

Background: Fe65 is a neuronal adaptor with essential roles in neuronal cells.
Results: Fe65 regulates promoter recruitments of the estrogen receptor α and growth response of breast cancers to estrogens and tamoxifen.
Conclusion: Fe65 is a positive regulator of estrogen actions in breast cancer cells.
Significance: The studies define a novel role for a neuronal adaptor in estrogen actions in breast cancer cells.
Keywords: Cancer Biology; Cell Growth; DNA Transcription; Endocrinology; Gene Transcription; Hormone Receptors; Hormones; Nuclear Receptors; Breast Cancer, Estrogens, Estrogen Receptor, Fe65, Tamoxifen
Abstract
Fe65 is a multidomain adaptor with established functions in neuronal cells and neurodegeneration diseases. It binds to the C terminus of the Aβ amyloid precursor protein and is involved in regulating gene transcription. The present studies show that Fe65 is expressed in breast cancer (BCa) cells and acts as an ERα transcriptional coregulator that is recruited by 17β-estradiol to the promoters of estrogen target genes. Deletion analyses mapped the ERα binding domain to the phosphotyrosine binding domain 2 (PTB2). Ectopic Fe65 increased the transcriptional activity of the ERα in a PTB2-dependent manner in reporter assays. Fe65 knockdown decreased, whereas its stable expression increased the transcriptional activity of endogenous ERα in BCa cells and the ability of estrogens to stimulate target gene expression, ERα, and coactivator recruitment to target gene promoters and cell growth. Furthermore, Fe65 expression decreased the antagonistic activity of tamoxifen (TAM), suggesting a role for Fe65 in TAM resistance. Overall, the studies define a novel role for the neuronal adaptor in estrogen actions in BCa cells.
Introduction
Estrogens are the female sex steroid hormone with established roles in reproduction and development as well as the biology and pathogenesis of many tissues including those of the central nervous, the cardiovascular, and the skeletal systems, etc. (1, 2). The best defined estrogen target tissues include the mammary gland of which both the normal development and epithelial tumorigenesis are subjected to estrogen control. Due to the retained sensitivity of the majority of human BCa to estrogens, the inhibition of estrogen action at the tumor with synthetic antagonists such as TAM2 has been the preferred therapeutic treatment for decades (3, 4). The effects of estrogens and antiestrogens on mammary epithelial and cancer cells are predominantly mediated through the ERα that belongs to the steroid/thyroid nuclear receptor superfamily of ligand-regulated transcription factors (5, 6). The ERα contains an N-terminal A/B region, a DNA binding domain composed of two C2C2 zinc fingers, a hinge region, a ligand binding domain, and an F tail (5). It forms a homodimer and binds to estrogen response elements (ERE) to control target gene expression. In addition, the ERα also regulates target gene expression through “tethering” to other transcription factors (7, 8). Estrogens bind to the ERα and recruit coactivators (9) to induce the expression of growth-promoting genes. Estrogen antagonists, on the other hand, induce a distinct conformation suitable for the binding of corepressor complexes, thereby shutting off gene transcription (10, 11). TAM, which aims to block the ERα action, has mixed agonist/antagonist activity and may either stimulate or inhibit ERα activity, depending on the tissue and gene context (12). The use of TAM has significantly benefitted women with ERα-positive BCa, but the benefit has been limited by the issue of resistance.
Besides mammary gland development and tumorigenesis, another known target organ for estrogens is the brain. Estrogens have been shown to be neuroprotective, and their decrease after menopause is believed to contribute to the development of neurodegenerative diseases such as Alzheimer disease. APP plays important roles in the pathogenesis of Alzheimer disease, and recent studies have shown that its C-terminal fragment produced after the cleavage by γ-secretase, namely APPct or AICD, forms a multimeric complex with the nuclear adaptor protein Fe65 and stimulates transcription (13–15). Published studies have shown that 17β-estradiol inhibits the transcriptional activity of the APPct complex and impairs the ability of the complex to induce neuroblastoma cell apoptosis (16), providing a mechanism explaining the neuronal protective effects of estrogens. Both in vitro and in vivo immunological analyses have revealed that the ERα forms a complex with full-length APP or APPct and that the complex formation occurs between endogenous proteins in mouse brains, which is increased in transgenic mice expressing mutant presenilin 1 and APP (16). Mechanistic investigations have found that the functional interaction between the ERα and APP is indirectly mediated through Fe65, identifying it as a novel ERα interacting protein (16).
Fe65 is a multidomain adaptor protein containing an undefined N terminus, a group II tryptophan-tryptophan (WW) domain in the middle, and two C-terminal PTB domains, namely PTB1 and PTB2 (17). Through PTB2, it forms a multimeric complex with APP or APPct to stimulate transcription through the recruitment of the transcription factor CP2/LSF/LBP1 and the histone acetyl transferase Tip60 (13–15) to PTB1 as well as the nucleosome assembly factor SET to the WW domain (18). The PTB1 domain also interacts with two cell surface lipoprotein receptors, the low density lipoprotein receptor-related protein (19) and ApoEr2 (20), forming trimeric complexes with APP and establishing a biological linkage between APP and the lipoprotein receptors. Besides SET, the WW domain also binds to Mena (21), through which Fe65 regulates actin cytoskeleton, cell motility, and neuronal growth cone formation (22, 23).
There are two Fe65 isoforms produced by the alternative splicing of a 6-bp mini-exon encoding Arg-Glu dipeptide inserted in the PTB1 domain. The isoform with this mini-exon is expressed exclusively in neurons, whereas the isoform lacking the dipeptide exists in non-neuronal cells (24). Besides its neuronal functions in APP processing and Alzheimer disease biology, Fe65 has been reported to regulate other essential cellular functions such as DNA damage repair that goes beyond neuronal cells. Fe65 null mice are more sensitive to DNA damages induced by etoposide and ionizing radiations (25). Studies with Fe65 null mouse embryonic fibroblasts concluded that Fe65 was required for the efficient repair of DNA double-strand breaks, a function dependent on its interaction with Tip60 and APPct (26, 27). However, functions of Fe65 in non-neuronal cells are largely undefined, and nothing is known about its involvement in estrogen actions in BCa.
In the present study we demonstrate for the first time that Fe65 is expressed in mammary epithelial cells and that its expression is increased in BCa cells and human breast tumor samples. Fe65 is recruited by estrogens to the promoters of estrogen target genes in BCa cells and potentiates the recruitment of the ERα and its coactivators to the promoters. It increases the agonistic activity of 17β-estradiol and decreases the antagonistic activity of TAM. The studies define Fe65 as a positive ERα regulator that increases the growth of human BCa cells and contributes to TAM resistance.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
17β-Estradiol, anti-FLAG affinity gels, and 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) were purchased from Sigma. Fetal bovine serum (FBS), charcoal stripped FBS, and Lipofectamine 2000 were from Invitrogen. Anti-hemagglutinin (anti-HA.11) antibody was obtained from Covance (Princeton, NJ). Anti-Fe65, anti-c-Myc, anti-cyclin D1 were from Cell Signaling (Boston, MA). Anti-APPct was from Calbiochem. The following antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA): anti-ERα (F10), anti-Fe65 (H-260), anti-β-actin (AC-15), anti-HSP60 (H-1), HDAC1 (H-11), anti-Tip60 (N-17), anti-histone H1 (N-16). Fe65 (siFe65) (sequence 5′-CUACGUAGCUCGUGAUAAG-3′), siERα (sequence 5′-GCCAGCAGGUGCCCUACUA-3′), and scrambled control (siCtrl) siRNA oligonucleotides were synthesized by Dharmacon/Thermo Scientific (Waltham, MA). The ECL Western blotting substrates were from Thermo Scientific. Luciferase assay substrates were from Promega Corp. (Madison, WI). Chip assay kit (EZ-ChipTM) was from Millipore (Billerica, MA). Breast invasive ductal carcinoma tissue array slides were purchased from US Biomax Inc. (Rockville, MD), and their usage was in compliance with policies of the institutional review board at University of South Florida.
