

Background: Isomerase activity for Synechocystis apocarotenoid oxygenase (ACO) has not been verified biochemically.
Results: ACO exclusively produces all-trans-retinal from all-trans-apo-8′-carotenol and is inhibited by linear polyoxyethylene detergents.
Conclusion: ACO cleaves but does not isomerize all-trans-8′-apocarotenol.
Significance: This study clarifies the activity of and influence of detergent on ACO, a classical non-isomerizing member of the carotenoid cleavage enzyme family.
Keywords: Carotene, Carotenoid, Enzyme Catalysis, Enzyme Inactivation, Enzyme Mechanisms, Enzyme Structure, Vitamin A
Abstract
Carotenoid cleavage enzymes (CCEs) constitute a group of evolutionarily related proteins that metabolize a variety of carotenoid and non-carotenoid substrates. Typically, these enzymes utilize a non-heme iron center to oxidatively cleave a carbon-carbon double bond of a carotenoid substrate. Some members also isomerize specific double bonds in their substrates to yield cis-apocarotenoid products. The apocarotenoid oxygenase from Synechocystis has been hypothesized to represent one such member of this latter category of CCEs. Here, we developed a novel expression and purification protocol that enabled production of soluble, native ACO in quantities sufficient for high resolution structural and spectroscopic investigation of its catalytic mechanism. High performance liquid chromatography and Raman spectroscopy revealed that ACO exclusively formed all-trans products. We also found that linear polyoxyethylene detergents previously used for ACO crystallization strongly inhibited the apocarotenoid oxygenase activity of the enzyme. We crystallized the native enzyme in the absence of apocarotenoid substrate and found electron density in the active site that was similar in appearance to the density previously attributed to a di-cis-apocarotenoid intermediate. Our results clearly demonstrated that ACO is in fact a non-isomerizing member of the CCE family. These results indicate that careful selection of detergent is critical for the success of structural studies aimed at elucidating structures of CCE-carotenoid/retinoid complexes.
Introduction
Carotenoids are a ∼700 member group of diverse, fat-soluble isoprenoid compounds (mostly C40) with up to 15 conjugated double bonds. The most widespread color pigments in nature, carotenoids exist in all kingdoms of life. The conjugated polyene chain of these compounds endows them with important biochemical properties including visible light absorption and antioxidant activity (1–3).
Apocarotenoids are biologically important molecules generated by the oxidative cleavage of carotenoids at specific double bond sites. A group of proteins called carotenoid cleavage enzymes (CCEs)4 are the main enzymes responsible for catalyzing cleavage reactions (4). These enzymes employ a non-heme iron cofactor to activate molecular oxygen for insertion into a carbon-carbon double bond of the carotenoid polyene. The issue of whether these enzymes are mono- or dioxygenases remains contentious and data supporting both cleavage mechanisms exist in literature for different family members (5, 6).
Carotenoid cleavage activity was first identified in the 1930s (7), but it took another 70 years before the first CCE called VP14 was molecularly identified (8). Plants express two CCE subgroups: carotenoid cleavage dioxygenases metabolize various (apo)carotenoids, and their products are important pigments, flavor molecules, and signaling compounds (9, 10); 9-cis-epoxycarotenoid dioxygenases specifically act on 9-cis-epoxycarotenoids to produce xanthoxin, the immediate precursor for abscisic acid biosynthesis (8, 11). Cyanobacterial CCEs produce retinal chromophore required for the formation of type 1 holo-opsins (12, 13). Two human CCEs, BCO1 and BCO2, are key enzymes involved in dietary carotenoid metabolism. BCO1 symmetrically cleaves β,β-carotene and other pro-retinoid carotenoids to yield all-trans-retinal, which is a key intermediate in the production of visual chromophore in the retina, as well as for generation of the signaling molecule all-trans-retinoic acid (14–18). BCO2 is more promiscuous in its substrate preference and is thought to regulate levels of bodily carotenoids (19, 20), which, in excess, can be toxic (21, 22).
Interestingly, certain members of the CCE family can also catalyze double bond isomerization concurrently with carotenoid/retinoid cleavage. Such activity was first identified in a retinal pigment epithelium-specific protein named RPE65, which is the retinoid isomerase of the visual cycle (16, 23, 24). This enzyme is unique among CCEs in that it catalyzes the non-oxidative cleavage and isomerization of all-trans-retinyl esters to form 11-cis-retinol in an unusual nucleophilic substitution reaction (25). The elucidation of RPE65, as the visual cycle isomerase, stimulated numerous efforts to understand catalysis by CCEs, in particular the molecular mechanisms of non-photochemical polyene geometrical isomerization (26–35). Research in this area led to the discovery that CCEs besides RPE65 also isomerize their substrates during oxidative cleavage (4, 36). In some insects, CCEs simultaneously cleave and isomerize all-trans-zeaxanthin to yield all-trans- and 11-cis-3-hydroxyretinal, the latter being used to synthesize visual pigments under dark conditions (37–39). Cleavage of 9-cis-β,β-carotene by human BCO1 resulted in a lower than expected production of the 9-cis-retinal product, indicating that this enzyme can catalyze cis to trans isomerization (40, 41). Isomerase activity has also been posited for a cyanobacterial member of the CCE family called apocarotenoid oxygenase (ACO) (42).
ACO was the first CCE enzyme to have its three-dimensional structure determined by x-ray crystallography, which revealed a 7-bladed β-propeller architecture with a 4 His-coordinated iron cofactor at its center as the basic CCE-fold (42). In this structural study, a kinked electron density feature was observed in the active site of iron-reconstituted crystals obtained from mother liquor that contained 3-hydroxy-8′-apocarotenol substrate and the detergent tetraethylene glycol monooctyl ether (C8E4). This density was attributed to bound substrate but a good fit of the apocarotenoid to the map could only be obtained if the compound was converted to a 13,14′-di-cis configuration. Notably, density for the characteristic β-ionone moiety of the molecule was not observed, which made the identity of the compound giving rise to the density uncertain (10, 42). The putative di-cis configuration led to the proposal that ACO isomerizes and cleaves its natural apocarotenoid substrate to generate 13-cis-retinal. However, this activity was not biochemically verified.
To date, crystal structures of ACO, RPE65, and VP14 have been determined (Fig. 1A) but ACO is the only one for which a purported intact enzyme substrate complex has been structurally described (33, 42, 43). The bound apocarotenoid model has been used in several studies aimed at elucidating the mechanisms of carotenoid cleavage and carotenoid/retinoid isomerization (30, 35). As a cyanobacterial enzyme, ACO is an ancient, archetypical CCE that can be regarded as a primitive scaffold onto which additional or modified structural elements and/or enzymatic activities have been added over the course of evolution. The possibility that (apo)carotenoid isomerase activity was acquired early in the evolution of CCEs is intriguing and has important implications in understanding the mechanism of isomerization by other members of this family, such as the visual cycle retinoid isomerase RPE65 (Fig. 1B). Therefore, it was of interest to advance our understanding of ACO biochemistry and its catalytic mechanism of apocarotenoid oxidation and isomerization.
FIGURE 1.

CCE substrate binding pockets and proposed isomerase activity. A, clipped views of the substrate binding clefts of Synechocystis ACO (PDB 2BIW), bovine RPE65 (PDB 3FSN), and maize VP14 (PDB 3NPE). Arrows indicate substrate entry sites for each CCE. The iron centers are shown as orange spheres. B, RPE65, the all-trans- to 11-cis-retinoid isomerase of the vertebrate visual cycle, is an atypical member of the CCE family that catalyzes a coupled ester cleavage/isomerization reaction rather than oxidative cleavage. NinaB, the sole CCE expressed in insects, cleaves and isomerizes zeaxanthin and related carotenoids to generate all-trans- and 11-cis-retinoids in a ∼1:1 ratio. ACO, a prototypical CCE expressed in cyanobacteria, was suggested to possess both oxidative cleavage and carotenoid isomerase activity. Red wavy lines indicate the location of the scissile bond for each cleavage reaction. Dashed double bonds in each substrate indicate known or potential sites of geometric isomerization. Percentages indicate yields for each product.
