GEM also resulted in enhanced bacterial clearance in aged lung (Figure 7G-H: two-way ANOVA, p<0.0001) and decreased levels of cellular apoptosis (Supplemental Figure 4B) when compared to infected, saline treated controls. In sum, treatment with GEM resulted in a significant decrease in Atg9a expression and a subsequent enhancement of STING mediated IFNβ production by aged macrophages in response to S. pne infection.
Figure 7. Gemcitabine HCl Decreases Atg9A Mediated Inhibition of STING and Enhances IFNβ Production during S. pneumoniae Infection.
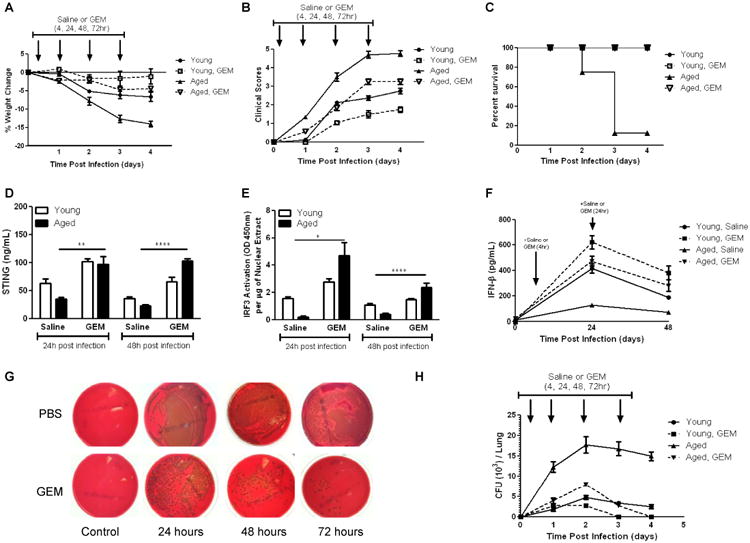
Young (2 months) and aged (19 months) male and female BALB/c mice received 1×103 CFU of S. pne (ATCC 6303) or saline via intranasal instillation. Starting at 4 hours post infection, animals received a 100μL intraperitoneal injection of gemcitabine HCl (0.3mg/mL) or saline control. (A-C) Weights (A: two-way ANOVA, p<0.0001), clinical scores (B: two-way ANOVA, p<0.0001), and survival (C: Mantel Cox test, p=0.0001) were assessed twice daily. (D-H) Lung tissue was isolated and homogenized at select time points post treatment. (D-F) STING expression, IRF3 activation, and IFNβ production were assessed by ELISA (D-E: t-test, p<0.05; F: two-way ANOVA, p<0.0001). (G) Representative bacterial clearance in aged lung samples during S. pne infection. (H) Bacterial titers were assessed and CFU were calculated (two-way ANOVA, p<0.0001). Similar results were obtained from three or more independent experiments. The values are representative of five or more mice per group and are expressed as the mean ± SEM.
Discussion
While the increased burden of Streptococcus pneumoniae (S. pne) infections in the elderly (>65 years) population is well know, the molecular mechanisms that influence this increased susceptibility have not been fully elucidated. In the current study, we examined the impact of aging on IFNβ production during S. pne infection. We found that STING mediated activation of IFNβ by aged macrophages is decreased during S. pne infection. Decreased IFNβ production was associated with enhanced morbidity as well as increased S. pne titers and bacterial lytic gene expression in lungs of aged mice early during infection. This was not due to impaired cellular recruitment to the lung, as we detected similar total cell number in both aged groups at 24 hours post infection, with significantly higher numbers of macrophages present in aged lung. Examination of aged macrophages illustrated that decreased IFNβ production was not due to alterations in phagocytosis or bacterial replication and was consistent with decreased IFNβ in response to other IFNβ stimulatory agents, such as TBD, MPLA-SM, and ADP (data not shown).
