

Abstract
Background
Individuals with hereditary retinoblastoma (RB) are at very high risk of developing subsequent malignant neoplasms (SMN) of which osteosarcoma (OS) is one of the most common. We hypothesized that annual surveillance using whole-body magnetic resonance imaging (WB-MRI) in asymptomatic survivors of hereditary RB would detect SMN of the bone and soft tissues at an early stage.
Procedure
Retrospective review of the results of a WB-MRI screening program in hereditary RB survivors from February 2008 – August 2012. The primary outcome was to determine the sensitivity and specificity of WB-MRI in detecting SMNs.
Results
Twenty-five patients had at least one WB-MRI performed (range: 1 – 5). First WB- MRI was performed at a median age of 16 years (range: 8 – 25 years). WB-MRI detected new osseous abnormalities suspicious for malignancy in 5 patients: 2 were diagnosed with localized high-grade OS of the extremity and 3 were found to have benign osseous abnormalities after dedicated imaging (n=5/5) and/or biopsy (n=3/5). One patient was diagnosed with secondary OS three months after a normal screening WB- MRI exam. Among a total of 41 WB-MRI screening tests performed in survivors of hereditary RB, the sensitivity of detecting SMN was 66.7% and the specificity was 92.1%.
Conclusions
Preliminary results suggest that annual WB-MRI surveillance detects SMN in survivors of hereditary RB, but with modest sensitivity. Further study is needed to assess the performance of annual surveillance WB-MRIs and whether this modality decreases SMN-related mortality in RB survivors.
Keywords: Retinoblastoma, survivors, screening, whole-body MRI, pediatric oncology
INTRODUCTION
Retinoblastoma (RB) is the most common primary intra-ocular malignancy of childhood with five-year survival rates in the Unites States exceeding 95% [1]. Survivors of the hereditary form of RB, however, are known to have a significantly increased lifetime risk of subsequent malignant neoplasms (SMN) including sarcomas, malignant melanomas, and central nervous system (CNS) tumors [2–5] with significant excess mortality [6,7]. Treatment with external beam radiotherapy (EBRT) further amplifies this risk [8].
Due to this well-established risk of SMN, it would seem that targeted surveillance for second and subsequent tumors may be warranted in survivors of the genetic form of RB. However, no established guidelines exist for screening this population. Data are lacking on which modality or modalities are optimal in detecting SMNs in this setting.
We implemented a radiologic surveillance program using whole-body magnetic resonance imaging (WB-MRI) to determine if such a program would facilitate detection of SMN of the bone and soft tissues at a localized stage in survivors of hereditary RB. WB-MRI provides a non-invasive, ionizing radiation-free screening tool with the potential to detect subsequent osseous and soft tissue malignancies [9,10]. The primary outcome was to determine the sensitivity and specificity of WB-MRI in detecting SMNs.
METHODS
We performed a single-institutional, retrospective review of WB-MRI screening results in survivors of hereditary RB. Among patients who participated in the screening program, WB-MRIs were intended to be ordered annually. All data were obtained from review of the Memorial Sloan-Kettering Cancer Center (MSKCC) medical record. The retrospective review of WB-MRI surveillance results was approved by the MSKCC Institutional Review Board/Privacy Board.
Eligible patients were pre-adolescent, adolescent, and young adult survivors of hereditary RB who were seen for ophthalmologic follow-up at MSKCC between February 2008 and August 2012. Patients either had bilateral disease at diagnosis, a positive family history, or a known RB1 mutation. All patients were thought to be free of disease at the time of WB-MRI scheduling. Presence of a pacemaker, aneurysm clip or any other condition that would warrant avoidance of a strong magnetic field resulted in exclusion from the study. Twenty-five patients with hereditary RB had at least one screening WB-MRI performed during the designated time period. Results are reported as of October 15, 2012.
Whole-body Magnetic Resonance Imaging (WB-MRI)
WB-MRI was performed on one of several 1.5T scanners (GE Healthcare, Milwaukee, Wisconsin). The entire body was imaged in the axial plane utilizing T1 and FSE T2 short-tau inversion recovery (STIR) technique. Using the same technique, the spine and central base of the skull was imaged in the sagittal plane. Additionally, axial diffusion-weighted imaging was performed in the axial plane (b-value 0 and 1,000 s/mm2). Overlapping acquisition field of views were confirmed to ensure complete anatomic coverage of the entire body, including the axial and appendicular skeleton and the adjacent soft tissues. Qualitative image evaluation was performed by dedicated onco-radiologists. On average, WB-MRI scanning studies lasted 90 minutes. Cost was covered by insurance for all patients.