Here, we assessed the ACO isomerase hypothesis using a combination of biochemical and structural approaches. To facilitate these studies, we developed a novel expression and purification protocol for ACO that overcame the difficulties associated with previously described refolding methods and allowed recombinant production of native, soluble ACO in Escherichia coli in quantities sufficient for biochemical, structural, and spectroscopic investigations. Our data highlighted the necessity of careful detergent selection in structural studies of CCEs and the binding mode of their substrates.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
The bacterial expression plasmid pET3a containing the coding sequence of ACO (Diox1, GenBankTM BAA18428.1) from Synechocystis sp. PCC 6803 was transformed into the T7 express BL21 E. coli strain (New England Biolabs, Ipswich, MA). Cells were grown at 37 °C to an A600 nm of ∼0.6 at which time protein expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (Roche Applied Science) to a final concentration of 10 μm. After a 4-h incubation at 28 °C cells were collected by centrifugation and either flash-frozen and stored at −80 °C or used immediately.
All purification procedures were carried out at 4 °C. Harvested cells were lysed by three passes through a French press in lysis buffer consisting of 25 mm HEPES-NaOH, pH 7.0. The lysate was clarified by centrifugation at 186,000 × g for 30 min. Solid ammonium sulfate powder (U.S. Biochemical Corp., Cleveland, OH) was slowly added within 1 h to the supernatant with continuous stirring to obtain 20, 30, 40, or 50% saturated solutions. Protein precipitation usually occurred within 40 min depending on the ammonium sulfate concentration. The suspension then was stirred for an additional 1 h. The suspension from 40% saturated solution was centrifuged at 46,000 × g for 20 min, the supernatant was discarded and the pellet was resuspended in lysis buffer. The sample was gently rocked for 2 h at 4 °C to allow dissolution of the pellet and then centrifuged at 186,000 × g for 30 min to remove any remaining debris. The supernatant was then loaded onto a 120-ml Superdex 200 gel filtration column (GE Healthcare) equilibrated with a buffer consisting of 25 mm HEPES-NaOH, pH 7.0, and 1 mm dithiothreitol. Fractions containing pure, enzymatically active ACO were pooled, concentrated 20 mg/ml, flash frozen in liquid nitrogen, and stored at −80 °C for further use.
Crystallization
For crystallization, ACO samples purified by the above method were loaded onto a 25-ml Superdex 200 gel filtration column (GE Healthcare) equilibrated with buffer containing 25 mm HEPES-NaOH, pH 7.0, 1 mm dithiothreitol, and 0.8% (w/v) hexaethylene glycol monooctyl ether (C8E6). ACO eluted in a single symmetrical peak at ∼12 ml. The fractions were pooled and concentrated to 10 mg/ml. Crystallization was carried out by the hanging drop vapor diffusion method by mixing 1 μl of purified ACO with 1 μl of reservoir buffer containing 0.1 m BTP-HCl, pH 6.0, 22–23% (w/v) PEG 3350, 0.2 m NH4Cl, and 1 mm MnCl2. To crystallize ACO in Triton X-100, 0.02% (w/v) of this detergent was used instead of C8E6 in the gel filtration buffer. Crystallization conditions for the sample containing Triton X-100 were the same as those for samples prepared in C8E6. Drops were prepared at room temperature and then incubated at 8 °C. Crystals with a tapered, rod-shaped morphology typically appeared within 3–4 days. Mature crystals were cryoprotected by soaking in the reservoir solution and flash cooled in liquid nitrogen before x-ray exposure.
Enzymatic Assay and High Performance Liquid Chromatography (HPLC) Analysis
The all-trans-8′-apocarotenol substrate was generated by NaBH4 reduction of all-trans-8′-apocarotenal (Sigma) in ethanol. Excess NaBH4 was eliminated by addition of water, and the apocarotenoid product was extracted with hexane. The solvent was removed in a SpeedVac, and the all-trans-8′-apocarotenol was redissolved in ethanol. The concentration of product was spectrophotometrically determined using a molar extinction coefficient of 120,000 m−1 cm−1 at 425 nm (44). Enzymatic activity of the ACO sample was assayed according to a previously published method with some modifications (12). The enzyme was sensitive to the Triton X-100 concentration and displayed the highest activity at 0.05% (w/v), which was the concentration used for enzymatic assays. Two μl of purified ACO at a concentration of 1 mg/ml was added to 200 μl of reaction buffer consisting of 20 mm BisTris HCl, pH 7.0, and 0.05% (w/v) Triton X-100. All-trans-8′-apocarotenol in ethanol was then added to initiate the reaction. The reaction mixture was incubated in a Thermomixer (Eppendorf, Hamburg, Germany) at 28 °C with 500 rpm shaking for 3 min and then quenched by addition of 300 μl of methanol. To transform the aldehyde products into oximes, 100 μl of 2 m hydroxylamine, pH 7.0, was added, and the mixture was incubated for 5 min at room temperature. The products and remaining substrate were extracted in 500 μl of hexane and analyzed by HPLC on a ZORBAX SIL (5 μm, 4.6 × 250 mm) normal phase column (Agilent, Santa Clara, CA). One-hundred μl of hexane phase extract was injected into the column and eluted isocratically with hexane/ethyl acetate (4:1, v/v) at a flow rate of 1.4 ml/min. The assays were carried out in both ambient light and in a dark room to assess the occurrence of photoisomerization reactions.
For analysis of ACO steady-state kinetics, apocarotenol substrate was added to the reaction tubes to achieve final concentrations ranging from 5 to 200 μm. A burst phase in the kinetics was observed over the first 15 s of the reaction followed by a linear phase up to 2 min for all substrate concentrations tested. Therefore, the reactions were quenched at 15 s, 30 s, 45 s, and 1 min for each substrate concentration, and initial velocities were derived by linear regression. Reactions were performed in triplicate for each substrate concentration. The all-trans-retinal product was converted to the oxime derivative prior to analysis by HPLC. Analysis of the kinetic data were performed with SigmaPlot (Systat Software, Inc., San Jose, CA). The effects of detergents on ACO activity were evaluated by dose-response assays performed in the presence of 0.05% (w/v) Triton X-100.
Purification and Mass Spectrometric Analysis of Apocarotenoid Product
The apocarotenoid cleavage reaction was performed with 200 μl of reaction mixture containing 10 mg/ml of ACO. The reaction was quenched after a 30-min incubation in the dark, and apocarotenoids were extracted with hexane/diethyl ether in a 1:4 (v/v) ratio without prior derivatization with hydroxyamine. Solvent was removed by evaporation in a SpeedVac. The sample was redissolved in hexane/ethyl acetate (4:1, v/v) and subjected to HPLC analysis. A hexane/ethyl acetate (4:1, v/v) mobile phase at a flow rate of 1.4 ml/min was used for the separation protocol consisting of: 1) hexane/ethyl acetate (4:1, v/v) for 12 min; 2) a 20–100% linear ethyl acetate gradient developed over 2 min; and 3) 100% ethyl acetate for 10 min. The peak that eluted at ∼17 min was collected and analyzed by LC-MS. The sample was re-injected into a Zorbax Sil (5 μm, 4.6 × 250 mm) normal phase HPLC column (Agilent) and eluted with a mixture of hexane/ethyl acetate in a 1:4 (v/v) ratio. The eluate was directed into an APCI source of an LXQ linear ion trap mass spectrometer (Thermo Scientific). Mass spectra were analyzed with the Xcalibur 2.0.7 software package.