To construct tagged Fe65, cDNA of the non-neuronal Fe65 (Thermo Scientific) was amplified by PCR using forward (GCGGGATCCATGTCTGTTCCATCATCACTG) and reverse (GAGGTCGACTCATGGGGTATGGGCCCC) primers. Myc-Fe65 was constructed by cloning the amplified cDNA into the Bam H1 and Sal1 of p-CMV-3Tag-2a-Myc plasmid (Agilent Technologies, Santa Clara, CA). To construct the expression plasmids of HA-Fe65 and deletion constructs, Fe65 cDNA or fragments were amplified by PCR using the Myc-Fe65 as the template, digested with BamH1 and Sal1, and ligated into pCMV-HA plasmid generated by replacing the FLAG tag with HA of pCMV-3Tag-1A (Agilent Technologies). The primers used for the constructions of Fe65 and its deletion mutants are as follows: full-length: GCGGGATCCATGTCTGTTCCATCATCACTGAGC (forward) and GAGGTCGACTCATGGGGTATGGGCCCCAGCCG (reverse); N-terminal 128 amino acid deletion (dN128): GCGGGATCCATGAACCGAGGCCTACGAGGACCT (forward) and GAGGTCGACTCATGGGGTATGGGCCCCCAGCCG (reverse); N-terminal 242 amino acid deletion (dN242): GCGGGATCCATGTTCTGGAACCCCAACGCCTTC (forward) and GAGGTCGACTCATGGGGTATGGGCCCCCAGCCG (reverse); WW deletion (dWW): TTCACCGGTCAAGAGGAGTCCCAGCTCAC (forward) and CTTACCGGTGTCGGAATCCGTCTCGAAG (reverse); PTB1 deletion (dPTB1): CCCAAGAGGAGGAGAAGTGCTTGGTAAATGGACT (forward) AGTCCATTTACCAAGCACTTCTCCTCCTCTTGGG (reverse); PTB2 deletion (dPTB2): CTTGTGGATGTCCCTTTCCAATCCCAGGCCTC (forward) and GAGGCCTGGGATTGGAAAGGGACATCCACAAG (reverse); C-terminal 182 amino acid deletion (dC182): GCGGGATCCATGTCTGTTCCATCATCACTGAGC (forward) GAGGTCGACTCATTGGAAAGGGACATCCACAAG (reverse); C-terminal 42 amino acid deletion (dC182): GCGGGATCCATGTCTGTTCCATCATCACTGAGC (forward) and GAGGTCGACACGGGCATCCAGACACTTCTGGTA (reverse).
FLAG-VDR was generated by sub-cloning VDR cDNA in-frame with 3×FLAG epitope in the pCMV vector. FLAG-ERα plasmid was a gift from Dr. Muyan (28). pGEX-ERα(297–595) was provided by Dr. Corbo (29), and AIB1 was provided by Dr. Meltzer (30). FLAG-AR (31), pLENβgal (16, 32), EREe1bLuc (16, 32), ARE2e1bLuc (31), and p23luc (33) plasmids had been used in previous studies.
Cell Culture
MCF-7, T47D, MDA-MB-231, and MDA-MB-361 cells were cultured in Dulbecco's modified Eagle's Medium (DMEM) containing 2 mm l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% FBS. BT474 and ZR75–1 cells were cultured in RPMI 1640 medium supplemented with 2 mm l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% FBS. MCF-10A cells were maintained in DMEM/F-12 medium containing 10 μg/ml insulin, 20 ng/ml EGF, 100 ng/ml cholera toxin, 0.5 μg/ml hydrocortisone, 100 units/ml penicillin, 100 μg/ml streptomycin, and 5% FBS. For all experiments involving estrogen and androgen treatment, cells were starved in phenol red-free medium containing 3% stripped FBS for 3 days to remove steroids in the assay system, and all steroid treatments were carried out under the same steroid-free conditions.
Transfections, Fe65 Knockdown, and Reporter Assays
For reporter assays, cells were steroid-starved for 2 days in DMEM containing 3% charcoal stripped FBS, and transfections were performed with Lipofectamine 2000. One day post transfections, cells were treated with vehicle (ethanol (EtOH)) or 17β-estradiol (E2) for 48 h. Cellular extracts were prepared by directly adding lysis buffer containing 25 mm Tris-phosphate (pH 7.8), 2 mm dithiothreitol, 2 mm 1, 2-diaminocyclohexane-N,N,N′,N′-tetraacetic acid, 10% glycerol, and 0.2% Triton X-100. Luciferase and β-galactosidase (β-gal) activity was determined as previously described (32, 34).
For Fe65 knockdown, cells were transfected with Fe65 (siFe65) or control (siCtrl) siRNA, and 24 h post-transfection, Fe65 knockdown was verified by immunoblotting (IB). For Fe65 stable expression, MCF-7 cells were transfected with Myc-Fe65 or empty vectors, and stable clones were selected in the presence of 1 μg/ml puromycin. Individual clones were isolated, and Fe65 expression was confirmed by IB. For ERα target gene expression, transfected cells were estrogen-starved for 3 days and treated with EtOH or E2 for 2 h, and the expression of selective estrogen target genes was measured by IB and quantified by ImageJ (National Institutes of Health, rsb.info.nih.gov).
GST Pulldown Assays
pGEX-ERα(297–595) in which GST is fused with ERα ligand binding domain (29) was transformed into the BL21(DE3) Escherichia coli strain. Transformed bacteria were cultured at 37 °C until the optical density at 600 nm reached 0.8. The culturing was continued for 4 h in the presence of 0.5 mm isopropylthiogalactopyranoside. The bacteria were then harvested and lysed by sonication in buffer containing 50 mm Tris (pH 7.5), 150 mm NaCl, 1 mm EDTA, 6 mm MgCl2, 1 mm dithiothreitol, and 1 mm phenylmethylsulfonyl fluoride. GST fusion proteins were purified using glutathione-Sepharose 4B beads (GE Healthcare). Then 50 μg of bead-bound GST-ERα were incubated overnight at 4 °C with extracts of 293T cells transfected with Fe65 constructs and washed 3 times with the lysis buffer containing 0.5% Nonidet P-40. Fe65 protein bound to the beads was released by boiling in sample buffer, resolved in an 8% SDS-polyacrylamide gel (SDS-PAGE), and detected by IB analyses.
Real Time RT-PCR
Total RNA was extracted from cells using TRIzol reagent and cDNA template synthesized from 2 μg of RNA using Oligo(dT)12–18 primer and Superscript II reverse transcriptase. Taqman probes (Invitrogen) were used to measure human cyclin D (assay ID number Hs00277039_m1), c-Myc (assay ID number Hs00153408_m1), and GAPDH (assay ID number Hs02758991_g1) mRNA expression. PCR reactions were performed in a 20-μl mixture containing 150 ng of cDNA, 10 μl of 2× TaqMan PCR master mixes, and 1 μl of corresponding TaqMan probes. The reactions were run as described (35) on the ABI Prism 7900 Fast Real-time PCR system in triplicate as follows: 95 °C for 10 min, 45 cycles of a 15-s denaturing at 95 °C, and 1 min annealing at 60 °C. Cyclin D and c-Myc mRNA expression levels were normalized with cognate GAPDH by subtracting the cycle threshold (Ct) value of GAPDH from Ct value of the target genes to produce a ΔCt. The -fold of induction over vehicle control was calculated based on the formula 2−[ΔCt (17β-estradiol) − ΔCt (vehicle)].
Subcellular Fractionation
Subcellular fractionation was carried out as previously described (36) with minor modifications. Briefly, cell pellets were resuspended in lysis buffer containing 10 mm HEPES-NaOH (pH 7.9), 10 mm KCl, 0.1 mm EDTA, 0.1 mm EGTA, and 1 mm dithiothreitol and incubated on ice for 15 min. To 400 μl of the suspension solution, 12.5 μl of 10% (v/v) Nonidet P-40 was added followed by agitation for 10 s and centrifugation at 13,000 rpm for 1 min. The supernatant was collected as cytoplasmic proteins, and the pellet (crude nuclear fraction) was washed 3 times with lysis buffer containing 0.625% (v/v) Nonidet P-40 and resuspended in extraction buffer containing 10 mm HEPES-NaOH (pH 7.9), 400 mm NaCl, 1 mm EDTA, and 1 mm EGTA. The suspension was incubated on ice for 30 min with a brief vortex every 5 min followed by centrifugation for 5 min at 13,000 rpm. The supernatant was then collected as nuclear proteins.
Immunological Analyses
For immunoprecipitations (IPs), cells suspended in lysis buffer containing 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm EDTA, 1% (v/v) Nonidet P-40, 1 mm PMSF, and protease inhibitor mixture were lysed by sonication for 6 s with two repeats and incubation on ice for 10 min. After centrifugation, cellular extracts were incubated with primary antibodies overnight at 4 °C and with beads for 2 h. The beads were washed with cold lysis buffer three times and boiled in SDS-PAGE sample buffer to release the precipitates.
For IB analyses, precipitates or cellular extracts were separated in SDS-PAGE, transferred to a nitrocellulose membrane, and probed with cognate antibodies. Horseradish peroxidase-linked secondary antibodies and enhanced chemiluminescence reagents (Thermo Scientific, Waltham, MA) were used to detect proteins.
