

Abstract
Wolf-Hirschhorn syndrome (WHS) represents an archetypical example of a contiguous gene deletion disorder – a condition comprising a complex set of developmental phenotypes with a multigenic origin. Epileptic seizures, intellectual disability, growth restriction, motor delay and hypotonia are major co-morbidities in WHS. Haploinsufficiency of LETM1, which encodes a mitochondrial inner-membrane protein functioning in ion transport, has been proposed as an underlying pathomechanism, principally for seizures but also for other core features of WHS, including growth and motor delay. Growing evidence derived from several model organisms suggests that reduced LETM1 expression is associated with some element of mitochondrial dysfunction. Surprisingly, LETM1-dependent mitochondrial functional deficits have not previously been described in cells from individuals with WHS. Here, using a unique panel of WHS-patient-derived cell lines with deletions of differing sizes, incorporating LETM1 or not, we show, for the first time, that LETM1 expression is reduced in mitochondria isolated from WHS-patient cells. Furthermore, we show that this is associated with distinct mitochondrial phenotypes, including altered intracellular [Ca2+] levels, dysfunctional mitochondrial transition-pore opening, hyperpolarization and superoxide leakage from resting mitochondria. Interestingly, we find that these phenotypes segregate with seizures in our WHS cohort. Our findings identify novel cellular phenotypes in WHS attributable to a 50% reduction in LETM1 expression level; these phenotypes could underlie and/or contribute to some of the core clinical features of this condition.
KEY WORDS: LETM1, Wolf-Hirschhorn syndrome, Mitochondria
INTRODUCTION
Wolf-Hirschhorn syndrome (WHS) is a contiguous gene deletion disorder caused by hemizygous deletion within chromosome 4p16.3 (Bergemann et al., 2005; Hirschhorn, 2008). The core clinical features of WHS consist of microcephaly, growth retardation, intellectual disability, atrial and ventricular septal defects, skeletal abnormalities, a characteristic facial dysmorphology, hypotonia, and epileptic seizures (Hirschhorn and Cooper, 1961; Hirschhorn et al., 1965; Wolf et al., 1965). The spectrum and severity of these clinical features typically correlate with deletion size (Battaglia et al., 2008; Maas et al., 2008; Van Buggenhout et al., 2004; Zollino et al., 2000). WHS is generally regarded as a multigenic disorder, although two critical regions have been described: WHSCR1 and WHSCR2, for WHS critical region 1 and 2, respectively (see Fig. 1). These critical regions are based on the demarcation of the minimum region of overlap in individuals exhibiting WHS-like phenotypes. WHSCR1 incorporates part of the WHS candidate gene WHSC1 and the entire WHSC2 gene (White et al., 1995; Wright et al., 1997). WHSCR2 incorporates LETM1 and part of WHSC1 but not WHSC2 (Rodríguez et al., 2005; Zollino et al., 2003) (Fig. 1). It is thought that haploinsufficiency of WHSC1 and WHSC2 accounts for many of the core phenotypes in WHS.
Fig. 1.
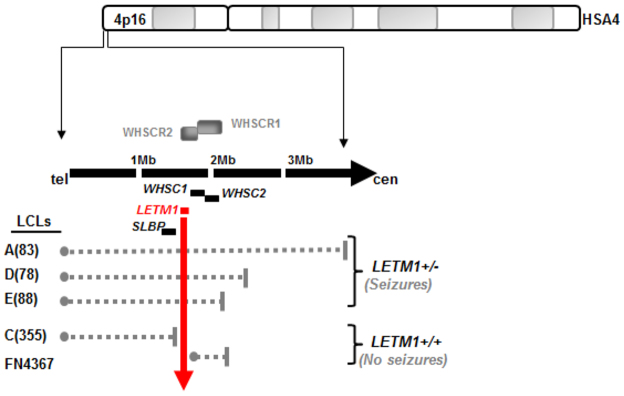
An ideogram of human somatic autosome (HSA) 4, focusing on 4p16.3 and showing the relative positions of the WHS critical regions WHSCR1 and WHSCR2. The relative position of certain key genes within and adjacent to these regions are also shown, with LETM1 highlighted in red. The various LCLs used in this study are listed on the bottom left-hand side adjacent to the dashed grey lines, which depict the position and extent of their specific deletions. The vertical red arrow is to denote which LCLs exhibit LETM1 haploinsufficiency and which do not. The LCLs A(83), D(78) and E(88) are from patients described in Maas et al. (Maas et al., 2008). Deletion sizes are: A(83), tel-3.85Mb; D(78), tel-2.42Mb; and E(88), tel-2.161Mb. The LCL C(355) has a terminal deletion up to and including SLBP and is clinically described in Engbers et al. (Engbers et al., 2009), whereas FN4367 is described in Rauch et al. (Rauch et al., 2001). tel, telomere; cen, centromere.
WHSC1, also known as NSD2 (nuclear receptor SET domain containing) and MMSET (multiple myeloma SET domain containing), encodes a putative histone methyltransferase with features of a transcriptional co-repressor (Kim et al., 2008; Marango et al., 2008). WHSC1 (NSD2/MMSET) controls histone H3-trimethylation on lysine-36 (H3-K36-me3), a modification associated with actively transcribed regions (Li et al., 2009; Nimura et al., 2009). WHSC2, also known as NELF-A, encodes a component of the negative elongation factor (NELF) complex (Narita et al., 2007). This complex induces promoter-proximal pausing by inhibiting RNA polymerase II progression early during elongation, thereby altering expression of its target genes in a positive and negative manner (Chiba et al., 2010; Gilchrist et al., 2008). Although haploinsufficiency of both WHSC1 and WHSC2 are thought to underlie many of the core clinical features of WHS, other genes additionally contribute to features such as facial dysmorphology, microcephaly and growth retardation, reinforcing the multigenic nature of this disorder (Engbers et al., 2009; Hammond et al., 2012; Hannes et al., 2012; South et al., 2007; South et al., 2008; Van Buggenhout et al., 2004). Untangling the complex functional relationships between the various genes that are haploinsufficient in WHS is essential to fully understand the underlying pathophysiology of this condition.
Over 90% of WHS patients with haploinsufficiency of the typical WHS critical regions exhibit severe generalised tonic-clonic epileptic seizures within the first 3 years of life (Battaglia et al., 2008; Battaglia et al., 2009). This represents a major clinical challenge in these individuals (Battaglia and Carey, 1999). Although generally improving with age, seizures can also be managed with valproate and/or phenobarbital (Battaglia et al., 2009). Haploinsufficiency of LETM1, which encodes the mitochondrial protein leucine-zipper EF-hand-containing transmembrane protein 1 (LETM1), is thought to contribute to seizure development in WHS (Dimmer et al., 2008; Endele et al., 1999; Hasegawa and van der Bliek, 2007; McQuibban et al., 2010; Nowikovsky et al., 2004; Tamai et al., 2008). This contribution is largely based on precisely defining the location and content of overlapping deletions in WHS and WHS-like patients with and without seizures. Nevertheless, there is additional evidence to suggest that other genes, aside from LETM1, might also play a role in some cases (Andersen et al., 2013; Misceo et al., 2012; South et al., 2007).
TRANSLATIONAL IMPACT.
Clinical issue
Wolf-Hirschhorn syndrome (WHS) is a genetic disorder that is caused by contiguous hemizygous deletions (deletions that affect only one copy of a chromosome pair) within chromosome 4p16.3. It is a complex disorder but its core clinical features consist of microcephaly, growth restriction, intellectual disability, cardiac and skeletal abnormalities, facial dysmorphology, hypotonia and epileptic seizures. The presence of only one functional copy of specific genes located in chromosome 4p16.3 (haploinsufficiency) has been implicated in the pathogenesis of this condition. To fully understand WHS, it is necessary to untangle the complex relationships between the size of the hemizygous deletion and the spectrum and severity of the condition’s clinical features. Haploinsufficiency of LETM1, which encodes a mitochondrial protein that is involved in ion transport, is thought to underlie the seizures and some other features of classical WHS, but how LETM1 haploinsufficiency contributes to seizure presentation or indeed whether individuals with WHS exhibit any LETM1-dependent phenotypes has not been fully elucidated.
Results
In this study, the authors use a unique panel of cell lines derived from individuals with WHS to show that LETM1 expression is reduced in cell extracts and mitochondria isolated from those with LETM1 haploinsufficiency. They catalogue several interdependent newly identified mitochondrial phenotypes that are associated with reduced LETM1 expression, including altered intracellular calcium levels, dysfunctional mitochondrial transition pore opening, hyperpolarisation and superoxide leakage. Using siRNA and cDNA transfection approaches, they show that these phenotypes are a direct consequence of reduced LETM1 expression. Finally, the authors report that the LETM1-dependent mitochondrial phenotypes segregate with seizures in the small cohort of patients from whom the cell lines were derived.