Raman Spectroscopy
For the Raman spectroscopic study, 100 μl of 2.5 mg/ml of purified ACO sample in 25 mm Tris-HCl, pH 8.0, were mixed with an equal volume of buffer containing 25 mm Tris-HCl, pH 8.0, 0.1% (w/v) Triton X-100, and 50 μm all-trans-8′-apocarotenol. The reaction was incubated in a Thermomixer at 28 °C with 500 rpm shaking at times ranging from 5 s to 60 min and then terminated by submerging the tube into liquid nitrogen. Experiments were performed in the dark or under ambient light. Frozen samples were lyophilized in the dark overnight, and Raman spectra were recorded from the resulting powders. Ab initio quantum mechanical calculations of the Raman scattering parameters for substrate and potential products/intermediates were performed on the Case Western Reserve University high performance computing cluster using Gaussian 03 (45).
X-ray Data Collection, Structure Determination, Refinement, and Analysis
Diffraction data were collected at APS ID-24-C and NSLS X29 beamlines. Data for crystals grown in the presence of MnCl2 were collected at wavelengths above and below the iron K-edge to assess the active site iron occupancy, whereas other data sets were obtained at wavelengths where x-ray flux was optimal. Data sets were processed with XDS (46). The crystals belonged to space group P212121 and were isomorphous to the previously reported ACO crystal structure (PDB accession code 2BIW) with four monomers in the asymmetric unit (42). Most crystals examined suffered from epitaxial twinning evident from the presence of two distinct lattices in the diffraction pattern that were rotated 180° about the a − b axis with respect to each other. In many cases the two lattices were sufficiently well resolved that their associated intensities could be separately indexed, integrated, and then scaled together. Structures were determined either by direct refinement or by molecular replacement using the previously determined Synechocystis ACO structure (42) as the starting model in the program Phaser (47). Initial models were then subjected to multiple rounds of manual model rebuilding and updating in Coot (48) followed by restrained refinement in the program Refmac (49). Refmac input files were prepared with the CCP4 interface (50). Bulk solvent parameters were determined using the “solvent optimize” keyword in Refmac and fixed during subsequent rounds of refinement. Non-crystallographic symmetry restraints were applied during refinement and gradually loosened or omitted as the model converged. For the “C8E6 ACO” structure, external distance restraints were applied to the Fe-Nϵ bonds during refinement to enforce a length of ∼2.15 Å. Translation libration screw refinement of the atomic B-factors (one translation libration screw group per monomer) was performed near the end of the refinement, which further reduced Rfree (51). The stereochemical quality of the model was assessed with the Molprobity server (52). Anomalous difference maps were computed using the program ANODE (53). Conformational differences between structures were assessed using the program ESCET (54). All structural figures were prepared with PyMOL (Schrödinger).
RESULTS
ACO Purification and Enzymatic Analysis
The previous structural study of ACO used a protein sample generated by refolding recombinant protein expressed in E. coli inclusion bodies (42). ACO has also been successfully expressed as a soluble GST fusion protein in E. coli (12). In both cases, the protein yields were not well documented, and multiple purification steps were required to achieve a homogeneous protein preparation. Thus, we sought to develop a novel, efficient expression and purification method that would generate large quantities of soluble, native enzyme for structural and biophysical studies.
Initially, we observed that native, untagged ACO tends to form large amounts of inclusion bodies when expressed under standard conditions (37 °C, 0.3 mm isopropyl 1-thio-β-d-galactopyranoside) in E. coli. However, reducing the expression temperature as well as the isopropyl 1-thio-β-d-galactopyranoside concentration dramatically increased the level of soluble ACO without compromising the total protein yield. Several types of chromatography media were tested for purification of ACO from the supernatant. Interestingly, ACO does not readily bind to common ion exchange or hydrophobic interaction chromatography media under the conditions tested, which prompted us to consider alternative purification methods. We found that ACO could be semiselectively precipitated from the supernatant by ammonium sulfate fractionation. As shown in Fig. 2A, ACO precipitates in a stepwise manner with increasing salt concentration, whereas many contaminants remain soluble. A 40% saturated ammonium sulfate solution was found to be optimal for the fractionation. The precipitate from this step was redissolved and then further purified by gel-filtration chromatography. A symmetric peak at ∼73 ml, corresponding to a mass intermediate between an ACO monomer and dimer that contained apparently pure ACO was collected (Fig. 2B). A portion of ACO was aggregated and appeared in the void volume but was not used for further experiments. The typical yield was 3–6 mg of purified protein per liter of E. coli culture (Table 1).
FIGURE 2.

Purification, enzymatic, and spectroscopic properties of native ACO. A, SDS-PAGE analysis of fractions precipitated by ammonium sulfate at different percent saturation levels. Proteins were separated on a 4–20% BisTris/glycine gradient gel and visualized by Coomassie R-250 staining. The arrow indicates the position, at about 54 kDa, where ACO migrates. B, the 40% saturation precipitate shown in panel A was redissolved and further purified by gel filtration chromatography. Protein eluting within the peak indicated by an arrow was pooled, concentrated, and used for experiments. The purity of the final sample was evaluated by SDS-PAGE followed by Coomassie R-250 staining (inset). C, initial velocity versus the all-trans-8′-apocarotenol concentration curve for the purified ACO sample showing typical Michaelis-Menten kinetics. Km and kcat values derived from the curve are shown in the inset. D, top, the UV-visible absorbance spectrum of a purified ACO sample at a concentration of 1 mg/ml (upper panel) was recorded immediately after the purification. Bottom, HPLC analyses of the purified protein sample. Hexane was used to extract any possible apo/carotenoids from 200 μl of purified ACO at 10 mg/ml. Traces a and b were monitored at 360 nm and traces c and d were monitored at 425 nm. Negative control samples in which the protein sample was omitted from the extraction procedure are shown in traces b and d. No apo/carotenoids were detectable at the wavelengths tested in this assay.
TABLE 1.
Purification of ACO from 6 liters of E. coli culture
| Step | Volume | Activitya | Protein concentration | Total activity | Specific activity | Recovery |
|---|---|---|---|---|---|---|
| ml | units/ml | mg/ml | units | units/mg | % | |
| Supernatant | 120 | 0.18 | 10 | 21.6 | 0.02 | 100 |
| (NH4)2SO4 fraction | 12 | 1.6 | 26 | 19.2 | 0.06 | 89 |
| Superdex 200 | 11 | 1.4 | 3 | 15.5 | 0.47 | 72 |
a One unit is defined as the amount of enzyme that causes the appearance of 1 μmol of all-trans-retinal product per min at 28 °C.
Our previous x-ray absorption spectroscopy studies on this as-isolated sample suggested that the ACO catalytic iron was in the ferrous state (32). To confirm that the as-isolated sample obtained by our protocol was enzymatically active, we measured its steady-state kinetic parameters. Values derived from a Michaelis-Menten plot were similar to those previously reported for ACO expressed as a GST fusion protein (12) (Fig. 2C). The UV-visible absorbance spectrum contained a single peak centered at ∼280 nm confirming that no (apo)carotenoids or other chromophores co-purified with ACO (Fig. 2D, upper panel). Additionally, HPLC analysis of a hexane extract of the purified ACO did not reveal any detectable co-purified carotenoids/retinoids as evidenced by the absence of peaks in the 360 and 425 nm chromatograms (Fig. 2D, lower panel).