Decreased IFNβ production by aged macrophages post culture with cyclic dinucleotides, c-di-AMP and c-di-GMP, was also consistent with in vitro S. pne infection. Cyclic dinucleotides are produced by nearly all bacterial species and are critical for the regulation of bacterial motility, adhesion, biofilm formation, and pathogenicity and may serve as a more specific target for innate immune recognition of bacteria (18). Within eukaryotic cells, cyclic dinucleotides are recognized as ‘danger signals’ and result in production of IFNβ (18, 46). In response to purified c-di-AMP or c-di-GMP there was robust production of IFNβ by both young and aged macrophages. While IFNβ production by aged macrophages was significantly lower than young in response to the purified cyclic dinucleotides, these levels remained augmented when compared to IFNβ production during in vitro S. pne infection. In addition to cyclic dinucleotides, cytosolic DNA sensors, such as DAI and IFI16 as well as cGAS, an enzyme that catalyzes the synthesis of cGAMP, play an important role in mediating IFNβ production in response to bacterial dsDNA (47, 48). 3′-3′ cGAMP has a strong binding affinity for STING and thereby, is a highly potent stimulator of IFNβ (46). Results of our current study illustrate that there was also an age associated decrease in IFNβ production in response to 3′-3′ cGAMP. It is important to note that in response to decreased ER stress, IFNβ production by aged macrophages stimulated with 3′-3′ cGAMP was significantly increased. Further, reduced ER stress post treatment with TUDCA resulted in decreased IRE1 mediated production of Atg9a and enhanced STING expression and IRF3 activation. Based upon these findings, it may be possible that in addition to Atg9a mediated inhibition of STING, recognition of bacterial DNA by cytosolic DNA sensors during an active S. pne infection may also be altered by enhanced ER stress in aged hosts. Examination of the relationship between cyclic dinucleotide and DNA sensing in aged macrophages as well as the role of Atg9a on their function will need to be evaluated in future studies.
Enhanced IRE1/XBP1 mediated acetylation of Atg9a in aged macrophages and lung during S. pne infection resulted in decreased STING mediated IFNβ production. Augmented Atg9a mRNA and protein levels contribute to an age-associated decrease in STING, TBK1, IRF3, and IFNβ in response to S. pne. In accordance with previously published reports, decreased Atg9a expression resulted in enhanced STING expression in response to S. pne, leading to heightened IFNβ production (19). Given the importance of Atg9a for STING mediated IFNβ production as well as autophagy, it is possible that Atg9a function may play a key role in determining how a cell responds to S. pne infection. As a host ages, mechanisms that mediate cellular responsiveness to increased basal ER stress levels may become more tolerant so as to prevent overly enhanced inflammatory signaling from occurring. In response to highly augmented ER stress post treatment with IRE1 activator SM there is increased Atg9a expression and decreased IFNβ production by aged macrophages. Decreased levels of ER stress post TUDCA treatment resulted in decreased BIP/GRP78, IRE1, XBP1, and Atg9a expression and increased IFNβ production by aged macrophages in response to S. pne infection. It is plausible that augmented Atg9a expression may have evolved in aged hosts to play an important role in protecting cells from pro-inflammatory signaling and apoptosis in response to increased basal ER stress levels that occur as the host ages. Enhanced Atg9a expression and acetylation may function as a means to ensure that as a host ages a tight balance on ER homeostasis is maintained and its expression and signaling can be adjusted depending on the status of the ER. Augmented Atg9a levels may be beneficial to regulate cellular responses in aged hosts during non-lethal forms of ER stress and in response to elevated mitochondrial mediated production of ROS. Future work will need to be performed to further examine the delicate balance between ER stress and Atg9a expression during the process of aging.
Enhanced ER stress levels in aged hosts in response to S. pne contribute to CHOP mediated induction of caspase-12 mediated apoptosis. Based upon our current results, we hypothesize that in addition to increased intracellular bacterial growth, the death of macrophages from aged mice greatly influences the increased number of bacterial counts present in cell culture supernatants. Additionally, increased ER stress mediated apoptosis in aged lung may contribute to enhanced dissemination of S. pne and underlie an early increase in morbidity and enhanced mortality in aged hosts during infection. As innate effector cells, such as dendritic cells and macrophages, play an important role in activating adaptive immunity, it is possible that enhanced ER stress mediated induction of apoptosis in aged lung impairs innate immune responses by decreasing the number of non-apoptotic effectors present. Our current results illustrate that in vitro treatment with GEM, which decreased apoptosis in response to S. pne infection, resulted in a significant decrease in bacterial counts present in the cell culture supernatants isolated from aged macrophages. Further, daily treatment with GEM resulted in decreased levels of apoptosis and reduced S. pne present in aged lung and post culture with aged macrophages. It is important to note that treatment with GEM did not completely inhibit apoptosis and thereby, residual levels of apoptosis in aged lung may still alter effector cell function and initiation of adaptive immune response to S. pne. Future work to investigate the impact of GEM upon these responses will need to be performed.