Implications and future directions
These findings provide the first direct evidence that cells with LETM1 haploinsufficiency obtained from individuals with WHS exhibit reduced LETM1 expression and that mitochondrial dysfunction is a consequence of the 50% reduction in LETM1 expression, thereby providing new pathomechanistic insights into WHS. Although studies in mouse models that have reduced Letm1 expression strongly suggest a link between reduced Letm1 expression and seizures, recent work in individuals with WHS suggests that LETM1 haploinsufficiency might also contribute to growth restriction, feeding difficulties, and motor and speech delays; the mitochondrial dysfunctions revealed here could conceivably contribute to these other characteristic features of WHS. Finally, these novel findings raise the possibility that mitochondrial-based therapeutic interventions could be of benefit in the management of WHS and should therefore be investigated.
LETM1 is a highly attractive contributing candidate for seizures in WHS principally because of its demonstrated role in various aspects of mitochondrial homeostasis (Dimmer et al., 2008; Endele et al., 1999; Hasegawa and van der Bliek, 2007; McQuibban et al., 2010; Nowikovsky et al., 2004; Tamai et al., 2008). Impaired mitochondrial function, abnormal ion-buffering and elevated mitochondrially derived reactive oxygen species (ROS) are all established interrelated pathomechanisms of seizures (Folbergrová and Kunz, 2012; Kang et al., 2013). Reduced expression of LETM1 and its orthologues have variously been identified as impacting upon mitochondrial morphology (increased swelling and elongation) and consequently upon function by controlling K+/H+ ion exchange, and also upon mitochondrial Ca2+ buffering by acting as a Ca2+/H+ antiporter (Jiang et al., 2009; Nowikovsky et al., 2012).
The precise role of LETM1 in mitochondrial ion transport is currently an area of intense debate (Nowikovsky et al., 2012; Nowikovsky et al., 2009). Nevertheless, recent studies modelling reduced Letm1 expression in mice and rats demonstrated a reduced threshold for chemically induced seizures (Jiang et al., 2013; Zhang et al., 2013). Furthermore, in the Letm1 rat study, the authors reported increased mitochondrial swelling and decreased mitochondrial cytochrome b levels (Zhang et al., 2013). In the mouse study, an associated impairment in mitochondrial Ca2+ buffering and consequent mitochondrial function were documented in Letm1+/− animals (Jiang et al., 2013).
Surprisingly, specific LETM1-dependent functional mitochondrial defects have not been demonstrated previously in WHS-patient-derived cell lines. In fact, in one study, although reduced LETM1 expression was observed in WHS-patient-derived fibroblasts, levels were found to be unaffected in the corresponding patient lymphoblastoid cell lines (LCLs) (Dimmer et al., 2008). Here, using a unique panel of WHS-patient-derived LCLs from individuals with deletions either incorporating LETM1 or not, we set out to identify and characterise LETM1-dependent mitochondrial phenotypes.
RESULTS
The patient LCLs used in this study and their respective deletions are shown in Fig. 1. All of these lines are derived from patients previously described in the literature (Engbers et al., 2009; Hannes et al., 2010; Maas et al., 2008; Rauch et al., 2001). Of note, FN4367 is an LCL derived from the patient described by Rauch and colleagues involving one of the smallest deletions catalogued in 4p16.3 in a patient exhibiting mild WHS phenotypes, but without seizures (Rauch et al., 2001). This is an interstitial 191.5 kb deletion encompassing WHSCR1, and not involving LETM1 (Rauch et al., 2001). LCL C(355) is from a WHS-like individual with a terminal deletion up to and including SLBP, thereby preserving LETM1 and the WHSCRs (Engbers et al., 2009; Hannes et al., 2010). This individual has not presented with seizures (Engbers et al., 2009).
WHS-patient LCLs haploinsufficient for LETM1 exhibit reduced LETM1 expression
Surprisingly, reduced LETM1 expression has not previously been consistently and unequivocally demonstrated in WHS-patient-derived cells despite LETM1 haploinsufficiency being proposed to contribute to seizure development in this condition (Dimmer et al., 2008; Hasegawa and van der Bliek, 2007). To conclusively address this issue, we interrogated LETM1 expression in our cohort of genomically characterised WHS and WHS-like patient LCLs. Using urea-denatured whole-cell extracts (WCEs) prepared from our panel of patient LCLs with and without deletions incorporating LETM1, we found that haploinsufficiency of LETM1 is in fact associated with an ~50% reduction in LETM1 expression (Fig. 2A,B). LCLs derived from patients A(83), D(78) and E(88) all showed reduced LETM1 expression associated with LETM1 haploinsufficiency. This is in contrast to extracts prepared from C(355) and FN4367, two lines with normal LETM1 copy number (Fig. 2A,B).
Fig. 2.

LETM1 expression in WHS-patient LCLs. (A) Urea-based whole cell extracts (WCEs) were titrated (2.5, 5, 10 μg) for each WHS-patient LCL and run alongside those from wild-type (WT) control extracts from a normal individual and blotted for LETM1 expression. Membranes were re-probed for β-tubulin to confirm loading. (B) LETM1 expression for each WHS cell line relative to WT were quantified (a.u., arbitrary units) using ImageJ software from scanned blots. Each quantitation represents the mean ± s.d. from at least three separate determinations. Measurements were also taken directly from membranes during enhanced chemiluminescence (ECL) development using the Image Quant LAS 4000 luminescent image analyser and analysed with Image Quant TL7.01 software, which yielded identical comparative changes as ImageJ processing. A(83), D(78) and E(88) exhibit reduced LEMT1 expression compared with WT, C(355) and FN4367 (P<0.05, Student’s t-test). (C) Urea-extracts were prepared from anti-TOM22 affinity-isolated mitochondria using the Miltenyi Biotech magnetic capture kit. Hexokinase expression, as a mitochondrial marker, is shown for each LCL. Different amounts of urea-mitochondrial extract was subjected to western blot analysis for LETM1 expression. (D) Relative LETM1 expression of each line compared with WT were quantified using ImageJ software from scanned blots as in B. Each quantitation represents the mean ± s.d. from at least three separate determinations. A(83), D(78) and E(88) exhibit reduced LEMT1 expression compared with WT and C(355) (P<0.05, Student’s t-test).
LETM1 protein resides in the inner mitochondrial membrane (IMM) (Endele et al., 1999; Tamai et al., 2008). Considering our results obtained using urea-denatured WCEs, we reasoned that WHS-patient LCLs haploinsufficient for LETM1 would also exhibit reduced LETM1 expression in mitochondria. Using an anti-TOM22 affinity-based magnetic mitochondrial isolation system (Miltenyi Biotech), we isolated intact mitochondria from each of our WHS-patient LCLs. Similar to our results obtained from WCEs, we found that mitochondrial extracts prepared from LCLs from patients with LETM1 haploinsufficiency exhibited reduced LETM1 compared with patient-derived LCLs with normal LETM1 copy number (Fig. 2C,D). Therefore, we conclude that LETM1 haploinsufficiency in the context of WHS is consistently associated with reduced expression of LETM1 in mitochondria of patient LCLs.
In our patient cohort, reduced mitochondrial LETM1 expression seems to segregate with seizure development. To try and understand whether we could link LETM1 expression levels with a specific mitochondrial deficit or a set of deficits, we investigated different aspects of mitochondrial function in these patient LCLs.
LETM1 haploinsufficiency in WHS does not affect mitochondrial mass but is associated with elevated intracellular [Ca2+]
Using MitoTracker-Green to determine mitochondrial mass, we found that all of the WHS-patient LCLs exhibited a similar mitochondrial content to each other, irrespective of LETM1 copy number, and to LCLs from a normal wild-type (WT) control (Fig. 3A). Therefore, haploinsufficiency of LETM1, although resulting in reduced mitochondrial LETM1 expression, does not seem to affect mitochondrial content within WHS-patient LCLs.
Fig. 3.

Mitochondrial mass and intracellular [Ca2+] in WHS-patient LCLs. (A) Mean relative fluorescence of MitoTracker-Green in WHS LCLs is shown relative to that of wild-type (WT) normal LCLs, the latter illustrated by the horizontal dashed line. Cells were treated with 250 nM MitoTracker-Green for 15 minutes and fluorescence quantified using the FACS Canto platform (FITC-A: area under the curve). All of the WHS LCLs, irrespective of LETM1 copy number, exhibit a comparable level of MitoTracker-Green fluorescence compared with WT. Data represents the mean ± s.d. from three separate experiments. (B) Calcium 1-AM profiles for the WHS LCLs. The profile for the wild-type (WT) normal LCLs is in blue. WHS LCLs with LETM1 haploinsufficiency are in green, whereas those with normal LETM1 copy number are depicted in red. (C) Mean relative Calcium 1-AM levels in WHS LCLs is shown relative to that of wild-type (WT) normal LCLs, the latter illustrated by the horizontal dashed line. Cells were treated with 10 μM Calcium 1-AM for 20 minutes and fluorescence analysed (excitation/emission 506/531) on the FACS Canto platform. Both A(83) and D(78) LCLs exhibit an elevated level of Calcium 1-AM relative to WT LCLs and the WHS LCLs C(355) and FN4367 (P<0.05, Student’s t-test). Both A(83) and D(78) exhibit LETM1 haploinsufficiency, whereas C(355) and FN4367 have normal LETM1 copy number. Data represents the mean ± s.d. from three separate experiments (FITC-A: area under the curve).