Data Analysis
Demographic data are reported with the use of range and median values for all continuous variables. Sensitivity was defined as the proportion of subjects with an SMN who were correctly identified as such by WB-MRI, or (true positives)/(true positives + false negatives). Using biopsy as the gold standard, and dedicated MRI as a surrogate when biopsy was not performed, true positives were defined as those with lesions suspicious for malignancy on WB-MRI which were confirmed as true malignancies on biopsy. False negatives were defined as those in whom a subsequent malignancy was diagnosed within 6 months of a normal WB-MRI.
Specificity was defined as the proportion of subjects without an SMN in whom WB-MRI was appropriately negative, or (true negatives)/(true negatives + false positives). True negatives were those in whom BM-MRI was correctly read as normal when no SMN was present. False positives were those in whom WB-MRI detected an abnormality suspicious for malignancy which was demonstrated to be benign based on dedicated imaging and/or biopsy results.
RESULTS
Patients
Baseline characteristics of study subjects are summarized in Table I. Twenty-four patients had bilateral disease at diagnosis and 1 had unilateral disease with a known RB1 mutation. Patients ranged from 8 – 25 years of age at the time of first WB-MRI (median age: 16) and had been followed for a median of 17 years (range: 10 – 24 years). Median time since last follow-up was three months (range: 1 – 28 months). At date of last contact, median patient age was 17 years (range: 11 – 26 years). Two patients had died of osteosarcoma (OS) at the time of data analysis. Both patients had localized, secondary OS detected on screening WB-MRI; however, one of the two patients died of complications related to a tertiary OS in another location, which was diagnosed after the patient presented symptomatically at a later date.
Table I.
Baseline Characteristics for all patients (n=25)
| Baseline Characteristics | |
|---|---|
| Sex | No. (%) |
|
| |
| Female | 13 (52) |
| Male | 12 (48) |
|
| |
| Race/ethnicity | No. (%) |
|
| |
| White | 14 (56) |
| Black | 8 (32) |
| Asian | 2 (8) |
| Other | 1 (4) |
|
| |
| Age at Diagnosis | |
|
| |
| Median (months) | 5 |
| Range (months) | 0–45 |
|
| |
| Laterality | |
|
| |
| Unilateral | 1 |
| Bilateral | 24 |
|
| |
| Type of Treatment Received | No. (%) |
|
| |
| Systemic Chemotherapy | 14 (58) |
| Radiation Therapy | 19 (79) |
| Autologous Transplant | 3 (13) |
| Enucleation(s) | 15 (58) |
| Local Therapy Alone | 3 (10) |
|
| |
| Age at First WB-MRI | |
|
| |
| Median (years) | 16 |
| Range (years) | 8–25 |
|
| |
| Number of WB-MRIs performed | |
|
| |
| Median | 1 |
| Range | 1–5 |
Abbreviations: No=number; WB-MRI=Whole-body MRI
Treatment History
Twenty-two of the 25 patients (87.5%) received systemic chemotherapy and/or external beam radiotherapy (EBRT) prior to enrolling in the study. The other three were treated with local therapy and/or enucleation(s) alone. Fifteen patients had at least one eye enucleated during their treatment course (unilateral =12, bilateral = 3). Treatment history for all patients is summarized in Table I.
WB-MRI Results
A total of 41 WB-MRIs were performed in 25 patients (14 patients had one WB-MRI, eight patients had two, two patients had three, and one patient had five WB-MRIs). Eight screening WB-MRIs in eight different patients detected a radiologic abnormality (see Figure 1 for flow diagram); among these, 50% (n= 4) were noted on the patient’s initial scan. Three marrow abnormalities were deemed likely to be benign and did not require further follow-up (diagnoses included probable hemangioma within the lumbosacral spine; minor marrow abnormality within the midshaft of the right tibia; multiple pelvic/uterine masses likely representing fibroids). Among the other five patients in whom an abnormality was detected on WB-MRI, all went on to receive dedicated MRIs of the area of concern. Three patients were found to have benign osseous lesions on dedicated imaging and/or biopsy (see Table II). Benign osseous findings included bone infarct (see Figure 2), chondroblastoma, and spondylolysis. The other two patients (see Figure 3) were found to have localized OS of the extremity after undergoing biopsy.
Figure 1.

Abnormal whole-body MRI (WB-MRI) screening results in all patients
Table II.