For chromatin IP (ChIP) assays, cells were plated in DMEM medium containing 10% FBS. One day after plating, the medium was changed to DMEM containing 3% charcoal stripped FBS for estrogen deprivation. For T47D cells, siRNA transfections were performed with Lipofectamine 2000 24 h after estrogen depletion. Then cells were cultured in DMEM medium containing 3% stripped FBS for another 48 h before estrogen treatment. For MCF-7 stable clones, cells were estrogen-starved for 72 h and treated with ethanol or 1 × 10−7 m 17β-estradiol for 45 min before cross-linking with 1% formaldehyde. All subsequent steps followed the company's instructions (EMD Millipore, Billerica, MA). ChIP assay primers for cathepsin D, c-Myc, and IGF-1 had been described previously (37). The primers for EBAG9 were ATTGTCTGCCCTTCGCCGT (forward) and TTTGGAGGCTGCGTGCTTT (reverse).
For ERα and Fe65 immunofluorescence staining and imaging, T47D cells in chamber slides were fixed in phosphate-buffered saline (PBS) containing 4% paraformaldehyde, permeabilized for 10 min with PBS containing 0.25% Triton X-100, and incubated in PBS containing 0.2% Tween 20 and 1% BSA for 30 min to block nonspecific binding. Then fixed cells were incubated overnight at 4 °C with primary antibodies in the same buffer in a humidified chamber, washed with PBS 3 times, and then incubated with secondary antibodies for 1 h at room temperature in the dark. After three more washes, the slides were mounted in medium with DAPI, and the coverslip was sealed with nail polish. Images were observed and captured under a Leica TCS SP2 confocal microscope in the USF Imaging Core.
Fe65 Immunohistochemistry (IHC) and Breast Tissue Microarray (TMA) Analyses
Fe65 expression was assessed by IHC of breast TMA slides containing paraffin sections from 70 cases of breast invasive ductal carcinomas and 2 cases of normal and 3 cases of adjacent breast tissue with duplicate cores per case. The TMA slides were deparaffinized before the antigen retrieval procedure was performed for 30 min in 10 mm citrate (pH 6.0). Endogenous peroxidase activity was quenched by incubating the slides with 3% H2O2 for 20 min. Then the slides were incubated overnight at 4 °C with primary antibody against Fe65 (Santa Cruz, H-260, SC33155, 1:50 dilution) in a humidified chamber and after 3 washes with biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA) at a 1:200 dilution for 1 h at room temperature. For detection, peroxidase-based Elite ABC (avidin/biotin complex) kit (Vector Laboratories) was used together with liquid 3,3′-diaminobenzidine, substrate and color development was monitored under the microscope. The slides were briefly counterstained in hematoxylin, dehydrated, cleared, and mounted in Permount medium (Fisher).
For automated quantitation of Fe65 IBC signals, the slides were scanned using the Aperio™ ScanScope XT system (Vista, CA) with a 200×/0.8 NA objective lens with a 2× doubler (0.265 μm/pixel) at a rate of 10 min/slide via Basler tri-linear-array detection. Whole slide images (.svs) were loaded into Tissue Studio v3.0 (Definiens, Munich, Germany) and segmented into individual TMA cores using the TMA mapping functionality. Individual cells were identified using hematoxylin thresholding (0.2) and an IHC threshold (0.5). The typical size of the nuclei was set to be 60 μm2, and the cells were grown (cell simulation at 2 μm) in every direction. The IHC classification was divided into negative, weak (<0.56), moderate, and strong (>0.64) staining for each cell and compiled into a summary data sheet using Microsoft Excel 2010. Percentages of positive nuclei were a sum of the percentage of the nuclei that were weak, moderate, and strong for Fe65. Statistical analyses were performed with Student's t test.
MTT Assays
Cells were seeded into 96-well plates at a density of 2 × 103 cells per well, estrogen-starved for 3 days, and treated with ethanol, 1 × 10−8 m E2, or 10−7 m TAM for various times. MTT reagent was added to each well to give a final concentration of 0.5 mg/ml and incubated for 3 h. After removing the medium, 100 μl of DMSO was added, and the absorption at 595 nm was determined with a MRX microplate reader (DYNEX Technologies, Chantilly, VA).
RESULTS
Fe65 Is a Positive ERα Transcriptional Regulator
Published studies have shown that estrogens regulate the transcriptional activity of APPct through a Fe65-ERα protein complex detected both in vitro and in the mouse brain (16). The complex formation makes it possible for Fe65 to regulate ERα activity. To test this idea, the ERα and Fe65 were ectopically expressed in HeLa cells, and ERα transcriptional activity was measured with the co-transfected EREe1bluc luciferase reporter gene. As shown in Fig. 1A, ectopic Fe65 expression in HeLa cells increased ERα activity induced by E2 in a dose-dependent manner. The increase was statistically significant and about half of what was induced by AIB1, a bona fide ER coactivator. In similar assays ectopic APPct expression did not increase ERα activity either in the absence (Fig. 1B) or presence (Fig. 1C) of Fe65. IB analyses (Fig. 1, right panels) showed that the ERα, Fe65, and APPct proteins were expressed in a manner dependent on the amounts of plasmids used in the transfections and that ERα protein expression was not significantly altered by Fe65. The data suggest that Fe65 increased ERα activity per molecule. The analyses identify Fe65 as a positive ERα regulator that stimulates ERα activity in a manner independent of its complex formation with APPct.
FIGURE 1.

Fe65 potentiates ERα activation by 17β-estradiol. A, HeLa cells were plated in 12-well plates and transfected with 0.1 μg of EREe1bluc, 0.1 μg of pLENβgal, 0.2 μg of FLAG-ERα, and indicated amounts of HA-Fe65 or 0.3 μg of AIB1 and treated with vehicle (EtOH) or 10−8 m E2 for 48 h. Luciferase activity was determined and normalized with β-gal activity. ERα activity was calculated by dividing the normalized luciferase activity with the activity of control cells transfected with empty vector and treated with EtOH. The levels of ERα, Fe65, and AIB1 expression were determined by Western blot with indicated antibodies. B, HeLa cells were transfected with ERα expression and reporter vectors as in panel A but with the indicated amounts of APPct. Cells were treated and ERα activity, and levels of protein expression were similarly determined. C, HeLa cells were transfected with ERα expression and reporter vectors as in panel A but together with indicated amounts of Fe65 and APPct. Cells were treated, and ERα activity and levels of protein expression were similarly determined. For all reporter assays, each data point represents analyses in triplicate performed in parallel, and each experiment was repeated three times. The error bars represent S.D. Statistical analyses were performed with Student's t test (n = 9).
In similar reporter assays (Fig. 2, left panels), Fe65 exhibited little effects on the transcriptional activity of the androgen (Fig. 2B) and vitamin D (Fig. 2C) receptors. The lack of an effect on the androgen and vitamin D receptors was not due to insufficient Fe65 expression because in parallel analyses (Fig. 2A) the same ectopic Fe65 expression increased ERα activity to levels comparable with that presented in Fig. 1, whereas IB analyses revealed a comparable amount of Fe65 protein expression (Fig. 2, right panels). These analyses show that Fe65 may be an ERα-selective transcriptional coactivator.
FIGURE 2.

Fe65 selectively increases ERα transcriptional activity. A, HeLa cells were transfected with 0.1 μg EREe1bluc, 0.1 μg of pLENβgal, and indicated amounts of FLAG-ERα and HA-Fe65. Cells were treated, and ERα activity and the levels of protein expression were determined with the indicated antibodies as in Fig. 1A. B, HeLa cells were transfected with 0.1 μg of AREe1bluc, 0.1 μg of pLENβgal, and the indicated amounts of FLAG-AR and HA-Fe65. Cells were treated with EtOH or 10−8 m R1881 and AR activity, and the levels of protein expression were determined with the indicated antibodies as in Fig. 1A. C, HeLa cells were transfected with 0.1 μg of p23luc reporter, 0.1 μg of pLENβgal, and the indicated amounts of FLAG-VDR and HA-Fe65. Cells were treated with EtOH or 10−8 m 1α,25-dihydroxvitamin D3 (VD3), and VDR activity and levels of protein expression were determined with the indicated antibodies as in Fig. 1A.