Mitochondria represent one of the key cellular Ca2+ buffering systems within the cell (Osellame et al., 2012; Szabadkai and Duchen, 2008). Because LETM1 has been described as a Ca2+/H+ antiporter, we investigated levels of intracellular Ca2+ using the fluorescent long-wave Ca2+ indicator, Calcium 1-AM, in the context of LETM1 haploinsufficiency (Jiang et al., 2009). Interestingly, we found elevated levels of intracellular [Ca2+] in WHS-patient LCLs with LETM1 haploinsufficiency [i.e. A(83), D(78) and E(88)], compared with those with normal LETM1 copy number [i.e. C(355) and FN4367], potentially suggestive of a problem in Ca2+ buffering in these cells (Fig. 3B,C). This phenotype has not previously been described in WHS-patient cells, but is consistent with reduced mitochondrial Ca2+ uptake recently demonstrated in cells from a Letm1+/− mouse model (Jiang et al., 2013).
LETM1 haploinsufficiency in WHS is associated with impaired mPTP dynamics
The mitochondrial permeability transition pore (mPTP) is a non-selective voltage-dependent mitochondrial channel proposed to reside in the IMM (Bernardi, 2013; Siemen and Ziemer, 2013). Opening of the mPTP increases the permeability of the IMM to solutes of >1.5 kDa in size. Under normal physiological conditions, this pore flickers between an open and closed state. However, various stimuli, such as elevated ROS levels and increased levels of mitochondrial calcium, promote the sustained opening of this pore. We investigated mPTP dynamics using the MitoProbe Transition Pore Assay Kit (Invitrogen-MitoProbes), which uses calcein fluorescence in the presence of CoCl2 as an indicator of the retention of calcein within mitochondria when the mPTP is in the closed state; CoCl2 quenches cytoplasmic calcein fluorescence, rendering the signal mitochondrial-specific. Firstly, we found that total calcein fluorescence load (i.e. in the absence of CoCl2) was comparable between all the patient-derived LCLs and WT cells (Fig. 4A). Interestingly, we found that WHS LCLs from patient A(83) and D(78) specifically exhibited elevated mitochondrial calcein fluorescence compared with WT LCLs and those from patient C(355) (Fig. 4B). This is suggestive of the mPTP favouring a predominantly closed state and/or insensitive ability to transition between open and closed conformations in resting mitochondria in A(83) and D(78) LCLs. These cell lines both exhibited LETM1 haploinsufficiency and decreased mitochondrial LETM1 levels compared with WT LCLs and those of patient C(355) (Figs 1, 2).
Fig. 4.

Mitochondrial transition pore (mPTP) dyamics and mitochondrial membrane polarization in WHS-patient LCLs. (A) Total cellular Calcein fluorescence is comparable between WT LCLs and WHS patient and WHS-like patient LCLs irrespective of LETM1 copy number. Calcein fluorescence was measured in the absence of CoCl2 and expressed as fold change relative to WT LCLs (a.u., arbitrary units). Data represents the mean ± s.d. from three separate experiments (FITC-A: area under the curve). (B) Mitochondrial calcein fluorescence levels (a.u., arbitrary units) indicate that the WHS LCLs A (83) and D(78) both exhibit elevated levels of mitochondrial calcein retention compared with WT LCLs and those of patient C(355). Here, calcein fluorescence is measured in the presence of CoCl2, which quenches the cytoplasmic (i.e. non-mitochondrially derived) signal. Data represent the mean ± s.d. from three separate experiments (FITC-A: area under the curve). (C) Mean relative level of mitochondrial calcein retention in WHS LCLs of differing LETM1 copy number compared with WT LCLs in the absence (black bar) or presence (white bar) of the Ca2+ ionophore, ionomycin. Both WT and C (355) exhibit a near 60% drop in calcein retention following ionomycin (500 nM) relative to CoCl2 treatment alone. This is in contrast to A(83) and D(78) LCLs, which only drop by about 20–30% under these conditions. This is suggestive of an insensitive mPTP favouring a closed conformation conducive to mitochondrial calcein retention. Data represents the mean ± s.d. from at least three separate experiments (FITC-A: area under the curve). (D) Mean relative fluorescence of MitoTracker-Red in WHS LCLs is shown relative to that of wild-type (WT) normal LCLs, the latter illustrated by the horizontal dashed line. Cells were treated with 250 nM MitoTracker-Red for 15 minutes and fluorescence quantified using the FACS Canto platform. WHS LCLs A(83), D(78) and E(88) exhibit elevated MitoTracker-Red fluorescence, suggestive of hyperpolarised resting mitochondria (elevated ΔΨmito), compared with WT LCLs and those from patients C(355) and FN4367 (P<0.05, Student’s t-test). Data represent the mean ± s.d. from three separate experiments (PE-A: area under the curve). Control experiments, using depolarising and hyperpolarising treatments, are shown in supplementary material Fig. S1A,B.
Treatment of cells with a Ca2+ ionophore such as ionomycin induces mitochondrial Ca2+ overload, typically resulting in mPTP opening and consequent reduction of calcein retention. Treatment of WT LCLs and LCLs from patient C(355) with ionomycin resulted in an ~60% decrease in mitochondrial calcein fluorescence, consistent with mPTP opening, as expected (Fig. 4C). In stark contrast, A (83) and D(78) patient-derived LCLs exhibited only a modest 25–30% relative reduction in mitochondrial calcein fluorescence under identical treatment conditions (Fig. 4C). These results are consistent with a [Ca2+]-insensitive mPTP adopting a predominantly closed conformation in WHS-patient LCLs with LETM1 haploinsufficiency, further indicative of altered mitochondrial-mediated [Ca2+] buffering in this context. This cellular phenotype has not previously been described in WHS-patient cells.
LETM1 haploinsufficiency in WHS is associated with mitochondrial membrane hyperpolarisation: elevated ΔΨmito
The mitochondrial membrane potential (ΔΨmito) is exquisitely sensitive to mitochondrial ion flux. In their identification of LETM1 as a mitochondrial Ca2+/H+ antiporter, Jiang and colleagues found that siRNA-mediated silencing of Letm1 in Drosophila S2 cells resulted in a modest although notable (~1.3-fold) increase in ΔΨmito (Jiang et al., 2009). Whether only a 50% reduction in LETM1 expression as a consequence of LETM1 haploinsufficiency could result in a similar effect upon ΔΨmito has not previously been investigated. Considering our findings regarding intracellular [Ca2+] and mPTP (Figs 3, 4), we examined ΔΨmito status in resting mitochondria in LCLs from our WHS cohort. Using MitoTracker Red-CMXRos fluorescence, we consistently found spontaneously elevated ΔΨmito (~1.4- to 1.6-fold) in WHS-patient LCLs with LETM1 haploinsufficiency [i.e. A(83), D(78), E(88)] compared with those with normal LETM1 expression [i.e. C(355), FN4367] and with WT LCLs (Fig. 4D). This suggests that LETM1 haploinsufficiency in the context of WHS is associated with spontaneous hyperpolarisation of resting mitochondria. This represents an additional novel mitochondrial phenotype associated with WHS and, in the context of this cohort, an additional phenotype segregating with seizure expression.
LETM1 haploinsufficiency in WHS is associated with elevated mitochondrial superoxide (O2−) production
Mitochondrial dysfunction is often associated with elevated superoxide (O2−) leakage (Kang and Pervaiz, 2012; Pieczenik and Neustadt, 2007). Using MitoTracker-SOX fluorescence as a direct measure of mitochondrial [O2−], we found a striking increase (~1.4-to 1.6-fold) in mitochondrial ROS in WHS-patient LCLs with LETM1 haploinsufficiency, specifically A(83), D(78) and E(88) patient LCLs, in contrast to WHS-patient cells with normal LETM1 copy number [i.e. C(355), FN4367] and to WT LCLs (Fig. 5A,B). In fact, the elevated level of mitochondrial O2− production in these LCLs was comparable to that of an LCL derived from a patient with myoclonus epilepsy with ragged-red fibres (MERRF) (Fig. 5B). MERFF is a primary mitochondrial disorder. Interestingly, elevated mitochondrially derived O2− was not associated with an overall elevated level of intracellular ROS in these cells, as determined by CellROX-Red fluorescence (Fig. 5C). These data further indicate that LETM1 haploinsufficiency in the context of WHS is associated with specific mitochondrial dysfunctions.
Fig. 5.