As GEM is a nucleoside metabolic inhibitor, it is possible that part of its function is to directly target S. pne biosynthesis. Despite its direct effect on S. pne, titers from GEM treated macrophage cultures were significantly lower, thereby illustrating a second potential mechanism of GEM in mediating S. pne clearance. Reduced S. pne replication may be a direct effect of GEM and decreased Atg9a expression may occur as part of an indirect secondary feedback mechanism that results in enhanced S. pne clearance from infected cells and tissues (summarized in Supplemental Figure 4C). Based upon our current results, we hypothesize that increased IFNβ synthesis is not the main effector mechanism of GEM and instead acts upstream of IRE1 to reduce Atg9a levels during S. pne infection and rescue STING mediated production of IFNβ. In addition to regulating STING mediated production of IFNβ, recent work has illustrated an important role of Atg9a in the induction of autophagy (20). As GEM treatment only partially suppresses Atg9a expression in lung during S. pne, it may not directly impact all of the downstream regulatory functions of Atg9a. Taken together, our current results illustrate GEM as a beneficial therapeutic to improve IFNβ production while allowing Atg9a to function in its regulatory capacity in other cellular processes.
In response to GEM treatment, despite a strong reduction in mortality, aged animals have significantly higher clinical scores and weight loss when compared to young, GEM treated counterparts. While our results illustrate that GEM treatment augments IFNβ production in aged lung and is necessary for survival, it does not decrease the production of inflammatory cytokines, such as TNFα and IL-6, at early time points post S. pne infection. Previous work has illustrated a detrimental effect for high expression of IL-6 and subsequent enhanced neutrophil recruitment in aged hosts in response to infectious stimuli (49). Examination of cellular infiltration illustrated a similar elevation in neutrophil recruitment in both saline and GEM treated mice at 24 hours post infection (data not shown). By 48 hours post infection, neutrophil numbers and mucus production in lung continued to significantly rise in saline treated aged mice (data not shown). In contrast, by 48 hours post infection, we did not detect a similar elevation in neutrophil infiltration or enhanced mucus production in GEM treated aged mice, with levels being similar to young, saline treated S. pne infected cohorts (data not shown). It is therefore possible that the observed early neutrophil influx and pro-inflammatory cytokine production in aged lung contributes to an early increase in morbidity and that an intervention, such as GEM, which results in decreased S. pne and increased IFNβ production, can improve morbidity and prevent mortality. To this extent, it may be necessary to examine combination treatment strategies using anti-inflammatory compounds in addition to GEM treatment to further improve morbidity in aged hosts during infection.
Recent work has illustrated an important role for the ER membrane transporter, SLC33AI/AT-1, in the regulation of Atg9a acetylation downstream of IRE/XBP1 signaling (20). It is possible that the indirect effect of GEM on Atg9a expression and acetylation may be due to an alteration in an upstream regulatory process, such as AT-1 mediated supply of acetyl-coA into the ER lumen. As Atg9a is an ER-based protein that appears to translocate out of the ER, the acetylation status of Atg9a may serve as an intracellular sensor for the activation of IFNβ responses to S. pne. Decreased Atg9a acetylation, as observed in young and GEM treated aged macrophages during S. pne infection, would result in the induction of STING mediated production of IFNβ while enhanced Atg9a acetylation, as observed in aged hosts, would result in decreased STING mediated production of IFNβ. Future studies will need to be performed to confirm the importance of AT-1 on regulating these responses in aged hosts.
ER stress is an important component of the innate immune response and is essential for enhancing signaling in response to TLR stimulation (44). Results of our current study illustrate that an age associated enhancement in ER stress contributed to increased XBP1 expression and production of pro-inflammatory cytokines, TNFα and IL-6, in response to S. pne infection. In response to TUDCA, there was a decrease in ER stress in aged macrophages that corresponded with enhanced IFNβ production in response to S. pne. It is possible that innate signaling during S. pne infection in aged macrophages is dependent on the status of the ER, with a highly stressed ER initiating apoptotic cascades and inhibiting STING mediated production of IFNβ. It is important to note that in response thapsigargin or tunicamycin, inhibitors of the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), IRF3 mediated production of IFNβ in response to LPS stimulation was enhanced (30, 50). It is therefore plausible, that as a host ages, there are differential thresholds for activation of ER stress mediated innate immune signaling cascades in response to pathogenic and non-pathogenic stimuli. As ER stress and initiation of the UPR involves multiple signaling cascades, it will be important to fully elucidate how aging and enhanced basal ER stress levels alter the regulation of these pathways and their impact innate immune responses to both infectious and non-infectious stimuli.