HPLC Analysis of the Cleavage Products
The proposal that ACO could possess isomerase activity was based on crystallographic data that suggested the presence of a 3-hydroxy-13,14′-di-cis-8′-apocarotenol intermediate in the ACO active site. However, this proposal was not further evaluated by rigorous biochemical studies. We reasoned that 13-cis-retinal should be a reaction product if the ACO isomerase hypothesis is correct (Fig. 1B). Indeed, a trace amount of 13-cis-retinal oxime was an observed ACO reaction product in a prior report (12). Consistent with this report, our HPLC analyses of retinal products generated from all-trans-8′-apocarotenol by ACO showed that the vast majority remained in an all-trans configuration (Fig. 3A, peak b), whereas only traces of 13-cis-retinal oxime were observed (Fig. 3A, peak a). To evaluate the possibility that 13-cis-retinal could be generated by photoisomerization of the all-trans-retinal product we performed the activity assay and HPLC analysis under dim light. Under these conditions 13-cis-retinal oxime was no longer detected, and all-trans-retinal oxime was the sole cleavage product (Fig. 3A), indicating that 13-cis-retinal oxime was generated by photoisomerization. To exclude the possibility that 13-cis-retinal could be isomerized to all-trans-retinal during its conversion to the oxime derivative, we also directly analyzed the retinal products by HPLC. In these experiments, photoisomerization was more pronounced, as indicated by the increased amount of 13-cis-retinal (Fig. 3B, peak a). Other retinal stereoisomers, 9- and 11-cis-retinal (Fig. 3B, peaks between a and b), were also observed. These isomerized products were not present in control reactions performed in the dark confirming that they indeed arose by photoisomerization. Thus, our data indicated that the generation of 13-cis-retinal during the ACO activity assay was caused by photoisomerization, rather than intrinsic ACO isomerase activity.
FIGURE 3.

HPLC analysis of the products generated from ACO-catalyzed cleavage of all-trans-8′-apocarotenol. A, the aldehyde products generated by ACO were converted into oxime derivatives by treatment with hydroxylamine prior to HPLC analysis. Exclusive production of all-trans-retinal was observed when the assay and HPLC analysis were performed under low illumination conditions. A small peak with a retention time of 8.7 min corresponding to syn-13-cis-retinal oxime was observed when the assay and analysis were conducted under ambient lighting conditions indicating photoisomerization of a small portion of all-trans-retinal product. Spectra for each peak are shown in the panels on the right. The analysis shown in panel B was conducted exactly as in A but reaction products were analyzed in their unaltered aldehyde forms. The presence of larger amounts of retinal geometric isomers in the sample generated under ambient lighting conditions indicates that photoisomerization occurs more readily in the retinal products as compared with the oxime derivatives. Note that peaks between a and b correspond to 11- and 9-cis-retinal, respectively. Peak a, syn-13-cis-retinal oxime (in A) or 13-cis-retinal (in B); peak b, syn-all-trans-retinal oxime (in A) or all-trans-retinal (in B); peak c, all-trans-8′-apocarotenol.
In Situ Analysis of ACO Reaction Products by Raman Spectroscopy
The above HPLC analysis strongly suggested that ACO only generated all-trans-retinal from all-trans-8′-apocarotenol, which argued against it possessing intrinsic isomerase activity. However, a previous study suggested that cis reaction products of ACO could readily convert back to their all-trans form (42). This possibility could account for the absence of detectible 13-cis-retinal in the HPLC assays, prompting us to employ other methods to test for the generation of this product more directly.
Structural differences in the length and geometrical configuration of the polyene backbone of carotenoids give rise to characteristic vibrational frequencies that can be monitored by Raman spectroscopy (55–57). We employed this technique to monitor changes in the levels of apocarotenoid reactants and products, including geometrical isomers, during the course of the reaction in situ with minimal additional sample workup to avoid potential back isomerization.
We measured Raman spectra for pure all-trans-8′-apocarotenol, all-trans- and 13-cis-retinal, and also for the second apocarotenoid product, 8′-hydroxy-15′-apocarotenal, which was obtained from ACO reaction mixtures and purified by HPLC (Table 2 and Fig. 4, A and B). Its identity was further confirmed by LC/MS (Fig. 4C). Because of difficulties associated with synthesis of the double-cis intermediate, we used the Gaussian package to predict the Raman spectrum of this species ab initio. We also calculated the signals for other molecules that could potentially appear during the reaction. As shown in Table 2, the calculated Raman peak wave numbers for each compound were in agreement with those determined experimentally.
TABLE 2.
Experimental and predicted Raman scattering peaks for all-trans-8′-apocarotenol and all-trans-retinal, as well as the di-cis intermediate and 13-cis-retinal product proposed to be generated during ACO-mediated oxidative carotenoid cleavage
FIGURE 4.

Purification, identification, and spectroscopic characterization of 8′-hydroxy-15′-apocarotenal, the secondary cleavage product of the ACO-catalyzed reaction. A, a 200-μl ACO sample at 10 mg/ml was incubated with 200 μm all-trans-8′-apocarotenol for 30 min in the dark. Apocarotenoids then were extracted and analyzed by HPLC. Peaks a and b eluted at 4.2 and 8.8 min and corresponded to all-trans-retinal product and substrate, respectively. Peak c eluted in 100% ethyl acetate at 17.3 min and corresponded to 8′-hydroxy-15′-apocarotenal on the basis of its absorbance spectrum (inset). This peak was collected for further analysis. B, a typical Raman spectrum of the 8′-hydroxy-15′-apocarotenal product. The collected peak c fraction was dried in a SpeedVac, and the resulting powder was dissolved in water prior to the Raman assay. The compound displayed a characteristic spectrum with prominent peaks at 1599 and 1635 cm−1. C, MS analysis of peak c revealed a major peak at m/z 167.2 corresponding to the protonated form of 8′-hydroxy-15′-apocarotenal. Tandem MS analysis of the 167.17 peak showed a major fragmentation product at an m/z of 149.17 representing loss of water from the parent ion.
Reactions were performed as described above and terminated at various time points by flash freezing. Following lyophilization, Raman spectra were directly measured on the resulting powders. To prevent any photoisomerization events, the assays were performed in the dark. Also, the 647.1 nm Raman excitation wavelength used in these experiments was far away from the absorbance bands of the polyene and not expected to induce photoisomerization. The all-trans-8′-apocarotenol substrate exhibited a characteristic peak at 1532 cm−1 (Fig. 5, light gray bar), which was noticeably reduced after a ∼10 min incubation, indicating the reaction occurs relatively slowly, a finding consistent with the HPLC data (Fig. 2C). The concurrent appearance of product peak at 1580 cm−1 (Fig. 5, dark gray bar) corresponding to all-trans-retinal confirmed that the substrate was being productively turned over. Raman peaks for the second apocarotenal product at 1610 and 1635 cm−1 started to appear at the same time as those for all-trans-retinal (Fig. 5, dark gray bar). Most of the substrate was consumed after the ∼60 min incubation. However, during the entire reaction, spectroscopic signals for 13-cis-retinal or the 13,14′-di-cis-8′-apocarotenol intermediate, which would be expected to appear around ∼1550–1555 cm−1 were not detected, indicating that no cis products were formed.
FIGURE 5.