Mitochondrial [O2−] and total cellular ROS levels in WHS-patient LCLs. (A) Representative fluorescence profiles following treatment of WT and WHS-patient LCLs with MitoTracker-SOX (250 nM for 15 minutes) as obtained from the FACS Canto platform. Elevated levels of mitochondrial-derived ROS (O2−) are indicated by the black triangle. The WHS LCLs with LETM1 haploinsufficiency show marked peaks towards the right of each profile (elevated fluorescence) compared with WT LCLs and those from the WHS patients with normal LETM1 copy number [C(355) and FN4367]. A control experiment using thalidomide, a drug known to produce reactive oxygen species, is shown in supplementary material Fig. S1C. (B) Mean relative fluorescence of MitoTracker-SOX in WHS LCLs is shown relative to that of wild-type (WT) normal LCLs, the latter illustrated by the horizontal dashed line. WHS LCLs A(83), D(78) and E(88) exhibit elevated MitoTracker-SOX fluorescence indicative of elevated O2− production from resting mitochondria, compared with WT LCLs (dashed line) and those from patients C(355) and FN4367 (P<0.05, Student’s t-test). Furthermore, a MERRF (myoclonus epilepsy associate with ragged-red fibres) patient-derived LCL also exhibited a similarly significantly elevated MitoTracker-SOX fluorescence to that of the LETM1+/− WHS LCLs. Data represent the mean ± s.d. from three separate experiments (PE-A: area under the curve). (C) Mean relative fluorescence of CellROX-Red in WHS LCLs is shown relative to that of wild-type (WT) normal LCLs, the latter illustrated by the horizontal dashed line. CellROX-Red fluorescence is a measure of total cellular ROS. Pre-treatment of WT LCLs with the oxidant tert-Butyl hydroperoxide (tBOOH; 100 μM, 1 hour) resulted in an ~fivefold increase in CellROX-Red mean fluorescence relative to untreated WT LCLs. All of the WHS LCLs, irrespective of LETM1 copy number, exhibit a comparable level of CellROX-Red fluorescence compared with WT. Data represent the mean ± s.d. from three separate experiments (PE-A: area under the curve).
Our analysis of WHS-patient LCLs has shown that haploinsufficiency of LETM1 is associated with reduced LETM1 expression within isolated mitochondria and this segregates with elevated intracellular [Ca2+], an insensitive mPTP adopting a predominantly closed conformation, spontaneously elevated ΔΨmito indicative of hyperpolarized mitochondria, as well as elevated mitochondrial ROS production in resting mitochondria. These represent novel cellular phenotypes for WHS. Interestingly, these phenotypes also seem to segregate with the WHS patients within our small cohort who have presented with seizures. Mindful of the fact that other genes are also deleted in our WHS lines and that other regions within 4p16.3 might also contribute to seizure development in this condition, we set out to establish whether reduced expression of LETM1 alone could recapitulate some of the key mitochondrial phenotypes that we have identified here.
Reduced LETM1 is associated with elevated ΔΨmito and elevated mitochondrial O2−
Using the mouse neuroblastoma cell line Neuro2A as a model system, we investigated whether siRNA-mediated reduction of Letm1 alone could recapitulate some of the novel mitochondrial phenotypes we have described here in LETM1-haploinsufficient WHS-patient LCLs. We carefully optimised our siRNA procedure (by titration) so as to only obtain an ~50% reduction in Letm1 to closer mimic the WHS-patient LCL situation. As shown in Fig. 6A, siRNA-mediated reduction of Letm1 to ~50% of control-transfected (Ctrl) Neuro2A cells resulted in elevated ΔΨmito (MitoTracker-Red) and mitochondrial O2− production (MitoTracker-SOX), whilst not affecting mitochondrial mass (MitoTracker-Green) (Fig. 6B,C). These phenotypes are identical to those identified here for WHS-patient LCLs haploinsufficient for LETM1 (Fig. 3A; Fig. 4C; Fig. 5A,B).
Fig. 6.

Knockdown of LETM1 recapitulates mitochondrial phenotypes of WHS-patient LCLs with haploinsufficiency of LETM1. (A) Western blot analysis of Letm1 expression following siRNA-mediated silencing of Letm1 in mouse Neuro2A cells. An ~50% reduction in Letm1 was observed 48 hours post-transfection. Ctrl, control-transfected. The membrane was re-probed for α-tubulin to confirm loading. (B) Representative fluorescence profiles following siRNA-mediated reduction of Letm1 to 50% in Neuro2A cells and incubation with MitoTracker-Red (upper panel) and MitoTracker-SOX (lower panel). The siRNA-treated profile for each MitoTracker is shown in red. Ctrl, control-transfected. Elevated fluorescence for each MitoTracker is indicated by a rightward shift in the profiles following siRNA of Letm1 relative to Ctrl. (C) Mean relative fluorescence of MitoTracker-Red, MitoTracker-Green and MitoTracker-SOX levels following siRNA in Neuro2A cells is shown relative to that of control-transfected (Ctrl) cells, the latter illustrated by the horizontal dashed line. The siRNA-mediated reduction of Letm1 to 50% results in elevated MitoTracker-Red and MitoTracker-SOX fluorescence specifically, not impacting upon MitoTracker-Green levels. Therefore, siRNA of Letm1 is associated with increased hyperpolarisation (elevated ΔΨmito) and elevated O2− production from resting mitochondria under these conditions, without impacting upon mitochondrial mass. Data represent the mean ± s.d. from three separate experiments (MitoT-Red and MitoT-Green; PE-A: area under the curve; MitoT-Green: FITC-A: area under the curve). (D) Western blot analysis for LETM1 from human T98G glioblastoma cells following either siRNA of LETM1 using a 3′UTR-directed oligonucleotide or transfection with a plasmid containing LETM1, or a combination of both as indicated. The membrane was re-probed for MCM2 to confirm loading. (E) Mean relative fluorescence of MitoTracker-SOX in T98G cells is shown relative to that of control-transfected cells (Ctrl), following silencing of LETM1 (siRNA), along with co-transfection with an siRNA-resistant LETM1 cDNA (siRNA + cDNA). Silencing of LETM1 using a 3′UTR-directed oligonucleotide directed against the endogenous gene results in a marked increase in MitoTracker-SOX levels indicative of elevated mitochondrial ROS production (Ctrl versus siRNA P<0.05, Student’s t-test). Co-transfection with an siRNA-resistant LETM1-encoding plasmid reduces the level of MitoTracker-SOX fluorescence to that of control-transfected cells (Ctrl). Data represent the mean ± s.d. from three separate experiments (PE-A: area under the curve).
To further reinforce the fact that the elevated levels of mitochondrial O2− observed under these conditions were directly attributable to reduced LETM1 levels, we performed a similar analysis, although in this case using the human T98G glioblastoma line, with the additional element of complementation with an siRNA-resistant cDNA encoding LETM1. The siRNA oligonucleotide was designed to the 3′UTR of LETM1 and, as expected, resulted in a significant reduction in LETM1, which was also associated with elevated mitochondrial O2− (MitoTracker-SOX) (Fig. 6D,E). In fact, under these conditions we observed an over threefold increase in mitochondrial O2− production (Fig. 6E).Importantly, co-transfection of the 3′UTR-directed oligo with the siRNA-resistant cDNA was associated with the consequent reduction in mitochondrial ROS (Fig. 6E). These data show that modestly reduced expression of LETM1 alone can cause mitochondrial dysfunction. Unfortunately, we were unable to generate a viable and stable WHS-patient LCL stably transduced with LETM1 from a lentivirus (CMV-driven pReceiver-Lv105 expression clone LP-W0230-Lv105-0200-S, Genecopoea).
In summary, using a panel of WHS and WHS-like patient LCLs with differing sized deletions incorporating LETM1 or not, we show that haploinsufficiency of LETM1 is associated with reduced LETM1 expression and a series of mitochondrial phenotypes. These include elevated intracellular [Ca2+], an insensitive mPTP and hyperpolarized mitochondria (elevated ΔΨmito) with elevated mitochondrial O2− production. Many of these mitochondrial deficits have been previously associated with seizures. In our patient cohort, these mitochondrial phenotypes segregate with seizure presentation. Our data strongly suggest that a 50% reduction in LETM1 expression as a consequence of LETM1 haploinsufficiency in WHS can result in overt mitochondrial phenotypes.