Fe65 Binds to the ERα and Stimulates Its Transcriptional Activity through the PTB2 Domain
Previous studies defined the Fe65 binding domain to the C-terminal ligand binding domain of the ERα (16). To define the Fe65 domain mediating its interaction with the ERα, Fe65 mutants lacking defined domains were generated (Fig. 3A), and their ability to bind to the ERα was assessed in co-IP analyses (Fig. 3B) and GST pulldown assays (Fig. 3C). In cells co-transfected with FLAG-ERα with either Myc- or HA-tagged full-length Fe65, Fe65 was detected in anti-FLAG precipitates together with the ERα. No Fe65 was detected in the precipitates when cells were transfected with Myc-Fe65 and the control vector, showing that the presence of Fe65 in the FLAG precipitates accurately measured the interaction between the ERα and Fe65. Deletion of Fe65 regions containing PTB2 eliminated its ability to co-precipitate with FLAG-ERα, whereas the deletion of other regions had little effects, showing that the interaction was mediated through PTB2. This conclusion was reaffirmed by subsequent GST pulldown assays (Fig. 3C), in which GST-ERα-C terminus (GST-ER-C) containing the ligand binding domain precipitated all tested Fe65 fragments except the one lacking PTB2. Overall, the co-precipitation analyses suggest that the interaction is mediated through direct interaction between Fe65 PTB2 domain and ERα ligand binding domain.
FIGURE 3.

Fe65 interacts with the ERα through its PTB2 domain, and the interaction occurs with endogenous proteins in BCa cells. A, schematic representations of the domain structure of full-length Fe65 and its deletion mutants. Numbers of amino acids of full-length Fe65 and its domains are marked. B, 293T cells were transfected with 1.5 μg of FLAG-ERα and Myc-tagged (left panels) or HA-tagged (right panels) Fe65 mutants as indicated. Cellular extracts were subjected to IP analyses followed by IB with indicated antibodies. C, GST and GST-ERα-C proteins produced in and purified from bacteria were incubated with lysates of cells transfected with HA-tagged Fe65 deletion mutants. GST pulldown assays were performed, and proteins in the precipitates were detected by IB with anti-HA antibody (lower panels). Coomassie blue (C.B.) staining was included to show that comparable amounts of GST and GST-ER-C were used in the pulldown assays. IB analyses of the lysates were also included to show the relative amounts of the Fe65 deletion mutants used in the pulldown assays. D, whole cell lysates of T47D cells were subjected to IP analyses with anti-ERα antibody followed by IB analyses with ERα and Fe65 antibodies as indicated. E, T47D cells were treated with EtOH or 10−7 m E2 for 1 h and subjected to DAPI (blue) and immunofluorescence staining with anti-ERα (green) and anti-Fe65 (red) antibodies. Images were captured under a confocal fluorescence microscope to show Fe65 and ERα location and co-localization. Images of MDA-MB-361 cells stably expressing control (Ctrl) or Fe65 (Fe65 KD) shRNA were included as a quality control for Fe65 staining (right panels).
In previous studies co-precipitation and immunofluorescence staining analyses revealed a complex formation between endogenous ERα and Fe65 in the mouse brain (16). Similar analyses were performed to test whether the complex formation occurred with endogenous proteins in BCa cells. As shown in Fig. 3D, anti-ERα, not control IgG, co-precipitated Fe65 together with the ERα in T47D cells, demonstrating that the complex formation also occurred between endogenous proteins in BCa cells. Similarly, confocal imaging revealed the presence of Fe65 in both the cytoplasm and the nucleus of T47D cells as well as its co-localization with the ERα (Fig. 3E). In the absence of E2 the co-localization was mainly detected in the cytoplasm, particularly areas around the plasma membrane. After E2 treatment the co-localization became predominantly nuclear accompanied with ERα nuclear localization. Fe65 knockdown significantly decreased the red fluorescence signal detected by the Fe65 antibody, showing the specificity of the staining to endogenous Fe65 (Fig. 3E). The data show that the co-localization was mainly determined by ERα localization in the cells.
Consistent with the mapping analyses, reporter assays showed that the PTB2 domain is essential for Fe65 to potentiate ERα transcriptional activation. As shown in Fig. 4A, although full-length Fe65 increased ERα activity in a dose-dependent manner, the PTB2 deletion mutant did not change ERα activity in parallel analyses. IB analyses showed that the PTB2 deletion mutant was expressed in levels comparable with full-length protein (Fig. 4B), ruling out the possibility that the lack of an effect of the PTB2 mutant was due to the insufficient protein expression. Levels of the ERα protein expression were not significantly altered by the full-length Fe65 or the PTB2 deletion mutant (Fig. 4B), showing that the reporter analyses reflected the effect of Fe65 on the specific activity of the ERα per molecule. The reporter analyses together with the domain mapping allow us to conclude that Fe65 binds to the ERα and potentiates its activity through its PTB2 domain.
FIGURE 4.
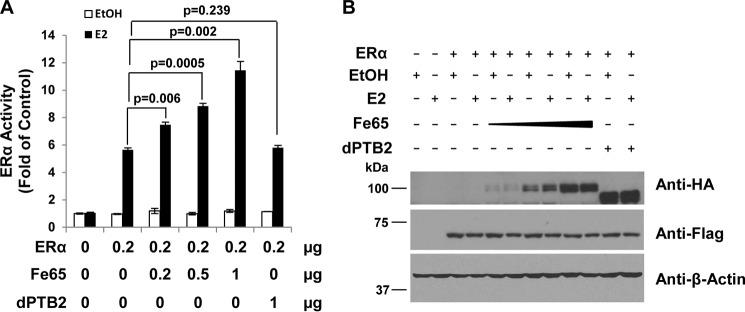
The PTB2 domain is essential for Fe65 to potentiate estrogen stimulation of ERα transcriptional activity. A, HeLa cells were plated and transfected as in Fig. 1A but together with the indicated amounts of HA-tagged Fe65 or PTB2 deletion mutant. Cells were treated with E2, and ERα activity was similarly determined. B, IB analyses were performed in parallel with lysates from transfected cells to show that Fe65 and the PTB2 deletion mutant were expressed at comparable levels and did not change ERα protein expression. β-Actin blot was included to show even loading.
Fe65 Is Expressed in Mammary Epithelial Cells, and the Expression Increased in BCa Cells and Breast Tumor Tissues
The ERα plays an important role in BCa formation and mediates the mitogenic activity of estrogens in stimulating BCa growth. The interaction between the ERα and Fe65 suggests a possible role for Fe65 in estrogen actions in BCa. Consistent with this idea, Fe65 was detected in the non-tumorigenic MCF-10A human breast epithelial cells, and its expression was found to be increased in human BCa cells as well as two mouse mammary tumor cells: WT145 and VDR-KO (Fig. 5A). The increased expression was detected in both ERα-negative and positive cells with the exception of MCF-7. Subcellular fractionation revealed the presence of Fe65 in the nucleus of all tested BCa cells, including MCF-7 (Fig. 5B), which appeared absent in MCF-10A cells. Consistent with the cell line data, IHC staining of breast tissue sections with anti-Fe65, not IgG control, detected Fe65 expression in both the cytoplasm and the nucleus of epithelial and stromal cells, with a stronger nuclear signal in the tumor than the control samples (Fig. 5C). Quantitative IHC analyses of breast TMA slides containing 140 breast tumor sections and 10 normal and adjacent controls showed that the nuclear Fe65 in breast tumors was higher than controls and that the difference was statistically significant (p = 0.0015) (Fig. 5D). The increased Fe65 expression in BCa cells, particularly the increase in the nucleus, supports the idea that Fe65 is a positive regulator of ERα actions in tumor cells that may promote breast tumorigenesis. It is important to point out that although the IB analyses in BCa cell lines indicated a higher Fe65 expression in ERα-negative than in ERα-positive cells (Fig. 5A), the TMA analyses did not detect a significant difference between ERα-positive and negative tumors (data not shown), showing that the Fe65 expression data from cultured cells did not reflect the situation in tumors. Our data mining and bioinformatics did not reveal a consistent increase (or decrease) in Fe65 mRNA expression in breast tumors as compared with controls (data not shown), indicating that the increased Fe65 protein expression in tumors may be due to an increase at the rate of protein synthesis or a decrease in protein degradation or both.
FIGURE 5.