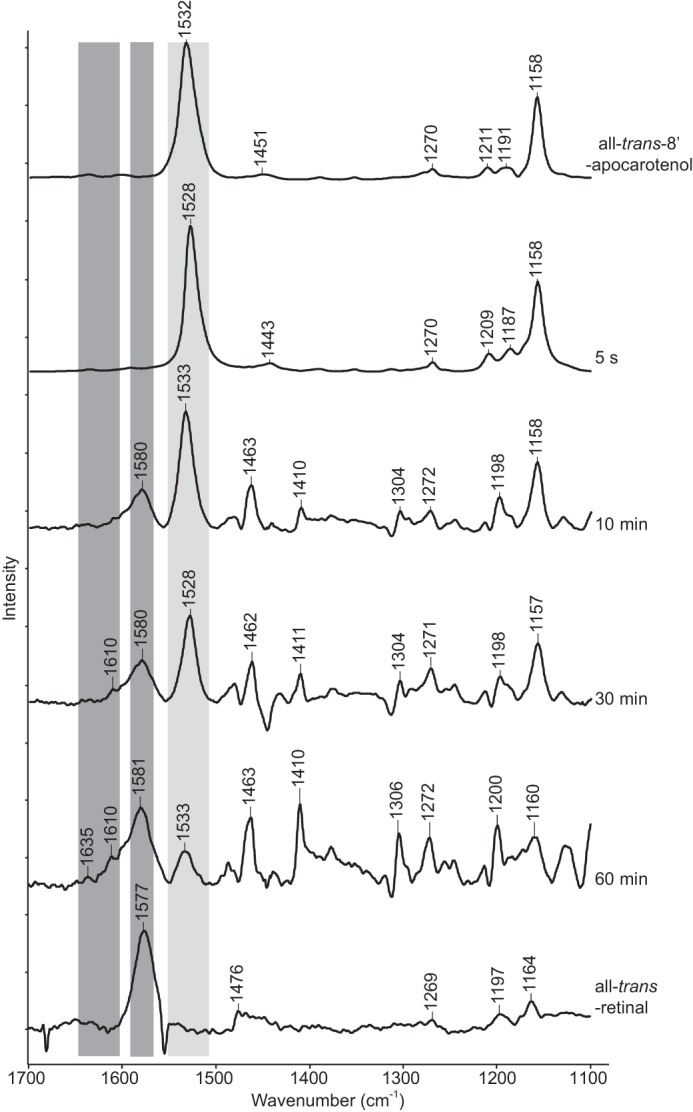
In situ Raman spectroscopy analysis of ACO reaction products generated from all-trans-8′-apocarotenol. Reactions were quenched at 5 s, 10 min, 30 min, and 60 min by emersion in liquid nitrogen. Samples were then lyophilized, and Raman difference spectra were measured. All-trans-8′-apocarotenol and all-trans-retinal gave rise to prominent peaks at 1532 cm−1 (light gray bar) and 1580 cm−1 (right dark gray bar), respectively. Peaks arising from the secondary product at 1610 and 1635 cm−1 (left dark gray bar) become detectable at 30 min. The reaction was nearly complete after the 60-min incubation. Notably, the spectra did not exhibit peaks at 1552 cm−1 (calculated) and 1555 cm−1 where the hypothesized 13,14′-di-cis-apocarotenoid intermediate and 13-cis-retinal products were expected to appear.
Detergents and PEG Affect ACO Activity
Our HPLC and spectroscopy findings did not support the prior hypothesis that ACO possesses intrinsic isomerase activity, which was based on crystallographic data (42). Because ACO crystallization in this study was carried out in the presence of PEG and the polyoxyethylene detergent C8E4, we examined the effects of these compounds on ACO catalytic function. PEG 3350 had little influence on activity up to a concentration of ∼10% (w/v) (Fig. 6A), but at higher concentrations the activity progressively declined, probably due to the ability of PEGs to cause protein aggregation. By contrast, both C8E4 and C8E6 strongly inhibited ACO activity even at concentrations below their critical micelle concentrations (CMC) (Fig. 6B, arrows). To examine whether or not this inhibition was a general property of detergents, we measured ACO activity in the presence of other high CMC, non-polyoxyethylene detergents. In contrast to C8E4 and C8E6, CHAPS and CYMAL-4 had much less effect on ACO activity and only partial inhibition occurred at the highest concentrations tested (Fig. 6B). Interestingly, ACO even displayed an elevated activity in the presence of CHAPS at relatively low concentrations. These data suggest that the linear structure of C8E4 and C8E6 could be responsible for their pronounced inhibitory effects. Indeed, a previously determined structure of iron-free apo-ACO was modeled with a C8E4 molecule bound in its active site, indicating potential competitive inhibition.
FIGURE 6.

Effects of detergents and PEG 3350 on ACO activity. A, inhibitory effects on ACO exerted by PEG 3350, the precipitant used for the growth of ACO crystals, occur at relatively high concentrations. B, the activity of ACO in 0.05% (w/v) Triton X-100, the detergent normally used in ACO activity assays, was severely inhibited by addition of the linear detergents C8E6 and C8E4 used for crystallographic studies of this enzyme, even at concentrations below their CMC values. Conversely, bulkier non-linear detergents such as CYMAL-4 and CHAPS had a much less pronounced effect on ACO activity. Error bars represent S.D. computed from three independent experiments.
Next, we examined the mechanism of ACO inhibition by linear polyoxyethylene detergents. The steady-state kinetic data shown in Fig. 7A and Table 3 demonstrated that C8E6 exerted non-competitive inhibition toward ACO, as indicated by the reduced Vmax and unchanged Km values. These data suggested that C8E6 might act allosterically to inhibit ACO function although direct binding to the active site could not be excluded, especially at high concentrations used for crystallization.
FIGURE 7.
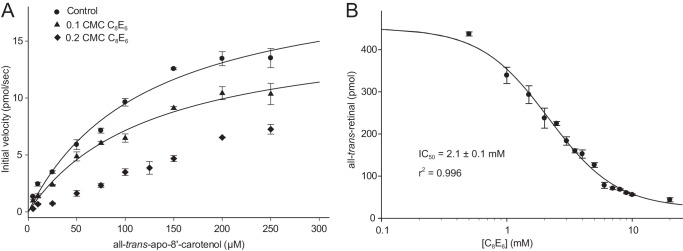
Polyoxyethylene detergent C8E6 non-competitively inhibits ACO catalytic function. A, a Michaelis-Menten kinetic plot of ACO in the presence of varying C8E6 concentrations. The decreased Vmax and unchanged Km (shown in Table 4) at a concentration of C8E6 equal to 0.1 times its CMC suggested a non-competitive mode of inhibition. Note that the reaction displayed more complex kinetics and the velocity did not plateau in the presence of C8E6 at a concentration of 0.2 times its CMC, which precluded derivation of Vmax and Km values from these data. B, C8E6 dose-dependently inhibited ACO activity. The IC50 for C8E6 was 2.1 mm, which corresponds to ∼0.22 times its CMC value. Error bars represent S.D. computed from triplicate measurements.
TABLE 3.
Inhibition kinetics
| Km(app) | Vmax(app) | r2 for fit | |
|---|---|---|---|
| μm | pmol/s | ||
| Control | 121 ± 24 | 21 ± 2 | 0.991 |
| 0.1 CMC C8E6 | 127 ± 21 | 16 ± 1 | 0.994 |
Structure of Native ACO in the Absence of Substrate
In the previously reported ACO structure (42) the proposed enzyme substrate binary complex was generated by crystallizing apo-ACO in the presence of substrate (all-trans-(3R)-3-hydroxy-8′-apocarotenol) and detergent (C8E4) and then soaking the resulting crystals in an iron(II)-containing solution. Electron density maps computed from x-ray data gathered from these crystals revealed a strong tube-shaped electron density feature in the active site that was attributed to the bound substrate in a di-cis configuration. However, this electron density assignment could not be confirmed because no isomorphous structure obtained in the absence of substrate was reported.
To validate these previous findings we crystallized native iron-bound ACO in the presence of the linear polyoxyethylene detergent C8E6 without the addition of any (apo)carotenoid molecules. C8E6 was used in place of C8E4, the detergent used in the original ACO crystallographic study (42), for our studies because it improved crystal reproducibility. Our crystals were isomorphous to the putative ACO-substrate complex crystals and were obtained under similar crystallization conditions. After refinement, the unbiased residual electron density map revealed a tube-like density that filled the substrate entry tunnel and extended past the iron in a curved fashion. Some additional peaks near the iron cofactor, which probably arose from bound water molecules, were also observed. The density was highly reminiscent of that reported by Kloer et al. (42). To more directly compare the two structures, apocarotenoid molecules in the deposited structure (PDB accession code 2BIW) were deleted, and residual electron density maps were calculated. As shown in Fig. 8, A and B, the residual active electron density was virtually identical between the two structures. The presence of iron in the active site was confirmed by computing Bijvoet-difference Fourier maps from data collected above and below the iron K-absorption edge (Fig. 8C). These data strongly indicated that the density previously attributed to isomerized apocarotenoid substrate arose instead from other component(s) of the crystallization mixture.