DISCUSSION
WHS is generally regarded as a multigenic condition with haploinsufficiency of the critical regions thought to explain many of the core clinical features, although other genes clearly have additional contributions (Engbers et al., 2009; South et al., 2007; South et al., 2008; Van Buggenhout et al., 2004). For example, we recently identified delayed S-phase progression and impaired chromatin remodelling in WHS-patient LCLs attributable to haploinsufficiency of SLBP and/or WHSC2 (NELF-A); phenotypes with implications for the maintenance of epigenetic memory, expansion of stem cell niches, and possibly microcephaly and growth retardation (Kerzendorfer et al., 2012). Assessing the contribution of haploinsufficiency of a single gene towards a specific phenotype or set of phenotypes characteristic of a contiguous gene deletion disorder can be complex. This needs to be considered in the context of variable expressivity, incomplete penetrance or the revelation of recessive alleles and/or complex positional effects (reviewed in Hart and O’Driscoll, 2013). Single-gene contributions are usually based on the existence of patients with rare atypical and/or very small deletions being placed in context of the more commonly sized deletions. Additionally, in some cases murine models might be available, enabling interpretation of the pathomechanistic impact of impaired function of a particular gene. For WHS, several mouse models for genes within human somatic autosome (HSA) 4p16.3 have been used as supportive evidence for the haploinsufficiency of certain genes in underlying specific clinical features of the condition. Examples include Whsc1 for cardiac and midline abnormalities, Fgfr3 and Ctbp1 for skeletal abnormalities, and Fgfr3, Ctbp1, Tacc3 and Hspx153 for growth retardation (Abrams and Jiao, 2009; Näf et al., 2001; Nimura et al., 2009; Simon and Bergemann, 2008).
When considering seizures in WHS, the situation is highly complex. The segregation of seizures with LETM1 haploinsufficiency does not seem to be absolute (Andersen et al., 2013; South et al., 2008). There are a modest although growing number of cases with sub-telomeric deletions not involving LETM1 that have been reported with seizures, whereas, conversely, there are also deletion cases incorporating LETM1 that are reported to be seizure free (Andersen et al., 2013; Bayindir et al., 2013; Engbers et al., 2009; Faravelli et al., 2007; Izumi et al., 2010; Misceo et al., 2012; South et al., 2008; Van Buggenhout et al., 2004). In fact, whether LETM1 haploinsufficiency plays any role whatsoever in seizure development has been questioned by some (Bayindir et al., 2013; Luo et al., 2011). Importantly, to our knowledge, neither LETM1 expression nor functional mitochondrial characterisation has been investigated in these situations (Bayindir et al., 2013; Luo et al., 2011). Furthermore, CTBP1 and/or CPLX1 haploinsufficiency, two genes telomeric to LETM1, have also been proposed to possibly underlie seizures in WHS, although, again, without any supportive, associative functional cellular characterization in WHS-patient cell lines (Misceo et al., 2012; Simon and Bergemann, 2008).
Attributing reduced LETM1 expression as the cause of or contributing factor to seizures in WHS has evolved from a combination of (i) cataloguing patients with different sized deletions and variable seizure expression, (ii) realising LETM1’s mitochondrial localisation and its role in ion transport, and (iii) the overt mitochondrial morphological changes observed in Saccharomyces cerevisiae, Drosophila, Caenorhabditis elegans and even HeLa cells, when LETM1 levels were reduced (Dimmer et al., 2008; Endele et al., 1999; Hasegawa and van der Bliek, 2007; McQuibban et al., 2010; Nowikovsky et al., 2004; Tamai et al., 2008). Furthermore, (iv) downregulation of CG4589, the LETM1 orthologue in Drosophila, also leads to reduced synaptic neurotransmitter release (McQuibban et al., 2010).
Recently, two interesting murine-based models of reduced Letm1 expression have further strengthened the pathomechanistic link between LETM1-induced mitochondrial dysfunction and seizures. Zhang and colleagues showed that stereotaxic intra-hippocampal injection of a Letm1-targeting shRNA lentivirus induced mitochondrial swelling and decreased mitochondrial cytochrome b expression in the brains of rats (Zhang et al., 2013). Furthermore, this was associated with reduced onset latency, and increased frequency and duration of pilocarpine-induced epilepsy (Zhang et al., 2013). These authors also reported lower LETM1 expression in the temporal neocortex of individuals with temporal lobe epilepsy compared with normal individuals (Zhang et al., 2013). Jiang and colleagues reported a gene-trap-based targeting of Letm1 in mice, which resulted in early embryonic lethality for the homozygous deletion and 50% loss of the heterozygotes before E13.5 (Jiang et al., 2013). The surviving Letm1 heterozygotes exhibited impaired brain-specific glucose metabolism and reduced ATP levels, as well as increased kainic-acid-induced seizure activity (Jiang et al., 2013).
Although the precise role of LETM1 in mitochondrial ion transport is still debated (i.e. its function as a Ca2+ antiporter versus K+/H+ exchange), there is growing evidence to suggest that reduced LETM1 expression can impact on different aspects of mitochondrial function (Nowikovsky et al., 2012; Nowikovsky et al., 2009). Altered mitochondrial function is strongly associated with seizure development (Folbergrová and Kunz, 2012; Kang et al., 2013). Indeed, several established mitochondrial disorders such as Kearns-Sayre syndrome and MERRF are strongly seizure-prone (Koopman et al., 2012). When considering LETM1 haploinsufficiency, the consequent chronic mitochondrial hyperpolarisation in the context of elevated mitochondrial O2− production and mPTP dysfunction could conceivably have catastrophic implications for normal neuronal function. Our work describes for the first time a set of mitochondrial phenotypes in WHS-patient-derived LCLs that also segregate with seizure development in the small cohort examined. This is notable because LCL C(355), which does not present with any of the aberrant mitochondrial phenotypes identified here, has a deletion that incorporates CTBP1 and CPLX1, yet this individual is seizure free (Hannes et al., 2010). Furthermore, we can model some of the mitochondrial phenotypes identified in the LCLs by manipulating LETM1 expression in cell lines of neuronal (Neuro2A) and glial (T98G) origin (Fig. 6). It would be fascinating to investigate the mitochondrial phenotypes we have identified here in cells from patients presenting with seizures in the context of normal LETM1 copy number. There is now growing evidence for a multigenic basis for seizures in individuals with deletions within 4p16.3 (Andersen et al., 2013; Misceo et al., 2012; South et al., 2007). This multigenic phenomenon is not without precedent in WHS. For example, we have recently characterised a multigenic basis for DNA replication and chromatin formation impairments in WHS (Kerzendorfer et al., 2013; Kerzendorfer et al., 2012).
The mitochondrial phenotypes we have described substantially build upon those already attributed to reduced LETM1 expression (Jiang et al., 2009; Jiang et al., 2013; Nowikovsky et al., 2012). The phenotypes described in our study are highly interdependent and likely a consequence of LETM1’s fundamental role in mitochondrial ion transport. We found elevated intracellular [Ca2+] and a [Ca2+]-insensitive mPTP in WHS LCLs with LETM1 haploinsufficiency. Each of these could underlie or contribute to the elevated ΔΨmito and elevated mitochondrial ROS we also observed in this context (Brookes et al., 2004). Importantly, we have described these findings in the clinically relevant setting of patient-derived cells.
Our work suggests that WHS-patient LCLs could serve as a tractable model platform to investigate potential therapeutically relevant routes into mitigating against the phenotypes that we have catalogued, phenotypes that are likely relevant to seizures or additional clinical features (Liu and Schubert, 2009). For example, in some instances treatment with nigericin, an ionophore that catalyses electroneutral K+/H+ exchange in mitochondria, has been reported to reverse LETM1-dependent mitochondrial morphological changes (Dimmer et al., 2008; Nowikovsky et al., 2004; Nowikovsky et al., 2009). Nevertheless, we found that treatment of WHS LCLs with nigericin (2 μM) for up to 24 hours had no impact on mitochondrial O2− levels (not shown).
We believe that our data provide supportive associative evidence for a contribution of reduced LETM1 expression and consequent mitochondrial dysfunction to the clinical presentation of WHS. Wider implications of LETM1-dependent mitochondrial dysfunction outside of seizures could include, for example, impacts upon optimal motor and muscle function with relevance to hypotonia in WHS. Furthermore, considering the high energy demands of the brain, LETM1-dependent mitochondrial dysfunction could impact upon other aspects of optimal neuronal function that might be relevant to intellectual disability herein. Collectively, our findings underscore the dramatic and newly identified impacts upon mitochondrial function that a modest 50% reduction in LETM1 expression has in WHS-patient-derived LCLs.
MATERIALS AND METHODS
Cell culture
Lymphoblastoid cell lines (LCLs) were cultured at 37°C in humidified incubators with 5% CO2 in RPMI 1640 supplemented with 2 mM L-glutamine, 500 U/ml penicillin, 50 μg/ml streptomycin and 15% fetal calf serum. The MERFF (myoclonus epilepsy associated with ragged-red fibres) LCL was obtained from the Coriell Cell Repository (Camden, NJ, USA). The line, GM11907, has a mutant tRNA-Lys, A>G transition at mtDNA nucleotide pair 8344/WT. Neuro-2A (N2A) cells were maintained in Dulbecco’s modified Eagle medium supplemented with 2 mM L-glutamine, 500 U/ml penicillin, 50 μg/ml streptomycin and 10% fetal bovine serum. T98G human glioblastoma cells were cultured in minimum essential medium supplemented with 10% fetal bovine serum, 1% non-essential amino acid (NEAA) and 1% sodium pyruvate, 2 mM L-glutamine, 500 U/ml penicillin and 50 μg/ml streptomycin.