In summary, our findings show that STING mediated production of IFNβ during S. pne infection is decreased in aged hosts. Enhanced ER stress during S. pne infection augmented IRE1/XBP1 mediated production of Atg9a and subsequent inhibition of STING/TBK1 activation of IRF3. Treatment with GEM resulted in decreased S. pne titers as well as enhanced STING mediated IFNβ production in lung and improved morbidity and mortality of aged animals during S. pne infection. Data presented in this study provide new evidence as to why older persons are more susceptible to S. pne infection and provide a possible mechanism to enhance these responses, thereby decreasing morbidity and mortality in this population. Future work will need to be performed to examine if GEM treatment may improve clinical outcomes in elderly to other highly pathogenic agents.
Supplementary Material
Acknowledgments
This work was supported by NIH K01AG034999-01A1 (H.W.S), NIH R21AG044755-01A1 (H.W.S) and by the LRRI/BWH Consortium for Lung Research.
Abbreviations
- S. pne
Streptococcus pneumoniae
- STING
stimulator of interferon genes
- IRF3
interferon regulatory factor 3
- ER
endoplasmic reticulum
- TBK1
TANK binding kinase 1
- Atg9a
autophagy related gene 9
- UPR
unfolded protein response
- IRE1
inositol-requiring protein
- ATF6
activating transcription factor 6
- XBP1
X-box binding protein 1
- GEM
gemcitabine HCl
- TUDCA
tauroursodeoxycholic acid
- SM
sunitinib malate
- IFNAR
type I interferon receptor
References
- 1.McBean AM, Hebert PL. New estimates of influenza-related pneumonia and influenza hospitalizations among the elderly. International journal of infectious diseases: IJID: official publication of the International Society for Infectious Diseases. 2004;8:227–235. doi: 10.1016/j.ijid.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 2.Heron M. Deaths: leading causes for 2007. National vital statistics reports: from the Centers for Disease Control and Prevention, National Center for Health Statistics, National Vital Statistics System. 2011;59:1–95. [PubMed] [Google Scholar]
- 3.Miyashita N, Kawai Y, Akaike H, Ouchi K, Hayashi T, Kurihara T, Kawanaka N, Okimoto N. Influence of age on the clinical differentiation of atypical pneumonia in adults. Respirology. 2012 doi: 10.1111/j.1440-1843.2012.02188.x. [DOI] [PubMed] [Google Scholar]
- 4.Weng NP. Aging of the immune system: how much can the adaptive immune system adapt? Immunity. 2006;24:495–499. doi: 10.1016/j.immuni.2006.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pawelec G, Akbar A, Caruso C, Solana R, Grubeck-Loebenstein B, Wikby A. Human immunosenescence: is it infectious? Immunol Rev. 2005;205:257–268. doi: 10.1111/j.0105-2896.2005.00271.x. [DOI] [PubMed] [Google Scholar]
- 6.Romero-Steiner S, Musher DM, Cetron MS, Pais LB, Groover JE, Fiore AE, Plikaytis BD, Carlone GM. Reduction in functional antibody activity against Streptococcus pneumoniae in vaccinated elderly individuals highly correlates with decreased IgG antibody avidity. Clin Infect Dis. 1999;29:281–288. doi: 10.1086/520200. [DOI] [PubMed] [Google Scholar]
- 7.Sousa D, Justo I, Dominguez A, Manzur A, Izquierdo C, Ruiz L, Nebot M, Bayas JM, Celorrio JM, Varona W, Llinares P, Miguez E, Sanchez E, Carratala J. Community-acquired pneumonia in immunocompromised older patients: incidence, causative organisms and outcome. Clinical microbiology and infection: the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2012 doi: 10.1111/j.1469-0691.2012.03765.x. [DOI] [PubMed] [Google Scholar]
- 8.Yokota S, Sato K, Kuwahara O, Habadera S, Tsukamoto N, Ohuchi H, Akizawa H, Himi T, Fujii N. Fluoroquinolone-resistant Streptococcus pneumoniae strains occur frequently in elderly patients in Japan. Antimicrob Agents Chemother. 2002;46:3311–3315. doi: 10.1128/AAC.46.10.3311-3315.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takahashi T, Morozumi M, Chiba N, Asami R, Kishii K, Murayama SY, Ubukata K. Co-infection with respiratory syncytial virus subgroup a and Streptococcus pneumoniae detected by a comprehensive real-time polymerase chain reaction assay in an elderly patient with community-acquired pneumonia. Journal of the American Geriatrics Society. 2009;57:1711–1713. doi: 10.1111/j.1532-5415.2009.02422.x. [DOI] [PubMed] [Google Scholar]