Fe65 is expressed in breast epithelial cells, and the expression is increased in BCa cells and breast tumor tissue samples. A, whole cell lysates of non-tumorigenic breast epithelial (MCF-10A) and BCa cells were subjected to IB analyses with the indicated antibodies. β-actin blot was included to show even loading. B, cytoplasmic and nuclear extracts were subjected to IB analyses with indicated antibodies. Hsp60 and HDAC1 blots were included to show the separation of cytoplasmic and nuclear extracts. C, representative images of normal breast and tumor tissue sections that were immunohistochemically stained with control IgG or anti-Fe65 as indicated. Pictures in the lower panels are enlarged (4×) versions of the marked areas of those in the top panels to show the bluish nuclei of normal ductal epithelial cells (middle panel), which indicated weak Fe65 signals, and the strong nuclear staining of breast tumor cells (right panels). D, Fe65 expression was assessed by IHC staining and automated quantification of TMA slides containing 140 cores of breast ductal carcinomas (T) and 10 cores of normal and adjacent breast tissues (N). The dot plot shows the distribution of nuclear Fe65 signals, whereas the tube plot compares the range and average nuclear Fe65 levels between N and T groups.
Fe65 Stimulates the Activity of Endogenous ERα in BCa Cells and Is Recruited by Estrogens to Target Gene Promoters, Which in Turn, Facilitates the Recruitment of the ERα and Its Coactivators
To test its effect on endogenous ERα, Fe65 was either stably expressed or knocked down in ERα-positive BCa cells, and the changes in the transcriptional activity of endogenous ERα was determined with transfected EREe1bluc reporters. The transcriptional analyses showed that stable Fe65 expression in MCF-7 cells significantly increased E2 stimulation of endogenous ERα activity in a dose-dependent manner (Fig. 6A), whereas Fe65 knockdown in both T47D (Fig. 6B) and BT474 (Fig. 6C) cells decreased ERα activity. Consistent with transient transfection studies, ERα expression was not significantly altered by Fe65 expression (Fig. 6A), supporting a positive effect of Fe65 on the specific activity of endogenous ERα per molecule. It is important to note that, under our assay conditions, the activity of endogenous ERα on transfected EREe1bluc reporter was much weaker than ectopic ERα expressed in HeLa cells, which may explain the less dramatic effects of Fe65 on endogenous ERα in BCa cells observed in reporter analyses, which was nevertheless statistically significant.
FIGURE 6.

Fe65 potentiates estrogen stimulation of endogenous ERα activity in BCa cells. A, MCF-7 cells stably transfected with empty vector (Ctrl) or Fe65 (Fe65 OE Clones) were transiently transfected with 0.3 μg of EREe1bluc and pLENβGal. B and C, T47D (panel B) and BT474 (panel C) cells were transfected with control (siCtrl) or Fe65 (siFe65) siRNA and 24 h later re-transfected with 0.3 μg of EREe1bluc and pLENβGal. Transfected cells were treated with E2, and luciferase activity was determined and normalized as in Fig. 1A. Fe65 overexpression and knockdown as well as its effect on ERα protein expression were monitored by IB analyses with cognate antibodies as indicated.
Consistent with the reporter-based transcriptional analyses, transient transfection of Fe65, but not the PTB2 deletion mutant, into MCF-7 cells increased E2 induction of c-Myc and cyclin D1 expression in a dose-dependent manner (Fig. 7A), showing that the positive effect of Fe65 on endogenous ERα observed in reporter assays was translated into changes in estrogen target gene expression and that the effect on the target genes involved PTB2-mediated complex formation with endogenous ERα. Similarly, stable Fe65 expression increased (Fig. 7B), whereas its knockdown in T47D (Fig. 7C) and BT474 (Fig. 7D) decreased the ability of E2 to induce c-Myc expression. The regulation of estrogen target genes by Fe65 overexpression (Fig. 7E) or knockdown (Fig. 7F) was also detected at mRNA levels, supporting a transcriptional control. The levels of endogenous ERα protein expression were not significantly altered by either Fe65 overexpression or knockdown, suggesting that the positive effect reflected an increase in ERα activity. The analyses identify Fe65 as a positive regulator of the endogenous ERα in BCa cells that exerts its positive effect through PTB2-mediated complex formation with the receptor.
FIGURE 7.

Fe65 potentiates the estrogen stimulation of ERα target gene expression in BCa cells. A, MCF-7 cells were transfected with empty vector (Ctrl), Fe65, or PTB2 deletion mutant and treated with EtOH or 10−8 m E2 for 2 h. Cellular extracts were subjected to IB analyses with the indicated antibodies. The expression levels of cyclin D1 and c-Myc were quantified with ImageJ and normalized with cognate β-actin signals. The normalized signals were divided by the signal of control cells that were transfected with empty vector or scrambled siRNA and treated with EtOH and are shown at the bottom of the blots. B, MCF-7 cells stably transfected with empty vector or Fe65 were treated, and levels of protein expression determined as in panel A. C and D, T47D (panel C) and BT474 (panel D) cells transiently transfected with siCrtl or siFe65 were treated, and levels of protein expression determined as in panel A. E, MCF-7 cells stably transfected with empty vector or Fe65 were treated as in panel A but for 1 h. Total RNA was extracted and subjected to real time RT-PCR analyses. The levels of c-Myc mRNA were normalized with GAPDH and are expressed as -fold of the EtOH control. F, T47D cells transiently transfected with siCrtl or siFe65 were treated as in panel A but for 1 h. Total RNA was extracted and subjected to real time RT-PCR. The levels of cyclin D1 and c-Myc mRNA were normalized with GAPDH and are expressed as -fold of the EtOH controls.
To further characterize the positive effect of Fe65 on endogenous ERα in BCa cells, ChIP assays were performed to test whether Fe65 was recruited to the promoters of defined estrogen target genes and, more importantly, whether changes in levels of Fe65 expression altered the promoter recruitment of the ERα and its coactivators. As shown in Fig. 8A, E2 recruited Fe65 to the promoters of four different estrogen target genes in T47D cells, and the amounts of Fe65 on the promoters were decreased by Fe65 siRNA. Interestingly, Fe65 knockdown also resulted in decreased recruitments of both the ERα and its coactivator AIB1, showing a role of Fe65 in determining the chromatin recruitment of the ERα and its coactivators. Such a role for Fe65 was further supported by the finding that the stable Fe65 expression in MCF-7 cells increased estrogen recruitment of the ERα and AIB1 to target gene promoters (Fig. 8B). More importantly, the ability of TAM to block E2-induced ERα and AIB1 recruitments to the promoters was significantly suppressed by Fe65 stable expression, suggesting a potential role for Fe65 in the development of TAM resistance.
FIGURE 8.

Fe65 is recruited to ERα target gene promoters by estrogens and required for optimal recruitment of the ERα and its coactivators. A, T47D cells were transfected with siCtrl or siFe65 and treated with EtOH or 10−7 m E2 for 45 min. ChIP assays were performed with the indicated antibodies. B, MCF-7 cells stably transfected with empty vector or Fe65 were treated with EtOH, 10−7 m E2, or 10−7 m E2 plus 10−7 m TAM (E2+TAM) for 45 min. ChIP assays were performed with the indicated antibodies. C, MCF-7 cells stably transfected with empty vector or Fe65 were treated with EtOH or 10−8 m E2 for 1 h. Cytoplasm and nucleus proteins were subjected to IB analyses with the indicated antibodies. Hsp60 and HDAC1 blots were included to show the separation of cytoplasmic and nuclear extracts.
The increased ERα and AIB1 recruitment to target gene promoters might be easily explained if Fe65 promoted estrogen-induced ERα nuclear localization. To test this, cellular distribution of the ERα and Fe65 was analyzed with nuclear and cytosolic extracts prepared from control and Fe65 stable clones of MCF-7 cells. As shown in Fig. 8C, the stable Fe65 expression resulted in a significant increase of its presence in both the cytosol and the nucleus but did not alter ERα localization to the nucleus induced by E2, showing a lack of Fe65 effect on ERα nuclear localization. Overall, the data suggest that the positive Fe65 effect on ERα recruitments to target gene promoters is likely due to a role of Fe65 in facilitating the ERα to interact with chromatin.
To test whether the recruitment of Fe65 to the target gene promoters by E2 depended on the ERα, ChIP assays were performed in T47D cells transfected with control or ERα siRNA (Fig. 9). As expected, ERα siRNA decreased ERα protein expression (Fig. 9A) as well as the amounts of the ERα and AIB1 coactivator on the target gene promoters (Fig. 9B, lower panels). The receptor siRNA did not alter the overall levels of Fe65 protein in the cells (Fig. 9A) but decreased its basal and E2-induced recruitment to the promoters of all tested estrogen target genes (Fig. 9B, top panels). The analyses show that Fe65 recruitment to estrogen target gene promoters depends on the ERα. Overall, the ChIP analyses support the existence of a mutual positive regulation between the ERα and Fe65 on their recruitment to estrogen target gene promoters.
FIGURE 9.