Antibodies
Anti-α-tubulin (T1568) was obtained from Sigma-Aldrich (Poole, Dorset, UK). Anti-hexokinase 1 (2024, C25C4) was obtained from Cell Signaling Technology (NEB, Hitchin, Hertfordshire, UK). Anti-β-tubulin (sc-9104), anti-MCM2 (sc-9839) and anti-LETM1 (sc-271232) were from Santa Cruz (Insight Biotec Ltd, Wembley, Middlesex, UK).
Extract preparation
Urea-based whole cell extracts (WCEs)
Cell pellets were washed in PBS then lysed in 50–100 μl of urea-based lysis buffer (9 M urea, 50 mM Tris-HCl at pH 7.5 and 10 mM 2-β-mercaptoethanol), followed by a 12-second sonication at 30% amplitude. Protein concentration was determined using the Bradford Assay. Samples were then stored at −20°C or immediately boiled in 2× SDS-loading buffer (5% SDS, 10% glycerol, 10% 2-β-mercaptoethanol, 125 mM Tris-HCl, pH 6.8 and 0.2% bromophenol blue) and loaded onto SDS-PAGE gels.
Mitochondrial isolation and extracts
Mitochondrial isolation was performed using the Miltenyi Biotec (Bisley, Surrey, UK) Mitochondrial Isolation Kit (Cat. no.: 130-094-532) according to the manufacturer’s instructions with slight modification as indicated. 107 cells were lysed using the lysis buffer provided and homogenised using a glass mini-strokes homogenizer, with 10–15 strokes per sample, on ice. Lysate was incubated with anti-TOM22 magnetic microbeads for 1 hour at 4°C with gentle shaking. An LS column was placed in the magnetic field of a QuadroMACS separation unit and the lysate was then applied to the LS column. Once the lysate had run through, the column was washed 3× with the supplied separation buffer before being removed from the magnetic field and placed onto a 1 ml collection tube. The magnetically retained mitochondria were eluted in 1 ml of the supplied separation buffer and subsequently centrifuged at 13,000 g for 2 minutes to pellet mitochondria. The mitochondrial pellet was resuspended in 60 μl of urea lysis buffer and sonicated for 15 seconds at 30% amplitude. 0.1–2 μl (for LETM1) or 8 μl (for hexokinase 1) of mitochondrial extract was then immediately boiled in 2× SDS-loading buffer (5% SDS, 10% glycerol, 10% 2-β-mercaptoethanol, 125 mM Tris-HCl, pH 6.8 and 0.2% bromophenol blue) and loaded onto SDS-PAGE gels.
Mitochondrial transition pore (mPTP) analysis
The mPTP kit (Life Technologies, Paisley, UK, Cat no.: M34153) was used according to the manufacturer’s instructions. LCLs were re-suspended in pre-warmed Hanks Balanced Salt Solution with Ca2+ at a final concentration of 1×106 cells/ml. 3×1 ml aliquots were prepared per cell line (tube 1,2,3). 10 nM calcein AM was added to each tube, 400 μM CoCl2 was added to tubes 2 and 3 and 500 nM ionomycin was added to tube 3. Samples were incubated for 15 minutes at 37°C, protected from light. Cells were pelleted by centrifugation, re-suspended in 500 μl PBS and filtered into FACS Falcon tubes (Becton Dickinson, Oxford, UK) for flow cytometry analysis using a Becton Dickinson (BD) FACS Canto. Samples were analysed using 488 nm excitation and emission filters appropriate for fluorescein with BD FACS Diva software. A sample without added reagents was used for instrument set up.
MitoTracker probes
LCLs were pelleted by centrifugation and resuspended in pre-warmed growth medium containing 250 nM MitoTracker-Red (M7512) or MitoTracker-Green (M7514) or MitoTracker-SOX (M36008), all of which were obtained from Molecular Probes, Life Technologies (Invitrogen, Paisley, UK). Cells were incubated for 15 minutes under growth conditions, protected from light. After treatment, LCLs were washed once in 1× PBS. Cells were then re-suspended in 500 μl PBS and filtered into FACS Falcon tubes for immediate flow cytometry analysis. Adherent cells (i.e. T98G and Neuro2A) were incubated with 250 nM MitoTracker probe for 15 minutes and washed once in 1× PBS. Cells were then trypsinised or detached using a cell scraper and filtered into FACS Falcon tubes for flow cytometry analysis. All data was collected using a BD FACS Canto Flow Cytometer (Oxford, UK) and analysed with BD FACS Diva software.
Total intracellular ROS determination
Cells were treated with CellROX-Red (Life Technologies, Paisley, UK, Cat no.: C10422) at a final concentration of 5 μM and incubated for 30 minutes at 37°C. The medium was then removed and cells were washed three times with PBS. Cells were then filtered into BD FACS Falcon tubes and analysed by flow cytometry using 640/665 nm excitation/emission filters. Data was collected using a BD FACS Canto flow cytometer (Oxford, UK) and analysed with the BD FACS Diva software.
Intracellular calcium determination
LCLs were incubated with 10 μM Calcium 1-AM probe (Life Technologies, Paisley, UK, Cat. no.: C3012) for 20 minutes under normal growth conditions. Cells were washed then re-suspended in 500 μl PBS and filtered into BD FACS Falcon tubes (Oxford, UK). Data was collected using a BD FACS Canto and subsequent analysis was performed using the BD FACS Diva software with 506/531 excitation/emission filters.
siRNA knockdowns
Mouse Letm1 (Neuro2A cell line)
ON-TARGETplus SMARTpool siRNAs against Letm1 (L-049478-01-005, mouse) were obtained from Thermo Fisher Scientific (Loughborough, UK) and re-suspended in DPEC-treated water to a final concentration of 5 nmol. Transfections were performed using 1×5 μl siRNA against Letm1 in the presence of 5 μl MetafectenePro (Cambio, Cambridge, UK). Cells were harvested 48 hours post-transfection. Sense target sequences are: (1) 5′-AGGUAGACAACAAGGCGAA-3′; (2) 5′-CCAACAACUUCCUGC -GUUU-3′; (3) 5′-CUAAAUAGUCGGGUGACAUA-3′; (4) 5′-CUGC -CUAAUUCAUGAGUAA-3′.
Human LETM1 (T98G cell line)
Stealth siRNAs (Life Technologies, Paisley, UK) were designed against the 3′UTR region of human LETM1 using the BLOCK-IT™ RNAi designer (Invitrogen Life Technologies, Paisley, UK). Oligos were re-suspended in 1 ml of DPEC-treated water to a final concentration of 20 nmol. Transfections were performed with 1× 5 μl siRNA against LETM1 in the presence of 5 μl MetafectenePro (Cambio, Cambridge, UK). Cells were harvested 24 hours post-transfection. LETM1 3′UTR target sequence is: 5′-CCACAG -AAUCGUGUCUGGAUCCACA-3′.
LETM1 overexpression
T98G human glioblastoma cells were transfected with 2 μg plasmid encoding LETM1 (pCMV6-XL4) purchased from Origene, in the presence of 5 μl MetafectenePro (Biontex) under normal growth conditions. Cells were harvested 24 hours post-transfection and prepared for flow cytometry or western blot analysis as described above.
LETM1 complementation
T98G cells were transfected with 5 μl siRNA against LETM1 in the presence of 5 μl MetafectenePro (Biontex) + 2 μg of LETM1-expression vector. The vector, pCMV6-XL4 containing human LETM1 (NM_012318), was obtained from Origene (Insight Biotec Ltd, Wembley, Middlesex, UK, Cat. no.: sc115448). At 24 hours post-transfection, cells were incubated with 250 nM MitoSOX for 15 minutes and processed as described above.
Supplementary Material
Acknowledgments
Special thanks to Ron Hochstenbach and Martin Poot of the University Medical Center, Utrecht, The Netherlands for LCL C(355).
Footnotes
Funding
The M.O’D. laboratory is programme funded by Cancer Research UK with additional support from the UK Medical Research Council (MRC) and Leukaemia Lymphoma Research. L.H. is an MRC Centenary Award (2013) recipient.
Competing interests
The authors declare no competing financial interests.
Author contributions
L.H. carried out the experimental work. A.R. and J.R.V. contributed essential characterised patient-derived cell lines. A.M.C., A.R. and J.R.V. provided critical evaluation of the draft manuscript. M.O’D. designed the study, supervised the experimental work, interpreted the data, drafted the figures with L.H. and wrote the paper.