- 10.Millar MR, Brown NM, Tobin GW, Murphy PJ, Windsor AC, Speller DC. Outbreak of infection with penicillin-resistant Streptococcus pneumoniae in a hospital for the elderly. The Journal of hospital infection. 1994;27:99–104. doi: 10.1016/0195-6701(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 11.Canton R, Unal S, Farrell DJ. Antibacterial resistance patterns in Streptococcus pneumoniae isolated from elderly patients: PROTEKT years 1-5 (1999-2004) International journal of antimicrobial agents. 2007;30:546–550. doi: 10.1016/j.ijantimicag.2007.07.025. [DOI] [PubMed] [Google Scholar]
- 12.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koppe U, Hogner K, Doehn JM, Muller HC, Witzenrath M, Gutbier B, Bauer S, Pribyl T, Hammerschmidt S, Lohmeyer J, Suttorp N, Herold S, Opitz B. Streptococcus pneumoniae stimulates a STING- and IFN regulatory factor 3-dependent type I IFN production in macrophages, which regulates RANTES production in macrophages, cocultured alveolar epithelial cells, and mouse lungs. J Immunol. 2012;188:811–817. doi: 10.4049/jimmunol.1004143. [DOI] [PubMed] [Google Scholar]
- 14.Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol. 2013;14:19–26. doi: 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
- 15.Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell reports. 2013;3:1355–1361. doi: 10.1016/j.celrep.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Science signaling. 2012;5:ra20. doi: 10.1126/scisignal.2002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McWhirter SM, Barbalat R, Monroe KM, Fontana MF, Hyodo M, Joncker NT, Ishii KJ, Akira S, Colonna M, Chen ZJ, Fitzgerald KA, Hayakawa Y, Vance RE. A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J Exp Med. 2009;206:1899–1911. doi: 10.1084/jem.20082874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, Yamamoto N, Kawai T, Ishii K, Takeuchi O, Yoshimori T, Akira S. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A. 2009;106:20842–20846. doi: 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pehar M, Jonas MC, Hare TM, Puglielli L. SLC33A1/AT-1 protein regulates the induction of autophagy downstream of IRE1/XBP1 pathway. J Biol Chem. 2012;287:29921–29930. doi: 10.1074/jbc.M112.363911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. The Journal of experimental medicine. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. In: Coligan John E, et al., editors. Current protocols in immunology. Chapter 14. 2008. p. Unit 14 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. American journal of respiratory cell and molecular biology. 2013;49:503–510. doi: 10.1165/rcmb.2013-0086MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stout-Delgado HW, Vaughan SE, Shirali AC, Jaramillo RJ, Harrod KS. Impaired NLRP3 inflammasome function in elderly mice during influenza infection is rescued by treatment with nigericin. J Immunol. 2012;188:2815–2824. doi: 10.4049/jimmunol.1103051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dimmock NJ, Marriott AC. In vivo antiviral activity: defective interfering virus protects better against virulent Influenza A virus than avirulent virus. J Gen Virol. 2006;87:1259–1265. doi: 10.1099/vir.0.81678-0. [DOI] [PubMed] [Google Scholar]
- 26.de Cunto Brandileone MC, Simonsen DVV, Tadeu Casagrande S, Cobo Zanella R, Leopoldo Silva Guerra ML, Pires Brandao A, de Andrade Melles CE, CP AC, Di Fabio JL, Austrian R. Characteristics of Isolates Streptococcus pneumoniae from Middle-Aged and Elderly Adults in Brazil: Capsular Serotypes and Antimicrobial Sensitivity to Invasive Infections. The Brazilian journal of infectious diseases: an official publication of the Brazilian Society of Infectious Diseases. 1998;2:90–96. [PubMed] [Google Scholar]
- 27.Kurtti P, Isoaho R, von Hertzen L, Keistinen T, Kivela SL, Leinonen M. Influence of age, gender and smoking on Streptococcus pneumoniae, Haemophilus influenzae and Moraxella (Branhamella) catarrhalis antibody titres in an elderly population. Scandinavian journal of infectious diseases. 1997;29:485–489. doi: 10.3109/00365549709011859. [DOI] [PubMed] [Google Scholar]