Treatment characteristics of patients with positive and/or false negative whole-body MRI (WB-MRI) findings
| Age at MRI | Sex | RB Treatment | WB-MRI Result | Dedicated MRI Result | Biopsy Result | Final Diagnosis | |
|---|---|---|---|---|---|---|---|
| Patient 1 | 21 | F | Chemotherapy; EBRT bilateral orbits | Small focal region of marrow abnormality within the distal right femoral metaphysis | Probable infarct | Not performed | Bony infarct |
| Patient 2 | 16 | M | Chemotherapy; EBRT right eye | New confluent marrow abnormality involving the distal left femur that is indeterminate | Small lesion in distal femoral epiphysis with marked surrounding edema pattern | Chondroblastoma | Chondroblastoma |
| Patient 3 | 12 | M | Chemotherapy; ASCR; EBRT right distal femur (metastatic RB) | New confluent marrow abnormality involving the proximal left tibia suspicious for neoplastic disease | Lesion in the left proximal tibia with bone formation suspicious for osteosarcoma | Osteosarcoma | Osteosarcoma |
| Patient 4 | 12 | F | Chemotherapy; EBRT right orbit & right tibia (metastatic RB); ASCR | Signal abnormality within the right proximal tibia, hypointense, minor edema | New osseous lesion with associated soft tissue mass in the proximal tibial metaphysis | Osteosarcoma | Osteosarcoma |
| Patient 5 | 19 | M | Chemotherapy; EBRT bilateral eyes; EBRT right orbit | No evidence of a secondary malignancy involving the osseous axial or proximal appendicular skeleton. No facial mass identified (June 2011) | N/A | Right maxillary gingiva: High-grade sarcoma (September 2011) | Osteosarcoma |
| Patient 6 | 14 | F | Chemotherapy | New marrow abnormality involving the posterior elements of L4 | Bilateral spondylolysis at L4 | N/A | Spondylolysis |
Abbreviations: RB = retinoblastoma; EBRT = external beam radiotherapy; ASCR = autologous stem cell rescue
Figure 2.
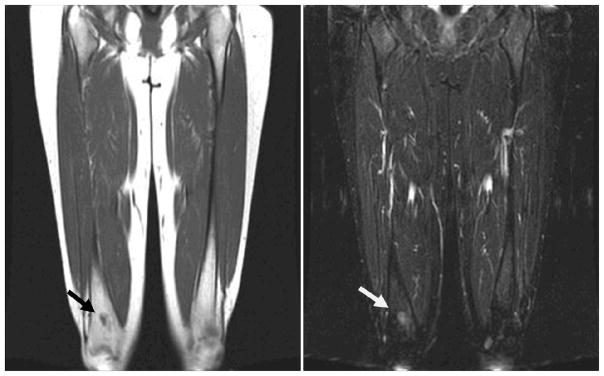
Twenty-one year old young women with history of bilateral retinoblastoma. Coronal T1 (left) and T2 STIR (right) images through the femurs from a whole body MRI show indeterminate marrow abnormality within the distal right femoral metaphysis. The patient was later determined to have a benign bone infarct on dedicated femur MRI.
Figure 3.
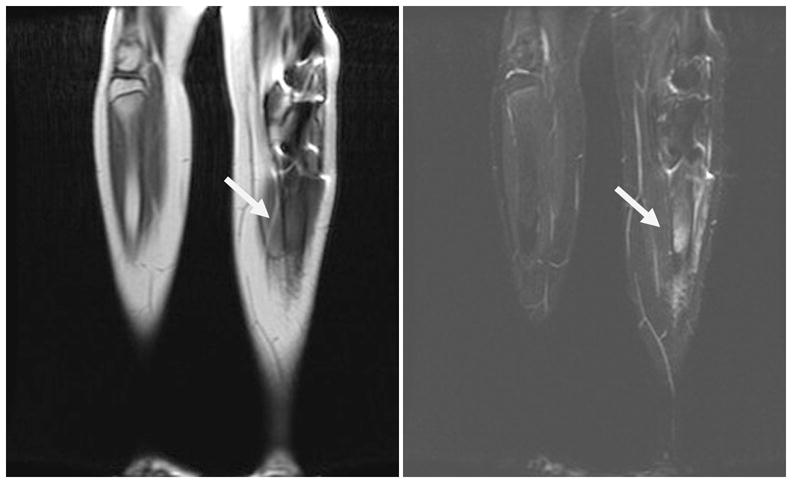
Twelve year old boy with history of bilateral retinoblastoma as well as previous metastatic disease to the right femur. Whole-body MRI (WB-MRI) showed new signal abnormality within the left tibial shaft on T1-weighted sequences (left) and T2 STIR (right). The patient later reported a several week history of left leg pain. Biopsy revealed a diagnosis of osteosarcoma of the left tibia.