Supplementary material
Supplementary material available online at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.014464/-/DC1
References
- Abrams J. M., Jiao Y. (2009). Keeping it simple: what mouse models of Wolf-Hirschhorn syndrome can tell us about large chromosomal deletions. Dis. Model. Mech. 2, 315–316 [DOI] [PubMed] [Google Scholar]
- Andersen E. F., Carey J. C., Earl D. L., Corzo D., Suttie M., Hammond P., South S. T. (2013). Deletions involving genes WHSC1 and LETM1 may be necessary, but are not sufficient to cause Wolf-Hirschhorn syndrome. Eur. J. Hum. Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia A., Carey J. C. (1999). Health supervision and anticipatory guidance of individuals with Wolf-Hirschhorn syndrome. Am. J. Med. Genet. 89, 111–115 [DOI] [PubMed] [Google Scholar]
- Battaglia A., Filippi T., Carey J. C. (2008). Update on the clinical features and natural history of Wolf-Hirschhorn (4p-) syndrome: experience with 87 patients and recommendations for routine health supervision. Am. J. Med. Genet. C. Semin. Med. Genet. 148C, 246–251 [DOI] [PubMed] [Google Scholar]
- Battaglia A., Filippi T., South S. T., Carey J. C. (2009). Spectrum of epilepsy and electroencephalogram patterns in Wolf-Hirschhorn syndrome: experience with 87 patients. Dev. Med. Child Neurol. 51, 373–380 [DOI] [PubMed] [Google Scholar]
- Bayindir B., Piazza E., Della Mina E., Limongelli I., Brustia F., Ciccone R., Veggiotti P., Zuffardi O., Dehghani M. R. (2013). Dravet phenotype in a subject with a der(4)t(4;8)(p16.3;p23.3) without the involvement of the LETM1 gene. Eur. J. Med. Genet. 56, 551–555 [DOI] [PubMed] [Google Scholar]
- Bergemann A. D., Cole F., Hirschhorn K. (2005). The etiology of Wolf-Hirschhorn syndrome. Trends Genet. 21, 188–195 [DOI] [PubMed] [Google Scholar]
- Bernardi P. (2013). The mitochondrial permeability transition pore: A mystery solved? Frontiers in Physiology 4, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes P. S., Yoon Y., Robotham J. L., Anders M. W., Sheu S.-S. (2004). Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. 287, C817–C833 [DOI] [PubMed] [Google Scholar]
- Chiba K., Yamamoto J., Yamaguchi Y., Handa H. (2010). Promoter-proximal pausing and its release: molecular mechanisms and physiological functions. Exp. Cell Res. 316, 2723–2730 [DOI] [PubMed] [Google Scholar]
- Dimmer K. S., Navoni F., Casarin A., Trevisson E., Endele S., Winterpacht A., Salviati L., Scorrano L. (2008). LETM1, deleted in Wolf-Hirschhorn syndrome is required for normal mitochondrial morphology and cellular viability. Hum. Mol. Genet. 17, 201–214 [DOI] [PubMed] [Google Scholar]
- Endele S., Fuhry M., Pak S.-J., Zabel B. U., Winterpacht A. (1999). LETM1, a novel gene encoding a putative EF-hand Ca(2+)-binding protein, flanks the Wolf-Hirschhorn syndrome (WHS) critical region and is deleted in most WHS patients. Genomics 60, 218–225 [DOI] [PubMed] [Google Scholar]
- Engbers H., van der Smagt J. J., van ’t Slot R., Vermeesch J. R., Hochstenbach R., Poot M. (2009). Wolf-Hirschhorn syndrome facial dysmorphic features in a patient with a terminal 4p16.3 deletion telomeric to the WHSCR and WHSCR 2 regions. Eur. J. Hum. Genet. 17, 129–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faravelli F., Murdolo M., Marangi G., Bricarelli F. D., Di Rocco M., Zollino M. (2007). Mother to son amplification of a small subtelomeric deletion: a new mechanism of familial recurrence in microdeletion syndromes. Am. J. Med. Genet. A. 143A, 1169–1173 [DOI] [PubMed] [Google Scholar]
- Folbergrová J., Kunz W. S. (2012). Mitochondrial dysfunction in epilepsy. Mitochondrion 12, 35–40 [DOI] [PubMed] [Google Scholar]
- Gilchrist D. A., Nechaev S., Lee C., Ghosh S. K. B., Collins J. B., Li L., Gilmour D. S., Adelman K. (2008). NELF-mediated stalling of Pol II can enhance gene expression by blocking promoter-proximal nucleosome assembly. Genes Dev. 22, 1921–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond P., Hannes F., Suttie M., Devriendt K., Vermeesch J. R., Faravelli F., Forzano F., Parekh S., Williams S., McMullan D., et al. (2012). Fine-grained facial phenotype-genotype analysis in Wolf-Hirschhorn syndrome. Eur. J. Hum. Genet. 20, 33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannes F., Hammond P., Quarrell O., Fryns J.-P., Devriendt K., Vermeesch J. R. (2012). A microdeletion proximal of the critical deletion region is associated with mild Wolf-Hirschhorn syndrome. Am. J. Med. Genet. A. 158A, 996–1004 [DOI] [PubMed] [Google Scholar]
- Hannes F., Van Houdt J., Quarrell O. W., Poot M., Hochstenbach R., Fryns J.-P., Vermeesch J. R. (2010). Telomere healing following DNA polymerase arrest-induced breakages is likely the main mechanism generating chromosome 4p terminal deletions. Hum. Mutat. 31, 1343–1351 [DOI] [PubMed] [Google Scholar]
- Hart L., O’Driscoll M. (2013). Causes and consequences of structural genomic alterations in the human genome. eLS 2013, a0024976 [Google Scholar]
- Hasegawa A., van der Bliek A. M. (2007). Inverse correlation between expression of the Wolfs Hirschhorn candidate gene Letm1 and mitochondrial volume in C. elegans and in mammalian cells. Hum. Mol. Genet. 16, 2061–2071 [DOI] [PubMed] [Google Scholar]
- Hirschhorn K. (2008). A short history of the initial discovery of the Wolf-Hirschhorn syndrome. Am. J. Med. Genet. 148C, 244–245 [DOI] [PubMed] [Google Scholar]
- Hirschhorn K., Cooper H. L. (1961). Apparent deletion of short arms of one chromosome (4 or 5) in a child with defects of midline fusion. Mammalian Chrom. Nwsl. 4, 14 [Google Scholar]
- Hirschhorn K., Cooper H. L., Firschein I. L. (1965). Deletion of short arms of chromosome 4-5 in a child with defects of midline fusion. Humangenetik 1, 479–482 [DOI] [PubMed] [Google Scholar]
- Izumi K., Okuno H., Maeyama K., Sato S., Yamamoto T., Torii C., Kosaki R., Takahashi T., Kosaki K. (2010). Interstitial microdeletion of 4p16.3: contribution of WHSC1 haploinsufficiency to the pathogenesis of developmental delay in Wolf-Hirschhorn syndrome. Am. J. Med. Genet. 152A, 1028–1032 [DOI] [PubMed] [Google Scholar]
- Jiang D., Zhao L., Clapham D. E. (2009). Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 326, 144–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D., Zhao L., Clish C. B., Clapham D. E. (2013). Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc. Natl. Acad. Sci. USA 110, E2249–E2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H.-C., Lee Y.-M., Kim H. D. (2013). Mitochondrial disease and epilepsy. Brain and Development 35, 757–761 [DOI] [PubMed] [Google Scholar]
- Kang J., Pervaiz S. (2012). Mitochondria: redox metabolism and dysfunction. Biochem. Res. Int. 2012, 896751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerzendorfer C., Colnaghi R., Abramowicz I., Carpenter G., O’Driscoll M. (2013). Meier-Gorlin syndrome and Wolf-Hirschhorn syndrome: two developmental disorders highlighting the importance of efficient DNA replication for normal development and neurogenesis. DNA Repair (Amst.) 12, 637–644 [DOI] [PubMed] [Google Scholar]
- Kerzendorfer C., Hannes F., Colnaghi R., Abramowicz I., Carpenter G., Vermeesch J. R., O’Driscoll M. (2012). Characterizing the functional consequences of haploinsufficiency of NELF-A (WHSC2) and SLBP identifies novel cellular phenotypes in Wolf-Hirschhorn syndrome. Hum. Mol. Genet. 21, 2181–2193 [DOI] [PubMed] [Google Scholar]
- Kim J.-Y., Kee H. J., Choe N.-W., Kim S.-M., Eom G.-H., Baek H. J., Kook H., Kook H., Seo S.-B. (2008). Multiple-myeloma-related WHSC1/MMSET isoform RE-IIBP is a histone methyltransferase with transcriptional repression activity. Mol. Cell. Biol. 28, 2023–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman W. J. H., Willems P. H. G. M., Smeitink J. A. M. (2012). Monogenic mitochondrial disorders. N. Engl. J. Med. 366, 1132–1141 [DOI] [PubMed] [Google Scholar]