- 28.Martens P, Worm SW, Lundgren B, Konradsen HB, Benfield T. Serotype-specific mortality from invasive Streptococcus pneumoniae disease revisited. BMC infectious diseases. 2004;4:21. doi: 10.1186/1471-2334-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, Tomasello F, Gambuzza M, Macri G, Ruggeri A, Leanderson T, Teti G. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol. 2007;178:3126–3133. doi: 10.4049/jimmunol.178.5.3126. [DOI] [PubMed] [Google Scholar]
- 30.Zeng L, Liu YP, Sha H, Chen H, Qi L, Smith JA. XBP-1 couples endoplasmic reticulum stress to augmented IFN-beta induction via a cis-acting enhancer in macrophages. J Immunol. 2010;185:2324–2330. doi: 10.4049/jimmunol.0903052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mayer M, Kies U, Kammermeier R, Buchner J. BiP and PDI cooperate in the oxidative folding of antibodies in vitro. J Biol Chem. 2000;275:29421–29425. doi: 10.1074/jbc.M002655200. [DOI] [PubMed] [Google Scholar]
- 32.Jha BK, Polyakova I, Kessler P, Dong B, Dickerman B, Sen GC, Silverman RH. Inhibition of RNase L and RNA-dependent protein kinase (PKR) by sunitinib impairs antiviral innate immunity. J Biol Chem. 2011;286:26319–26326. doi: 10.1074/jbc.M111.253443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie Q, Khaoustov VI, Chung CC, Sohn J, Krishnan B, Lewis DE, Yoffe B. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology. 2002;36:592–601. doi: 10.1053/jhep.2002.35441. [DOI] [PubMed] [Google Scholar]
- 34.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Molecular cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 35.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO reports. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes & development. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes & development. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell death and differentiation. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 39.Lin JH, Li H, Zhang Y, Ron D, Walter P. Divergent effects of PERK and IRE1 signaling on cell viability. PLoS One. 2009;4:e4170. doi: 10.1371/journal.pone.0004170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 41.Back SH, Schroder M, Lee K, Zhang K, Kaufman RJ. ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods. 2005;35:395–416. doi: 10.1016/j.ymeth.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 42.Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes & development. 2002;16:452–466. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith JA, Turner MJ, DeLay ML, Klenk EI, Sowders DP, Colbert RA. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur J Immunol. 2008;38:1194–1203. doi: 10.1002/eji.200737882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Denisova OV, Kakkola L, Feng L, Stenman J, Nagaraj A, Lampe J, Yadav B, Aittokallio T, Kaukinen P, Ahola T, Kuivanen S, Vapalahti O, Kantele A, Tynell J, Julkunen I, Kallio-Kokko H, Paavilainen H, Hukkanen V, Elliott RM, De Brabander JK, Saelens X, Kainov DE. Obatoclax, saliphenylhalamide, and gemcitabine inhibit influenza a virus infection. J Biol Chem. 2012;287:35324–35332. doi: 10.1074/jbc.M112.392142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takaoka A, Taniguchi T. Cytosolic DNA recognition for triggering innate immune responses. Advanced drug delivery reviews. 2008;60:847–857. doi: 10.1016/j.addr.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 48.Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 49.Stout-Delgado HW, Du W, Shirali AC, Booth CJ, Goldstein DR. Aging promotes neutrophil-induced mortality by augmenting IL-17 production during viral infection. Cell Host Microbe. 2009;6:446–456. doi: 10.1016/j.chom.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu YP, Zeng L, Tian A, Bomkamp A, Rivera D, Gutman D, Barber GN, Olson JK, Smith JA. Endoplasmic reticulum stress regulates the innate immunity critical transcription factor IRF3. J Immunol. 2012;189:4630–4639. doi: 10.4049/jimmunol.1102737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.