In addition to the five patients with WB-MRI screening results suspicious for malignancy, one other patient was diagnosed with a localized maxillary OS three months after having a normal WB-MRI. Even in retrospect, however, this lesion was not apparent on the WB-MRI.
Among a total of 41 WB-MRI screening tests performed in survivors of RB, the sensitivity of detecting a SMN was 66.7% with a specificity of 92.1%. The positive predictive value was 0.4 and the negative predictive value was 0.97.
DISCUSSION
Early detection of disease has become a diagnostic and therapeutic objective for a wide variety of cancer subtypes. With the goal of minimizing morbidity and mortality, the use of cancer screening modalities has been suggested for selected high-risk populations [11], including those with hereditary cancer predisposition syndromes [12–14]. Yet the development of optimal surveillance strategies which maximize benefit and minimize harm has been difficult and the source of considerable controversy [15].
The genetic form of RB, usually due to a germline mutation in the RB1 tumor suppressor gene, causes ocular malignancies in childhood and an increased lifetime risk of early-onset SMNs with cumulative incidence rates reported to be as high as 36% by 50 years after the initial diagnosis of RB [4,16,17]. Since this risk is further elevated by exposure to ionizing radiation [8,17–19], it seems prudent to minimize radiation exposure when performing routine testing or screening in patients with the germline RB1 mutation.
With these provisions in mind, we sought to create a clinical surveillance protocol that might enable early detection of long bone and soft tissue malignancies in survivors of hereditary RB. Previous reports have demonstrated the utility of WB-MRI screening for the early and accurate diagnosis of musculoskeletal diseases, including sarcomas [10]. Since RB survivors are at highest risk of developing OS of the long bones during periods of peak linear growth and early adulthood, we limited screening to survivors who were pre-adolescents, adolescents, and young adults.
Based on the results of this pilot study, WB-MRI appears to have limited sensitivity in detecting SMN in survivors of heritable RB. In our cohort, among the eight patients in whom WB-MRI detected osseous abnormalities, five required further dedicated imaging and three required biopsy of the suspicious lesion, which may have resulted in psychological distress for patients and their families.
Our study is limited by a small sample size and its retrospective design. Due to the small sample size, limited outcome data exist in our study. Both patients who were correctly diagnosed with localized OS on WB-MRI subsequently died during the study period. However, one of the two patients died from complications related to a tertiary OS that was diagnosed two years later after the patient presented with leg pain. The third patient who had a normal WB-MRI, followed by a diagnosis of SMN three months later, was receiving palliative care without active treatment at the time of last data analysis.
A larger cohort of patients would be required to more precisely determine the sensitivity and specificity of WB-MRI in detecting SMN in asymptomatic survivors of hereditary RB. Furthermore, a prospective study would be required to assess patient satisfaction and/or psychological burden in those undergoing routine surveillance for SMN. While it is possible that patients and their families experienced increased anxiety around the testing, it is equally plausible that they felt a sense of empowerment and reassurance after having whole-body screening performed [20].
While annual WB-MRI surveillance scans are technically feasible in this population, it is unclear at this time whether routine whole-body imaging decreases SMN-related mortality in RB survivors and whether the benefits outweigh the potential risks.
Acknowledgments
Research Support: This work was supported by The Perry Promise Fund and grant UL1TR000457 of the Clinical and Translational Science Center at Weill Cornell Medical College; Kevin Oeffinger is supported in part by the National Institutes of Health (K05CA160724)
The authors wish to thank Joe Olechnowicz for his editorial contributions.