FIGURE 8.
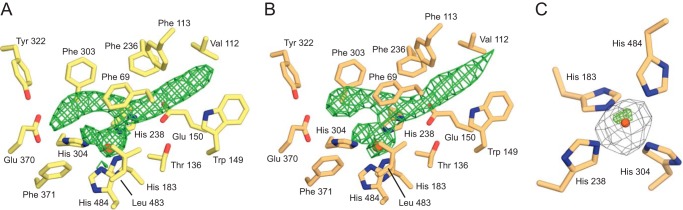
Electron density in the ACO active site. A, the 13,14′-di-cis-3-hydroxy-8′-apocarotenol molecules and water molecules in close proximity to the catalytic iron were removed from the deposited ACO structural model (PDB accession code 2BIW), and residual (mFo − DFc) electron density maps were calculated in REFMAC. The density was averaged over the four ACO monomers in the asymmetric unit using Coot. Density within 3 Å of the proposed substrate position is shown in green at a contour level of 5σ. Residues within 5 Å of the density are shown as sticks. The ferrous iron is shown as an orange sphere. B, a strikingly similar unbiased density feature is present in crystals grown in the absence of substrate. Representations are the same as in panel A. C, Bijvoet difference Fourier maps were calculated from data collected above (blue mesh) and below (green mesh) the iron K absorption edge, each contoured at the 6 σ level. The large difference in anomalous scattering confirms the presence of iron in the active site of “as-isolated” ACO.
ACO Structure in Triton X-100
The strong inhibitory effects of linear polyoxyethylene detergents on ACO activity suggested that the bent tube-like density observed in the ACO active site might represent bound C8E4 or C8E6 molecules, which could block substrate binding. Crystallization in the absence of detergent would be a straightforward way to address this possibility, but we were unable to generate crystals from detergent-free mother liquor. Thus, we tested a number of non-linear, ring-containing detergents with bulky hydrophobic moieties including CYMAL-4, CHAPS, and Triton X-100. Of these, only ACO samples containing Triton X-100 yielded diffraction quality crystals. Despite the change in detergent, these crystals were essentially isomorphous to crystals grown in C8E4 and C8E6 (Table 4). Crystals grown in Triton X-100 showed higher resolution diffraction compared with crystals grown in C8E6 with the best crystals diffracting up to 1.9-Å resolution. Additionally, twinning was much less pronounced in these crystals compared with those obtained in C8E6. Electron density maps computed after several rounds of refinement revealed that these crystals also contained residual active site density with a somewhat altered appearance compared with the density observed in the crystals grown in C8E6 (Fig. 9, A and B). As in the C8E6 crystals, a tube of density was present in the substrate entry tunnel that closely followed the contours of the protein structure. This density merged with a second punctate density feature in direct contact with the iron cofactor, which was best modeled as a coordinated H2O or hydroxide ligand. The tube-like density was somewhat variable in different protomers of the asymmetric unit. Examination of 2Fo − Fc maps at low contour levels revealed a bent density in the vicinity of the iron somewhat similar in appearance to that of the C8E6 structure residual map density. Notably, this active site density was quite variable from crystal to crystal.
TABLE 4.
X-ray crystallographic data collection and refinement statistics
| Data collectiona | ||
| Data set name | C8E6 crystal | Triton X-100 crystal |
| Beamline | X29 | NECAT 24-ID-C |
| Wavelength (Å) | 1.7308 | 0.9793 |
| Space group | P212121 | P212121 |
| Unit cell parameters (Å) | a = 118.56 | a = 118.47 |
| b = 125.12 | b = 125.39 | |
| c = 203.01 | c = 202.54 | |
| Resolution (Å) | 50-2.8 (2.97-2.8)a | 50-2.0 (2.05-2) |
| Unique reflections | 75,089 (11,789) | 200,469 (14,869) |
| Completeness (%) | 99.7 (98.2) | 98.7 (99.8) |
| Multiplicity | 11.7 (9.9) | 7.3 (5.9) |
| 〈I/σI〉 | 10.1 (1.4) | 11.74 (1.56) |
| RmergeI (%)b | 27.2 (168.5) | 8.6 (97.7) |
| RmeasI (%)b | 28.5 (177.7) | 9.3 (107.7) |
| CC½ (%)b | 99 (54.2) | 99.8 (54.5) |
| Wilson B-factor (Å2) | 45.9 | 46.4 |
| Refinement | ||
| Resolution (Å) | 48.6-2.8 | 48.6-2.0 |
| No observations | 71479 | 190607 |
| Rwork/Rfree (%)c | 21.1/23.6 | 17.7/20.5 |
| No. atoms | ||
| Protein | 15072 | 15154 |
| Water | 87 | 875 |
| Metal/ion | 4 Fe, 3 Cl | 4 Fe, 2 Cl |
| B-factors (Å2) | ||
| Protein | 51 | 47 |
| Water | 35 | 47 |
| Metal/ion | 30 (Fe), 55 (Cl) | 34 (Fe), 48 (Cl) |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.011 | 0.011 |
| Bond angles (°) | 1.41 | 1.44 |
| Ramachandran plotd | ||
| Favored/outliers (%) | 97.5/0 | 98/0 |
| PDB accession code | 4OU8 | 4OU9 |
a Values in parentheses are those for the highest resolution shell of data.
b As calculated in XDS.
c As calculated in REFMAC.
d As defined in MolProbity.
FIGURE 9.

Structural comparison of ACO crystallized in C8E6 or Triton X-100. Shown are unbiased mFc − DFc (green mesh) and 2Fo − Fc (blue mesh) electron density maps contoured at 3σ and 1σ, respectively, displayed within the active site regions of ACO crystallized in the presence of C8E6 (A) or Triton X-100 (B). The densities are similar in appearance with the exception of a break in the density after it passes the iron center in the Triton X-100 structure. Black arrows indicate residual electron density that likely represents an iron(II)-bound water or hydroxide ligand. C, difference distance matrix analysis of the C8E6 and Triton X-100 ACO structures. The triangular matrix below the diagonal is an error-scaled difference distance matrix generated by pairwise comparisons of non-crystallographic symmetry-related monomers using the program ESCET, whereas the upper matrix is an absolute difference distance matrix. Red and blue elements stand for structural expansion or contraction, respectively. The intensity of the color is proportional to the changes in distance, and gray elements indicate structural invariance. Secondary structural elements are shown beneath the matrix: open boxes are helices, black boxes are β-sheets. D, structural superposition of the C8E6 (yellow) and Triton X-100 (blue) showing a rigid body movement of the top face of propeller blade II in chain A of the structure. Consequently, the mouth of the protein is more open in the Triton X-100 structure compared the C8E6 structure in this particular monomer. Small changes were also evident in regions of the protein predicted to contact membranes (blue arrows). The catalytic iron is shown as an orange sphere. The green arrow delineates the substrate entry tunnel that leads to the catalytic center.
Comparison of these two models by error-scaled difference distance matrix analysis revealed little variability in Cα positions with a few exceptions. One notable difference occurs on the top face of blade II within monomer A, residues 205–212 and 229–234, where the end of the β sheet is shifted away from the substrate entry tunnel in the Triton X-100 versus the C8E6 structures (Fig. 9). Some modest ∼1 Å shifts in a few active site residue side chains including Phe-69 and Phe-113 were also observed between the two structures.