Fe65 recruitment to estrogen target gene promoters is ERα-dependent. A, T47D cells were transfected with siCtrl or siERα. Cellular extracts were subjected to IB analyses with indicated antibodies. B, T47D cells were transfected with siCtrl or siERα and treated with EtOH or 10−7 m for 45 min. ChIP assays were performed with indicated antibodies.
Fe65 Promotes Estrogen-induced BCa Cell Growth and Suppresses the Antagonistic Activity of TAM
Given the fact that ERα activity and the expression of c-Myc and cyclin D1 are often associated with cell growth in BCa cells, the positive effects of Fe65 on ERα activity and target gene expression are expected to be translated into a positive effect on BCa cell growth and its stimulation by estrogens. Indeed, our cell growth analyses showed that Fe65 knockdown significantly decreased the ability of E2 to stimulate the growth of T47D (Fig. 10A) and ZR75–1 (Fig. 10B) cells. Consistently, the ability of E2 to stimulate MCF-7 cell growth was significantly stronger in Fe65 stable clones than controls (Fig. 10C), and the increase in estrogen stimulation was positively correlated with Fe65 expression levels.
FIGURE 10.
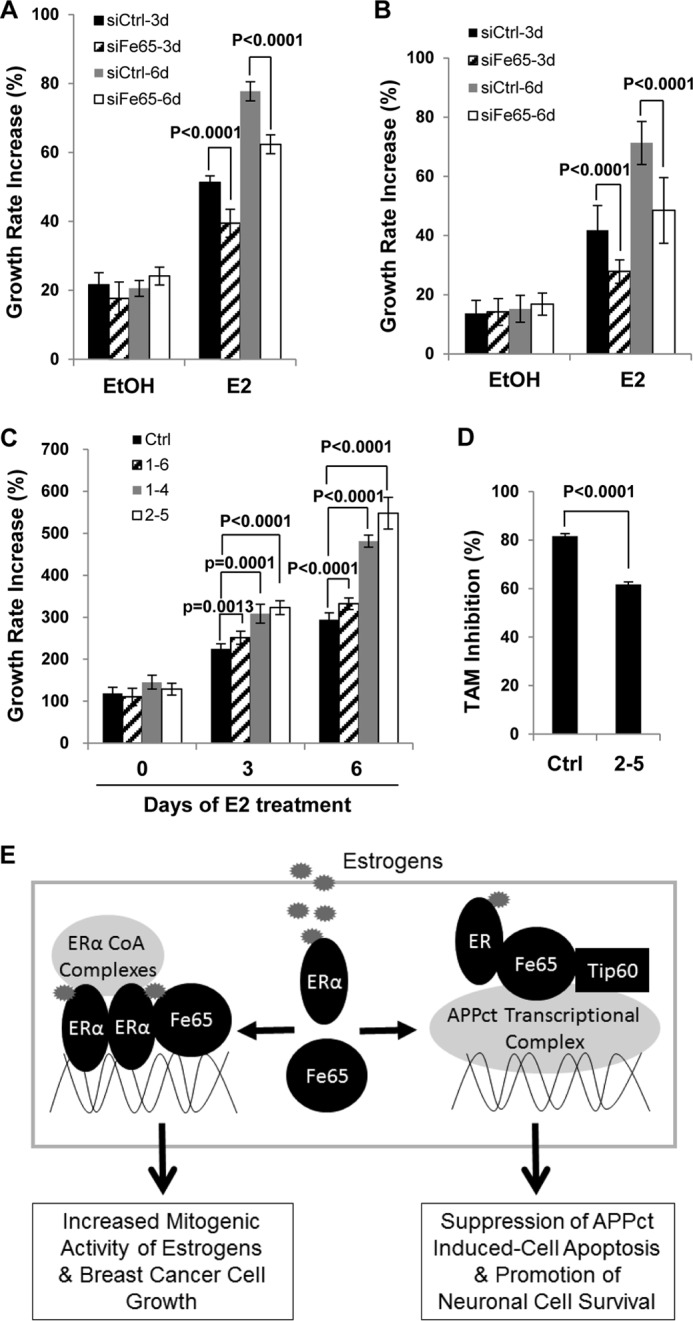
Fe65 promotes estrogen-induced BCa cell growth and suppresses the antagonistic activity of TAM. T47D (A) and ZR75–1 (B) cells were estrogen-starved and transfected with siCtrl or siFe65 and 48 h later re-transfected for another time. 24 h post the second transfection, the cells were plated in 96-well plates and treated with EtOH or 10−8 m E2 for 3 and 6 days. Cell growth was determined in MTT assays. The increase in cell growth rate was calculated by subtracting from and dividing A595 values with A595 of control cells (time zero). C, control MCF-7 and Fe65 OE clone (2–5) were estrogen-starved for 72 h and treated with 10−8 m E2 for 0, 3, and 6 days. E2 treatment was started at different times to allow the growth of all treated cells being measured in MTT assays at the same time. For example, for 0 day treatment, the cells were treated with EtOH for 6 days. The increase in cell growth rate was calculated as in panels A and B. D, control MCF-7 and Fe65 OE clone (2–5) were estrogen-starved for 72 h and treated with either EtOH, 10−7 m E2 or 10−7 m E2 plus 1 μm TAM for 3 days. Cell growth was determined in MTT assays. Percentages of TAM inhibition were calculated by subtracting from the E2-induced increase in growth rate (over day 0) with that induced by E2 plus TAM followed by division with E2-induced increase in growth rate. For all growth analyses, each data point represents 18 samples analyzed in parallel. Each experiment was repeated three times. p values were calculated by Student's t test. The error bars in all growth analyses represent the S.D. E, a model describing the role of Fe65 in estrogen actions in neurons and BCa cells. See the first paragraph of “Discussion” for details.
Consistent with the ChIP assay data presented in Fig. 8B, the ability of TAM to suppress E2-induced cell growth was significantly reduced by stable Fe65 expression (Fig. 10D). In the control clone, TAM suppressed E2-induced cell growth by ∼81.6%, whereas in the Fe65 overexpression clone, it was reduced to 61.7%. The data demonstrate that Fe65 promotes estrogen-induced cell growth and reduces the antiestrogenic activity of TAM in suppressing the estrogen stimulation.
DISCUSSION
Fe65 has been well studied in neuronal cells and in the pathogenesis of Alzheimer disease for its functions in mediating the trafficking and processing of APP as well as its participation in transcriptional regulation (38). Up to now there are essentially no reports about the function of Fe65 in cancer cells. The present studies are the first to clearly establish a role for Fe65 in BCa cells. Multiple lines of evidences are presented to support the novel role of Fe65 as a positive ERα regulator that potentiates estrogen stimulation of BCa cell growth. First, Fe65 bound to the ERα through the PTB2 domain and increased ERα activity in reporter assays in a PTB2-dependent manner. Second, Fe65 expression was detected in breast epithelial cells, and its expression was found to be increased in human BCa cells and breast tumor tissue samples. Third, E2 recruited Fe65 to the promoters of natural estrogen target genes. Finally, Fe65 stable expression increased and its knockdown decreased the activity of endogenous ERα in BCa cells as well as estrogen-induced target gene expression, recruitment of the ERα and coactivators to target gene promoters, and cell growth. Together with studies published previously (16), the present studies extend Fe65 actions from neuronal to BCa cells as presented in Fig. 10E. In neuronal cells, Fe65 brings the ERα to APPct transcriptional complex, allowing estrogens to protect neuronal cells from apoptosis induced by toxicity associated with APP cleavage. In BCa cells, Fe65 binds to the ERα and potentiates its chromatin binding at target gene promoters, which permits Fe65 to act as a stimulator of BCa cell growth. Although it remains to be determined, the increased Fe65 expression in breast tumor tissue samples suggests that the findings about Fe65 in BCa cell lines will most likely reflect an in vivo role for this neuronal adaptor in breast tumorigenesis.
Tip60 is a histone acetyl transferase that binds to PTB1 domain of Fe65 (13). It was also reported to be a coactivator for the ERα (39). It is a legitimate question whether the positive effect of Fe65 on the ERα involves Tip60. Our studies have yielded conflicting data regarding the role of Tip60 in ERα activation by Fe65. In ChIP assays, estrogens recruited Tip60 to ERα target gene promoters, which were enhanced by Fe65 overexpression (data not shown). The data are consistent with the fact that Tip60 is a common interacting protein for Fe65 and ERα. However, in reporter gene analyses, ectopic expression of Tip60, instead of synergizing with Fe65 in stimulating ERα activity as it would be expected for a coactivator, actually decreased the ability of Fe65 to activate the ERα (data not shown). The reporter data are consistent with the fact that APPct did not increase ERα activity or potentiate the positive effect of Fe65 on the ERα (Fig. 1). Tip60 has been shown to be a tumor suppressor of which the expression, particularly the expression in the nucleus, is decreased in breast tumors (40). Interestingly, it has been shown that estrogen-induced c-Myc and cyclin D1 mRNA expression was not affected by Tip60 depletion in MCF-7 cells (41) even though several non-growth related ERα target genes were altered, suggesting a selective involvement of Tip60 in ERα action in BCa cells.