- Li Y., Trojer P., Xu C.-F., Cheung P., Kuo A., Drury W. J., 3rd, Qiao Q., Neubert T. A., Xu R.-M., Gozani O., et al. (2009). The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J. Biol. Chem. 284, 34283–34295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Schubert D. R. (2009). The specificity of neuroprotection by antioxidants. J. Biomed. Sci. 16, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y., Hermetz K. E., Jackson J. M., Mulle J. G., Dodd A., Tsuchiya K. D., Ballif B. C., Shaffer L. G., Cody J. D., Ledbetter D. H., et al. (2011). Diverse mutational mechanisms cause pathogenic subtelomeric rearrangements. Hum. Mol. Genet. 20, 3769–3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas N. M. C., Van Buggenhout G., Hannes F., Thienpont B., Sanlaville D., Kok K., Midro A., Andrieux J., Anderlid B.-M., Schoumans J., et al. (2008). Genotype-phenotype correlation in 21 patients with Wolf-Hirschhorn syndrome using high resolution array comparative genome hybridisation (CGH). J. Med. Genet. 45, 71–80 [DOI] [PubMed] [Google Scholar]
- Marango J., Shimoyama M., Nishio H., Meyer J. A., Min D.-J., Sirulnik A., Martinez-Martinez Y., Chesi M., Bergsagel P. L., Zhou M.-M., et al. (2008). The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood 111, 3145–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuibban A. G., Joza N., Megighian A., Scorzeto M., Zanini D., Reipert S., Richter C., Schweyen R. J., Nowikovsky K. (2010). A Drosophila mutant of LETM1, a candidate gene for seizures in Wolf-Hirschhorn syndrome. Hum. Mol. Genet. 19, 987–1000 [DOI] [PubMed] [Google Scholar]
- Misceo D., Barøy T., Helle J. R., Braaten O., Fannemel M., Frengen E. (2012). 1.5Mb deletion of chromosome 4p16.3 associated with postnatal growth delay, psychomotor impairment, epilepsy, impulsive behavior and asynchronous skeletal development. Gene 507, 85–91 [DOI] [PubMed] [Google Scholar]
- Näf D., Wilson L. A., Bergstrom R. A., Smith R. S., Goodwin N. C., Verkerk A., van Ommen G. J., Ackerman S. L., Frankel W. N., Schimenti J. C. (2001). Mouse models for the Wolf-Hirschhorn deletion syndrome. Hum. Mol. Genet. 10, 91–98 [DOI] [PubMed] [Google Scholar]
- Narita T., Yung T. M. C., Yamamoto J., Tsuboi Y., Tanabe H., Tanaka K., Yamaguchi Y., Handa H. (2007). NELF interacts with CBC and participates in 3′ end processing of replication-dependent histone mRNAs. Mol. Cell 26, 349–365 [DOI] [PubMed] [Google Scholar]
- Nimura K., Ura K., Shiratori H., Ikawa M., Okabe M., Schwartz R. J., Kaneda Y. (2009). A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature 460, 287–291 [DOI] [PubMed] [Google Scholar]
- Nowikovsky K., Froschauer E. M., Zsurka G., Samaj J., Reipert S., Kolisek M., Wiesenberger G., Schweyen R. J. (2004). The LETM1/YOL027 gene family encodes a factor of the mitochondrial K+ homeostasis with a potential role in the Wolf-Hirschhorn syndrome. J. Biol. Chem. 279, 30307–30315 [DOI] [PubMed] [Google Scholar]
- Nowikovsky K., Pozzan T., Rizzuto R., Scorrano L., Bernardi P. (2012). Perspectives on: SGP symposium on mitochondrial physiology and medicine. The pathophysiology of LETM1. J. Gen. Physiol. 139, 445–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowikovsky K., Schweyen R. J., Bernardi P. (2009). Pathophysiology of mitochondrial volume homeostasis: Potassium transport and permeability transition. Biochim. Biophys. Acta 1787, 345–350 [DOI] [PubMed] [Google Scholar]
- Osellame L. D., Blacker T. S., Duchen M. R. (2012). Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 26, 711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parman T., Wiley M. J., Wells P. G. (1999). Free radical-mediated oxidative DNA damage in the mechanism of thalidomide teratogenicity. Nat. Med. 5, 582–585 [DOI] [PubMed] [Google Scholar]
- Pieczenik S. R., Neustadt J. (2007). Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol. 83, 84–92 [DOI] [PubMed] [Google Scholar]
- Rauch A., Schellmoser S., Kraus C., Dörr H. G., Trautmann U., Altherr M. R., Pfeiffer R. A., Reis A. (2001). First known microdeletion within the Wolf-Hirschhorn syndrome critical region refines genotype-phenotype correlation. Am. J. Med. Genet. 99, 338–342 [DOI] [PubMed] [Google Scholar]
- Rodríguez L., Zollino M., Climent S., Mansilla E., López-Grondona F., Martínez-Fernández M. L., Murdolo M., Martínez-Frías M. L. (2005). The new Wolf-Hirschhorn syndrome critical region (WHSCR-2): a description of a second case. Am. J. Med. Genet. 136A, 175–178 [DOI] [PubMed] [Google Scholar]
- Siemen D., Ziemer M. (2013). What is the nature of the mitochondrial permeability transition pore and what is it not? IUBMB Life 65, 255–262 [DOI] [PubMed] [Google Scholar]
- Simon R., Bergemann A. D. (2008). Mouse models of Wolf-Hirschhorn syndrome. Am. J. Med. Genet. 148C, 275–280 [DOI] [PubMed] [Google Scholar]
- South S. T., Bleyl S. B., Carey J. C. (2007). Two unique patients with novel microdeletions in 4p16.3 that exclude the WHS critical regions: implications for critical region designation. Am. J. Med. Genet. 143A, 2137–2142 [DOI] [PubMed] [Google Scholar]
- South S. T., Hannes F., Fisch G. S., Vermeesch J. R., Zollino M. (2008). Pathogenic significance of deletions distal to the currently described Wolf-Hirschhorn syndrome critical regions on 4p16.3. Am. J. Med. Genet. 148C, 270–274 [DOI] [PubMed] [Google Scholar]
- Szabadkai G., Duchen M. R. (2008). Mitochondria: the hub of cellular Ca2+ signaling. Physiology (Bethesda) 23, 84–94 [DOI] [PubMed] [Google Scholar]
- Tamai S., Iida H., Yokota S., Sayano T., Kiguchiya S., Ishihara N., Hayashi J., Mihara K., Oka T. (2008). Characterization of the mitochondrial protein LETM1, which maintains the mitochondrial tubular shapes and interacts with the AAA-ATPase BCS1L. J. Cell Sci. 121, 2588–2600 [DOI] [PubMed] [Google Scholar]
- Van Buggenhout G., Melotte C., Dutta B., Froyen G., Van Hummelen P., Marynen P., Matthijs G., de Ravel T., Devriendt K., Fryns J. P., et al. (2004). Mild Wolf-Hirschhorn syndrome: micro-array CGH analysis of atypical 4p16.3 deletions enables refinement of the genotype-phenotype map. J. Med. Genet. 41, 691–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargesson N. (2009). Thalidomide-induced limb defects: resolving a 50-year-old puzzle. Bioessays 31, 1327–1336 [DOI] [PubMed] [Google Scholar]
- White D. M., Pillers D. A., Reiss J. A., Brown M. G., Magenis R. E. (1995). Interstitial deletions of the short arm of chromosome 4 in patients with a similar combination of multiple minor anomalies and mental retardation. Am. J. Med. Genet. 57, 588–597 [DOI] [PubMed] [Google Scholar]
- Wolf U., Reinwein H., Porsch R., Schröter R., Baitsch H. (1965). [Deficiency on the short arms of a chromosome No. 4.] Humangenetik 1, 397–413 [PubMed] [Google Scholar]
- Wright T. J., Ricke D. O., Denison K., Abmayr S., Cotter P. D., Hirschhorn K., Keinänen M., McDonald-McGinn D., Somer M., Spinner N., et al. (1997). A transcript map of the newly defined 165 kb Wolf-Hirschhorn syndrome critical region. Hum. Mol. Genet. 6, 317–324 [DOI] [PubMed] [Google Scholar]
- Zhang X., Chen G., Lu Y., Liu J., Fang M., Luo J., Cao Q., Wang X. (2013). Association of mitochondrial Letm1 with epileptic seizures. Cereb. Cortex. [DOI] [PubMed] [Google Scholar]
- Zollino M., Di Stefano C., Zampino G., Mastroiacovo P., Wright T. J., Sorge G., Selicorni A., Tenconi R., Zappalà A., Battaglia A., et al. (2000). Genotype-phenotype correlations and clinical diagnostic criteria in Wolf-Hirschhorn syndrome. Am. J. Med. Genet. 94, 254–261 [DOI] [PubMed] [Google Scholar]
- Zollino M., Lecce R., Fischetto R., Murdolo M., Faravelli F., Selicorni A., Buttè C., Memo L., Capovilla G., Neri G. (2003). Mapping the Wolf-Hirschhorn syndrome phenotype outside the currently accepted WHS critical region and defining a new critical region, WHSCR-2. Am. J. Hum. Genet. 72, 590–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.