DISCUSSION
To facilitate the study of ACO we first developed a novel expression and purification protocol. In contrast to previous methods that relied on a fusion protein strategy (12) or refolding from E. coli inclusion bodies (42), we were able to express native, untagged ACO in a soluble form in E. coli and purify it to homogeneity by a simple two-step procedure involving ammonium sulfate fractionation and gel filtration chromatography. Through this procedure we could obtain quantities of pure ACO sufficient for future high resolution biophysical studies, which are in general lacking for this class of enzyme. The preparation was enzymatically active and the metal center was fully occupied by iron (32). We observed that freshly purified ACO samples display 2.6-fold higher activity but 2.7-fold decreased Km for its apocarotenoid substrate as compared with an aged, air-exposed sample (compare Figs. 2C (aged for 2 weeks at 4 °C) and 7A (fresh)). Ferrous iron is known to be required for the activity of CCEs (11, 58, 59). Because ACO was purified under aerobic conditions in the absence of reducing agent, we expect that the diminished activity could be the result of iron oxidation. Nevertheless, the ACO iron(II) center appears to be quite stable compared with many other non-heme iron(II)-dependent enzymes (60, 61). Analysis of ACO reaction products generated from the all-trans-apocarotenoid substrate both by standard HPLC as well as by in situ Raman spectroscopy revealed exclusive production of all-trans-retinal. We found that the previously reported traces of 13-cis-retinal produced during the assay could be attributed to photoisomerization of all-trans-retinal (12, 42). Surprisingly, we also found that the polyoxyethylene detergent, C8E4, used in the original structural study strongly inhibits ACO activity, indicating that the crystallized protein might be catalytically inactive. Indeed, using Raman spectroscopy we could not detect in crystallo production of all-trans-retinal from ACO crystallized in the presence of C8E4. Inhibition was observed for a related linear polyoxyethylene detergent, C8E6, but was absent or much less pronounced for some bulky detergents depending on their concentration.
To directly examine the possibility that the active site electron density in the previously reported structure represents bound apocarotenoid substrate or an isomerized intermediate, we crystallized native ACO under conditions similar to those previously reported except that apocarotenoids were not present during protein purification or crystallization. The active site electron density observed in these crystals was strikingly similar in appearance to that reported by Kloer et al. (42) despite the absence of added carotenoids demonstrating that the density was not attributable to bound substrate (Fig. 9A). We have observed that this density does not change in appearance when our ACO crystals are soaked with all-trans-8′-apocarotenol. A second structure obtained with Triton X-100, a detergent that supports ACO activity, substituted for C8E6 also displays residual active site density albeit with a slightly altered appearance (Fig. 9B). Given the similarity in density between the two structures, we feel that it is unlikely to represent bound detergent. Instead, this density might arise from another component of the crystallization mixture. It could also simply represent a chain of partially ordered, hydrogen bonded water molecules. The relatively narrow dimensions and hydrophobic nature of the substrate entry tunnel might cause an ordering of water molecules that could occupy the cavity to prevent formation of an energetically unfavorable vacuum (62, 63).
The structure reported by Kloer et al. (42) is the only CCE for which an experimental enzyme-substrate was proposed. In light of the data presented here the model of the bound substrate can only be considered an educated guess that is wrong in detail (i.e. the configuration of the substrate is incorrect). Thus, this model of the bound apocarotenoid should be used with caution when developing hypotheses of CCE substrate specificity and catalytic function. The kinetic data presented in this study emphasize that proper selection of detergent will be critical for obtaining a genuine CCE-substrate complex. CCEs possess a conserved hydrophobic patch on their surfaces near their substrate entry tunnels that facilitates extraction of lipophilic substrates from membranes (4, 25). Inclusion of detergent seems to be critical for the crystallization of ACO and RPE65, probably due to its ability to bind the hydrophobic patch and prevent nonspecific protein aggregation. Interestingly, the structure of the 9-cis-epoxycarotenoid dioxygenase, VP14, was determined in the absence of detergent (43). In this structure hydrophobic surfaces from symmetry-related molecules pack together and shield each other from the solvent. Despite inclusion of carotenoids in the crystallization solutions such compounds were not identified in the active site of this enzyme (43). In this case, delivery of highly hydrophobic carotenoids to the enzyme may have been impeded by the absence of detergent micelles.
In summary, using this highly purified, enzymatically active preparation we investigated the enzymatic activity of ACO by HPLC and Raman spectroscopy. Both methods revealed robust production of all-trans-retinal from all-trans-8′-apocarotenol by ACO. Production of trace cis isomers was observed only in reactions performed under ambient light suggesting partial photoisomerization of the retinal product. We observed a pronounced inhibitory effect of linear polyoxyethylene detergents used for ACO crystallographic studies on all-trans-retinal production by ACO. We showed that the ACO active site density previously attributed to an isomerized apocarotenoid intermediate originates from non-substrate molecule(s). Thus, obtaining an intact CCE-substrate complex remains a challenge for future studies.
Acknowledgments
We thank Dr. Leslie T. Webster, Jr., and members of the Palczewski laboratory (Case Western Reserve University) for valuable comments on the manuscript. We also thank Dr. Zhaoyang (John) Feng (Case Western Reserve University) for providing equipment used for this study. A portion of this work is based upon research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which is supported by National Institutes of Health Grant GM103403 from the National Center for Research Resources. Use of the Advanced Photon Source is supported by the United States Department of Energy, Office of Basic Energy Sciences, under Contract number DE-AC02-06CH11357. Some data for this study were measured at beamline X29 of the National Synchrotron Light Source.
This work was supported, in whole or in part, by National Institutes of Health Grants GM054072 (to P. R. C.) and EY009339 (to K. P.), and EY020551 (to J. v. L.), the Offices of Biological and Environmental Research, Basic Energy Sciences of the United States Department of Energy, National Center for Research Resources Grant P41RR012408, and NIGMS Grant P41GM103473.