Our data suggest that APPct is not involved in ERα activation by Fe65 (Fig. 1). Questions remain of what signals regulate Fe65 nuclear localization in BCa cells and its positive effect on the ERα. Under overexpression conditions, Fe65-ERα complex formation appeared to be constitutive and not altered by estrogen treatments (data not shown). However, ChIP assays revealed that Fe65 binding to estrogen target gene promoters was induced by E2 (Figs. 8 and 9). The analyses suggest that estrogens are part of the signals operating in BCa cells to regulate the actions of Fe65-ERα complex in the nucleus. Fe65 overexpression increased both cytoplasmic and nuclear Fe65 levels in MCF-7 cells (Fig. 8C), showing that a simple way of promoting nuclear Fe65-ERα activity is to increase overall Fe65 expression, which naturally occurs in breast tumors as suggested by data in Fig. 5, C and D. In addition to APP cleavage, Fe65 phosphorylation had been shown to induce Fe65 nuclear localization (42). Fe65 has also been reported to interact with the C terminus of Notch1 (43). It would be interesting to find out whether these signaling pathways known to operate in BCa cells regulate ERα activation through Fe65.
Although the present studies have focused on the effect of Fe65 on ERα actions, it is important to point out that Fe65 may have a much broad impact on BCa biology beyond estrogen stimulation of cell growth. For example, Fe65 has been described to be required for efficient repair of DNA double-strand breaks, a function that depends on its interaction with Tip60 and APPct (26, 27). There are also reports showing that the phosphorylated H2A.X is higher in Fe65 null cells than in the wild type under genotoxic damages (25) and that the phosphorylation somehow depends on Fe65 accumulation in the nucleus (44–46). Thus, through Fe65-ERα complex formation, estrogens may regulate DNA damage repair in breast epithelial cells. Furthermore, the WW domain of Fe65 is known to bind to Mena (21), through which it regulates the actin cytoskeleton, cell motility, and neuronal growth cone formation (22, 23). Fe65 thus may also control BCa invasion through Mena and allow estrogens to regulate BCa invasion through the Fe65-ERα complex. In this regard it is important to point out that significant amounts of Fe65 protein were detected in the cytoplasm of BCa cells and that Mena splice isoforms regulate BCa invasion (47), which is apparently a cytoplasmic activity mediated through actin filaments.
In summary, the present studies have extended the functions of the Fe65 neuronal adaptor protein to BCa cells and defined a positive role for Fe65 in estrogen actions and BCa cell growth. The existence of Fe65-ERα complex in BCa cells increases the complexity of the mechanisms underlying estrogen actions in BCa cells. For the reason that Fe65 is a multidomain protein that interacts with a variety of proteins with diverse functions, its interaction with the ERα may change the receptor activity both quantitatively and qualitatively by allowing more complex regulation patterns. Our finding that Fe65 expression suppressed the antiestrogenic activity of TAM implicates that Fe65 may be a new molecular target for BCa intervention and that targeted disruption of the ERα-Fe65 interaction may represent a new strategy to overcome TAM resistance.
Acknowledgments
We thank Dr. JoEllen Welsh for the mammary tumor cells and Drs. M. Muyan, L. Corbo, and P. S. Melzer for providing ERα and AIB1 plasmids, respectively. We also thank Mark C. Lloid and Agnieszka Kasprzak and the pathology core facility of Moffitt Cancer Center for the assistance with the TMA analyses.
This work was supported, in whole or in part, by National Institutes of Health Public Health Service Grant R01 CA111334. This work was also supported by an Idea Award from the Breast Cancer Research Program of United States Department of Defense Grant W81XWH-09-1-0574.
- TAM
- tamoxifen
- ERE
- estrogen response element
- MTT
- 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide
- E2
- 17β-estradiol
- IB
- immunoblotting
- IP
- immunoprecipitation
- IHC
- immunohistochemistry
- TMA
- breast tissue microarray
- BCa
- breast cancer
- APP
- amyloid precursor protein
- VDR
- vitamin D receptor
- AR
- androgen receptor.
REFERENCES
- 1. Couse J. F., Korach K. S. (1999) Estrogen receptor null mice. What have we learned and where will they lead us? Endocr. Rev. 20, 358–417 [DOI] [PubMed] [Google Scholar]
- 2. Le Romancer M., Poulard C., Cohen P., Sentis S., Renoir J. M., Corbo L. (2011) Cracking the estrogen receptor's posttranslational code in breast tumors. Endocr. Rev. 32, 597–622 [DOI] [PubMed] [Google Scholar]
- 3. Early Breast Cancer Trialists' Collaborative Group (EBCTCG) (2005) Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival. An overview of the randomised trials. Lancet 365, 1687–1717 [DOI] [PubMed] [Google Scholar]
- 4. Gradishar W. J. (2005) The future of breast cancer. The role of prognostic factors. Breast cancer research and treatment. Breast Cancer Res. Treat. 89, S17–S26 [DOI] [PubMed] [Google Scholar]
- 5. Tsai M. J., O'Malley B. W. (1994) Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu. Rev. Biochem. 63, 451–486 [DOI] [PubMed] [Google Scholar]
- 6. Mangelsdorf D. J., Thummel C., Beato M., Herrlich P., Schütz G., Umesono K., Blumberg B., Kastner P., Mark M., Chambon P., Evans R. M. (1995) The nuclear receptor superfamily. The second decade. Cell 83, 835–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Umayahara Y., Kawamori R., Watada H., Imano E., Iwama N., Morishima T., Yamasaki Y., Kajimoto Y., Kamada T. (1994) Estrogen regulation of the insulin-like growth factor I gene transcription involves an AP-1 enhancer. J. Biol. Chem. 269, 16433–16442 [PubMed] [Google Scholar]
- 8. Savouret J. F., Rauch M., Redeuilh G., Sar S., Chauchereau A., Woodruff K., Parker M. G., Milgrom E. (1994) Interplay between estrogens, progestins, retinoic acid, and AP-1 on a single regulatory site in the progesterone receptor gene. J. Biol. Chem. 269, 28955–28962 [PubMed] [Google Scholar]
- 9. Lonard D. M., O'Malley B. W. (2006) The expanding cosmos of nuclear receptor coactivators. Cell 125, 411–414 [DOI] [PubMed] [Google Scholar]
- 10. Shang Y., Hu X., DiRenzo J., Lazar M. A., Brown M. (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103, 843–852 [DOI] [PubMed] [Google Scholar]
- 11. Perissi V., Jepsen K., Glass C. K., Rosenfeld M. G. (2010) Deconstructing repression. Evolving models of co-repressor action. Nat. Rev. Genet 11, 109–123 [DOI] [PubMed] [Google Scholar]
- 12. Smith C. L., O'Malley B. W. (2004) Coregulator function. A key to understanding tissue specificity of selective receptor modulators. Endocr. Rev. 25, 45–71 [DOI] [PubMed] [Google Scholar]
- 13. Cao X., Südhof T. C. (2001) A transcriptionally (correction of transcriptively) active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293, 115–120 [DOI] [PubMed] [Google Scholar]
- 14. Zambrano N., Minopoli G., de Candia P., Russo T. (1998) The Fe65 adaptor protein interacts through its PID1 domain with the transcription factor CP2/LSF/LBP1. J. Biol. Chem. 273, 20128–20133 [DOI] [PubMed] [Google Scholar]
- 15. Kim H. S., Kim E. M., Lee J. P., Park C. H., Kim S., Seo J. H., Chang K. A., Yu E., Jeong S. J., Chong Y. H., Suh Y. H. (2003) C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3β expression. FASEB J. 17, 1951–1953 [DOI] [PubMed] [Google Scholar]