Footnotes
Disclaimers: None
References
- 1.Broaddus E, Topham A, Singh AD. Survival with retinoblastoma in the USA: 1975–2004. The British journal of ophthalmology. 2009;93(1):24–27. doi: 10.1136/bjo.2008.143842. [DOI] [PubMed] [Google Scholar]
- 2.Kleinerman RA, Tucker MA, Abramson DH, et al. Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. Journal of the National Cancer Institute. 2007;99(1):24–31. doi: 10.1093/jnci/djk002. [DOI] [PubMed] [Google Scholar]
- 3.MacCarthy A, Bayne AM, Draper GJ, et al. Non-ocular tumours following retinoblastoma in Great Britain 1951 to 2004. The British journal of ophthalmology. 2009;93(9):1159–1162. doi: 10.1136/bjo.2008.146035. [DOI] [PubMed] [Google Scholar]
- 4.Marees T, Moll AC, Imhof SM, et al. Risk of second malignancies in survivors of retinoblastoma: more than 40 years of follow-up. Journal of the National Cancer Institute. 2008;100(24):1771–1779. doi: 10.1093/jnci/djn394. [DOI] [PubMed] [Google Scholar]
- 5.Moll AC, Imhof SM, Bouter LM, et al. Second primary tumors in patients with retinoblastoma. A review of the literature Ophthalmic genetics. 1997;18(1):27–34. doi: 10.3109/13816819709057880. [DOI] [PubMed] [Google Scholar]
- 6.Eng C, Li FP, Abramson DH, et al. Mortality from second tumors among long-term survivors of retinoblastoma. Journal of the National Cancer Institute. 1993;85(14):1121–1128. doi: 10.1093/jnci/85.14.1121. [DOI] [PubMed] [Google Scholar]
- 7.Yu CL, Tucker MA, Abramson DH, et al. Cause-specific mortality in long-term survivors of retinoblastoma. Journal of the National Cancer Institute. 2009;101(8):581–591. doi: 10.1093/jnci/djp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong FL, Boice JD, Jr, Abramson DH, et al. Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk JAMA: the journal of the American Medical Association. 1997;278(15):1262–1267. doi: 10.1001/jama.278.15.1262. [DOI] [PubMed] [Google Scholar]
- 9.Lauenstein TC, Goehde SC, Herborn CU, et al. Whole-body MR imaging: evaluation of patients for metastases. Radiology. 2004;233(1):139–148. doi: 10.1148/radiol.2331030777. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt GP, Reiser MF, Baur-Melnyk A. Whole-body imaging of the musculoskeletal system: the value of MR imaging. Skeletal radiology. 2007;36(12):1109–1119. doi: 10.1007/s00256-007-0323-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saslow D, Solomon D, Lawson HW, et al. American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. CA: a cancer journal for clinicians. 2012;62(3):147–172. doi: 10.3322/caac.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Villani A, Tabori U, Schiffman J, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study. The lancet oncology. 2011;12(6):559–567. doi: 10.1016/S1470-2045(11)70119-X. [DOI] [PubMed] [Google Scholar]
- 13.Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. The Journal of clinical endocrinology and metabolism. 2001;86(12):5658–5671. doi: 10.1210/jcem.86.12.8070. [DOI] [PubMed] [Google Scholar]
- 14.Field M, Shanley S, Kirk J. Inherited cancer susceptibility syndromes in paediatric practice. Journal of paediatrics and child health. 2007;43(4):219–229. doi: 10.1111/j.1440-1754.2007.01027.x. [DOI] [PubMed] [Google Scholar]
- 15.Bach PB, Mirkin JN, Oliver TK, et al. Benefits and harms of CT screening for lung cancer: a systematic review. JAMA: the journal of the American Medical Association. 2012;307(22):2418–2429. doi: 10.1001/jama.2012.5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abramson DH, Melson MR, Dunkel IJ, et al. Third (fourth and fifth) nonocular tumors in survivors of retinoblastoma. Ophthalmology. 2001;108(10):1868–1876. doi: 10.1016/s0161-6420(01)00713-8. [DOI] [PubMed] [Google Scholar]
- 17.Kleinerman RA, Tucker MA, Tarone RE, et al. Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2005;23(10):2272–2279. doi: 10.1200/JCO.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 18.Rodjan F, Graaf P, Brisse HJ, et al. Second cranio-facial malignancies in hereditary retinoblastoma survivors previously treated with radiation therapy: Clinic and radiologic characteristics and survival outcomes. European journal of cancer (Oxford, England: 1990) 2013;49(8):1939–1947. doi: 10.1016/j.ejca.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 19.Kleinerman RA. Radiation-sensitive genetically susceptible pediatric sub-populations. Pediatric radiology. 2009;39 (Suppl 1):S27–31. doi: 10.1007/s00247-008-1015-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gopie JP, Vasen HF, Tibben A. Surveillance for hereditary cancer: does the benefit outweigh the psychological burden?--A systematic review. Critical reviews in oncology/hematology. 2012;83(3):329–340. doi: 10.1016/j.critrevonc.2012.01.004. [DOI] [PubMed] [Google Scholar]