- 16. Bao J., Cao C., Zhang X., Jiang F., Nicosia S. V., Bai W. (2007) Suppression of β-amyloid precursor protein signaling into the nucleus by estrogens mediated through complex formation between the estrogen receptor and Fe65. Mol. Cell. Biol. 27, 1321–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McLoughlin D. M., Miller C. C. (2008) The FE65 proteins and Alzheimer's disease. J. Neurosci. Res. 86, 744–754 [DOI] [PubMed] [Google Scholar]
- 18. Telese F., Bruni P., Donizetti A., Gianni D., D'Ambrosio C., Scaloni A., Zambrano N., Rosenfeld M. G., Russo T. (2005) Transcription regulation by the adaptor protein Fe65 and the nucleosome assembly factor SET. EMBO Rep. 6, 77–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trommsdorff M., Borg J. P., Margolis B., Herz J. (1998) Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J. Biol. Chem. 273, 33556–33560 [DOI] [PubMed] [Google Scholar]
- 20. Hoe H. S., Magill L. A., Guenette S., Fu Z., Vicini S., Rebeck G. W. (2006) FE65 interaction with the apoE receptor ApoEr2. J. Biol. Chem. 281, 24521–24530 [DOI] [PubMed] [Google Scholar]
- 21. Ermekova K. S., Zambrano N., Linn H., Minopoli G., Gertler F., Russo T., Sudol M. (1997) The WW domain of neural protein FE65 interacts with proline-rich motifs in Mena, the mammalian homolog of Drosophila enabled. J. Biol. Chem. 272, 32869–32877 [DOI] [PubMed] [Google Scholar]
- 22. Sabo S. L., Ikin A. F., Buxbaum J. D., Greengard P. (2001) The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J. Cell Biol. 153, 1403–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sabo S. L., Ikin A. F., Buxbaum J. D., Greengard P. (2003) The amyloid precursor protein and its regulatory protein, FE65, in growth cones and synapses in vitro and in vivo. J. Neurosci. 23, 5407–5415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu Q., Hearn M. G., Jin L. W., Bressler S. L., Martin G. M. (1999) Alternatively spliced isoforms of FE65 serve as neuron-specific and non-neuronal markers. J. Neurosci. Res. 58, 632–640 [DOI] [PubMed] [Google Scholar]
- 25. Minopoli G., Stante M., Napolitano F., Telese F., Aloia L., De Felice M., Di Lauro R., Pacelli R., Brunetti A., Zambrano N., Russo T. (2007) Essential roles for Fe65, Alzheimer amyloid precursor-binding protein, in the cellular response to DNA damage. J. Biol. Chem. 282, 831–835 [DOI] [PubMed] [Google Scholar]
- 26. Stante M., Minopoli G., Passaro F., Raia M., Vecchio L. D., Russo T. (2009) Fe65 is required for Tip60-directed histone H4 acetylation at DNA strand breaks. Proc. Natl. Acad. Sci. U.S.A. 106, 5093–5098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murr R., Loizou J. I., Yang Y. G., Cuenin C., Li H., Wang Z. Q., Herceg Z. (2006) Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 8, 91–99 [DOI] [PubMed] [Google Scholar]
- 28. Li X., Huang J., Yi P., Bambara R. A., Hilf R., Muyan M. (2004) Single-chain estrogen receptors (ERs) reveal that the ERα/β heterodimer emulates functions of the ERα dimer in genomic estrogen signaling pathways. Mol. Cell. Biol. 24, 7681–7694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Le Romancer M., Treilleux I., Leconte N., Robin-Lespinasse Y., Sentis S., Bouchekioua-Bouzaghou K., Goddard S., Gobert-Gosse S., Corbo L. (2008) Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol. Cell 31, 212–221 [DOI] [PubMed] [Google Scholar]
- 30. Anzick S. L., Kononen J., Walker R. L., Azorsa D. O., Tanner M. M., Guan X. Y., Sauter G., Kallioniemi O. P., Trent J. M., Meltzer P. S. (1997) AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 277, 965–968 [DOI] [PubMed] [Google Scholar]
- 31. Yang Y., Tse A. K., Li P., Ma Q., Xiang S., Nicosia S. V., Seto E., Zhang X., Bai W. (2011) Inhibition of androgen receptor activity by histone deacetylase 4 through receptor SUMOylation. Oncogene 30, 2207–2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee H., Bai W. (2002) Regulation of estrogen receptor nuclear export by ligand-induced and p38-mediated receptor phosphorylation. Mol. Cell. Biol. 22, 5835–5845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang F., Li P., Fornace A. J., Jr., Nicosia S. V., Bai W. (2003) G2/M arrest by 1,25-dihydroxyvitamin D3 in ovarian cancer cells mediated through the induction of GADD45 via an exonic enhancer. J. Biol. Chem. 278, 48030–48040 [DOI] [PubMed] [Google Scholar]
- 34. Li P., Nicosia S. V., Bai W. (2001) Antagonism between PTEN/MMAC1/TEP-1 and androgen receptor in growth and apoptosis of prostatic cancer cells. J. Biol. Chem. 276, 20444–20450 [DOI] [PubMed] [Google Scholar]
- 35. Kasiappan R., Shen Z., Tse A. K., Jinwal U., Tang J., Lungchukiet P., Sun Y., Kruk P., Nicosia S. V., Zhang X., Bai W. (2012) 1,25-Dihydroxyvitamin D3 suppresses telomerase expression and human cancer growth through microRNA-498. J. Biol. Chem. 287, 41297–41309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakaya T., Suzuki T. (2006) Role of APP phosphorylation in FE65-dependent gene transactivation mediated by AICD. Genes Cells 11, 633–645 [DOI] [PubMed] [Google Scholar]
- 37. Shang Y., Brown M. (2002) Molecular determinants for the tissue specificity of SERMs. Science 295, 2465–2468 [DOI] [PubMed] [Google Scholar]
- 38. Bórquez D. A., González-Billault C. (2012) The amyloid precursor protein intracellular domain-Fe65 multiprotein complexes. A challenge to the amyloid hypothesis for Alzheimer's disease? Int. J. Alzheimers Dis. 2012, 353145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brady M. E., Ozanne D. M., Gaughan L., Waite I., Cook S., Neal D. E., Robson C. N. (1999) Tip60 is a nuclear hormone receptor coactivator. J. Biol. Chem. 274, 17599–17604 [DOI] [PubMed] [Google Scholar]
- 40. Gorrini C., Squatrito M., Luise C., Syed N., Perna D., Wark L., Martinato F., Sardella D., Verrecchia A., Bennett S., Confalonieri S., Cesaroni M., Marchesi F., Gasco M., Scanziani E., Capra M., Mai S., Nuciforo P., Crook T., Lough J., Amati B. (2007) Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 448, 1063–1067 [DOI] [PubMed] [Google Scholar]
- 41. Jeong K. W., Kim K., Situ A. J., Ulmer T. S., An W., Stallcup M. R. (2011) Recognition of enhancer element-specific histone methylation by TIP60 in transcriptional activation. Nat. Struct. Mol. Biol. 18, 1358–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee E. J., Chun J., Hyun S., Ahn H. R., Jeong J. M., Hong S. K., Hong J. T., Chang I. K., Jeon H. Y., Han Y. S., Auh C. K., Park J. I., Kang S. S. (2008) Regulation Fe65 localization to the nucleus by SGK1 phosphorylation of its Ser-566 residue. BMB Rep. 41, 41–47 [DOI] [PubMed] [Google Scholar]
- 43. Fischer D. F., van Dijk R., Sluijs J. A., Nair S. M., Racchi M., Levelt C. N., van Leeuwen F. W., Hol E. M. (2005) Activation of the Notch pathway in Down syndrome. Cross-talk of Notch and APP. FASEB J. 19, 1451–1458 [DOI] [PubMed] [Google Scholar]
- 44. Nakaya T., Kawai T., Suzuki T. (2008) Regulation of FE65 nuclear translocation and function by amyloid β-protein precursor in osmotically stressed cells. J. Biol. Chem. 283, 19119–19131 [DOI] [PubMed] [Google Scholar]
- 45. Nakaya T., Kawai T., Suzuki T. (2009) Metabolic stabilization of p53 by FE65 in the nuclear matrix of osmotically stressed cells. FEBS J. 276, 6364–6374 [DOI] [PubMed] [Google Scholar]
- 46. Kawai T., Nakaya T., Suzuki T. (2010) Roles of the intramolecular regions of Fe65 in its trans-accumulation and in p53 stabilization in the nuclear matrix of osmotically stressed cells. FEBS Lett. 584, 765–769 [DOI] [PubMed] [Google Scholar]
- 47. Di Modugno F., Iapicca P., Boudreau A., Mottolese M., Terrenato I., Perracchio L., Carstens R. P., Santoni A., Bissell M. J., Nisticò P. (2012) Splicing program of human MENA produces a previously undescribed isoform associated with invasive, mesenchymal-like breast tumors. Proc. Natl. Acad. Sci. U.S.A. 109, 19280–19285 [DOI] [PMC free article] [PubMed] [Google Scholar]