The atomic coordinates and structure factors (codes 4OU8 and 4OU9) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CCEs
- carotenoid cleavage enzymes
- ACO
- apocarotenoid oxygenase
- BCO1
- β,β-carotene-15,15′-oxygenase
- BCO2
- β,β-carotene-9,10-oxygenase
- C8E4
- tetraethylene glycol monooctyl ether
- C8E6
- hexaethylene glycol monooctyl ether
- CMC
- critical micelle concentration
- PDB
- Protein Data Bank
- RPE65
- retinal pigment epithelium 65-kDa protein
- VP14
- viviparous 14
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Demmig-Adams B., Adams W. W., 3rd. (2002) Antioxidants in photosynthesis and human nutrition. Science 298, 2149–2153 [DOI] [PubMed] [Google Scholar]
- 2. von Lintig J. (2010) Colors with functions: elucidating the biochemical and molecular basis of carotenoid metabolism. Annu. Rev. Nutr. 30, 35–56 [DOI] [PubMed] [Google Scholar]
- 3. Britton G. (1995) Structure and properties of carotenoids in relation to function. FASEB J. 9, 1551–1558 [PubMed] [Google Scholar]
- 4. Sui X., Kiser P. D., von Lintig J., Palczewski K. (2013) Structural basis of carotenoid cleavage: from bacteria to mammals. Arch. Biochem. Biophys. 539, 203–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leuenberger M. G., Engeloch-Jarret C., Woggon W. D. (2001) The reaction mechanism of the enzyme-catalyzed central cleavage of β-carotene to retinal. Angew. Chem. Int. Ed. Engl. 40, 2613–2617 [DOI] [PubMed] [Google Scholar]
- 6. Schmidt H., Kurtzer R., Eisenreich W., Schwab W. (2006) The carotenase AtCCD1 from Arabidopsis thaliana is a dioxygenase. J. Biol. Chem. 281, 9845–9851 [DOI] [PubMed] [Google Scholar]
- 7. Moore T. (1930) Vitamin A and carotene: the absence of the liver oil vitamin A from carotene. VI. The conversion of carotene to vitamin A in vivo. Biochem. J. 24, 692–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tan B. C., Schwartz S. H., Zeevaart J. A., McCarty D. R. (1997) Genetic control of abscisic acid biosynthesis in maize. Proc. Natl. Acad. Sci. U.S.A. 94, 12235–12240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alder A., Jamil M., Marzorati M., Bruno M., Vermathen M., Bigler P., Ghisla S., Bouwmeester H., Beyer P., Al-Babili S. (2012) The path from β-carotene to carlactone, a strigolactone-like plant hormone. Science 335, 1348–1351 [DOI] [PubMed] [Google Scholar]
- 10. Auldridge M. E., McCarty D. R., Klee H. J. (2006) Plant carotenoid cleavage oxygenases and their apocarotenoid products. Curr. Opin. Plant Biol. 9, 315–321 [DOI] [PubMed] [Google Scholar]
- 11. Schwartz S. H., Tan B. C., Gage D. A., Zeevaart J. A., McCarty D. R. (1997) Specific oxidative cleavage of carotenoids by VP14 of maize. Science 276, 1872–1874 [DOI] [PubMed] [Google Scholar]
- 12. Ruch S., Beyer P., Ernst H., Al-Babili S. (2005) Retinal biosynthesis in Eubacteria: in vitro characterization of a novel carotenoid oxygenase from Synechocystis sp. PCC 6803. Mol. Microbiol. 55, 1015–1024 [DOI] [PubMed] [Google Scholar]
- 13. Scherzinger D., Ruch S., Kloer D. P., Wilde A., Al-Babili S. (2006) Retinal is formed from apo-carotenoids in Nostoc sp. PCC7120: in vitro characterization of an apo-carotenoid oxygenase. Biochem. J. 398, 361–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wyss A., Wirtz G., Woggon W., Brugger R., Wyss M., Friedlein A., Bachmann H., Hunziker W. (2000) Cloning and expression of β,β-carotene 15,15′-dioxygenase. Biochem. Biophys. Res. Commun. 271, 334–336 [DOI] [PubMed] [Google Scholar]
- 15. Paik J., During A., Harrison E. H., Mendelsohn C. L., Lai K., Blaner W. S. (2001) Expression and characterization of a murine enzyme able to cleave β-carotene. The formation of retinoids. J. Biol. Chem. 276, 32160–32168 [DOI] [PubMed] [Google Scholar]
- 16. Redmond T. M., Gentleman S., Duncan T., Yu S., Wiggert B., Gantt E., Cunningham F. X., Jr. (2001) Identification, expression, and substrate specificity of a mammalian β-carotene 15,15′-dioxygenase. J. Biol. Chem. 276, 6560–6565 [DOI] [PubMed] [Google Scholar]
- 17. Lindqvist A., Andersson S. (2002) Biochemical properties of purified recombinant human β-carotene 15,15′-monooxygenase. J. Biol. Chem. 277, 23942–23948 [DOI] [PubMed] [Google Scholar]
- 18. von Lintig J., Vogt K. (2000) Filling the gap in vitamin A research: molecular identification of an enzyme cleaving β-carotene to retinal. J. Biol. Chem. 275, 11915–11920 [DOI] [PubMed] [Google Scholar]
- 19. Eriksson J., Larson G., Gunnarsson U., Bed'hom B., Tixier-Boichard M., Strömstedt L., Wright D., Jungerius A., Vereijken A., Randi E., Jensen P., Andersson L. (2008) Identification of the yellow skin gene reveals a hybrid origin of the domestic chicken. PLoS Genet. 4, e1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berry S. D., Davis S. R., Beattie E. M., Thomas N. L., Burrett A. K., Ward H. E., Stanfield A. M., Biswas M., Ankersmit-Udy A. E., Oxley P. E., Barnett J. L., Pearson J. F., van der Does Y., Macgibbon A. H., Spelman R. J., Lehnert K., Snell R. G. (2009) Mutation in bovine β-carotene oxygenase 2 affects milk color. Genetics 182, 923–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Amengual J., Lobo G. P., Golczak M., Li H. N., Klimova T., Hoppel C. L., Wyss A., Palczewski K., von Lintig J. (2011) A mitochondrial enzyme degrades carotenoids and protects against oxidative stress. FASEB J. 25, 948–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lobo G. P., Isken A., Hoff S., Babino D., von Lintig J. (2012) BCDO2 acts as a carotenoid scavenger and gatekeeper for the mitochondrial apoptotic pathway. Development 139, 2966–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jin M., Li S., Moghrabi W. N., Sun H., Travis G. H. (2005) RPE65 is the retinoid isomerase in bovine retinal pigment epithelium. Cell 122, 449–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moiseyev G., Chen Y., Takahashi Y., Wu B. X., Ma J. X. (2005) RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc. Natl. Acad. Sci. U.S.A. 102, 12413–12418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kiser P. D., Palczewski K. (2010) Membrane-binding and enzymatic properties of RPE65. Prog. Retin. Eye Res. 29, 428–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Takahashi Y., Moiseyev G., Nikolaeva O., Ma J. X. (2012) Identification of the key residues determining the product specificity of isomerohydrolase. Biochemistry 51, 4217–4225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takahashi Y., Moiseyev G., Chen Y., Farjo K., Nikolaeva O., Ma J. X. (2011) An enzymatic mechanism for generating the precursor of endogenous 13-cis retinoic acid in the brain. FEBS J. 278, 973–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takahashi Y., Moiseyev G., Chen Y., Ma J. X. (2005) Identification of conserved histidines and glutamic acid as key residues for isomerohydrolase activity of RPE65, an enzyme of the visual cycle in the retinal pigment epithelium. FEBS Lett. 579, 5414–5418 [DOI] [PubMed] [Google Scholar]
- 29. Poliakov E., Gubin A. N., Stearn O., Li Y., Campos M. M., Gentleman S., Rogozin I. B., Redmond T. M. (2012) Origin and evolution of retinoid isomerization machinery in vertebrate visual cycle: hint from jawless vertebrates. PLoS One 7, e49975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chander P., Gentleman S., Poliakov E., Redmond T. M. (2012) Aromatic residues in the substrate cleft of RPE65 protein govern retinol isomerization and modulate its progression. J. Biol. Chem. 287, 30552–30559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Redmond T. M., Poliakov E., Kuo S., Chander P., Gentleman S. (2010) RPE65, visual cycle retinol isomerase, is not inherently 11-cis-specific: support for a carbocation mechanism of retinol isomerization. J. Biol. Chem. 285, 1919–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kiser P. D., Farquhar E. R., Shi W., Sui X., Chance M. R., Palczewski K. (2012) Structure of RPE65 isomerase in a lipidic matrix reveals roles for phospholipids and iron in catalysis. Proc. Natl. Acad. Sci. U.S.A. 109, E2747–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kiser P. D., Golczak M., Lodowski D. T., Chance M. R., Palczewski K. (2009) Crystal structure of native RPE65, the retinoid isomerase of the visual cycle. Proc. Natl. Acad. Sci. U.S.A. 106, 17325–17330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McBee J. K., Kuksa V., Alvarez R., de Lera A. R., Prezhdo O., Haeseleer F., Sokal I., Palczewski K. (2000) Isomerization of all-trans-retinol to cis-retinols in bovine retinal pigment epithelial cells: dependence on the specificity of retinoid-binding proteins. Biochemistry 39, 11370–11380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Borowski T., Blomberg M. R., Siegbahn P. E. (2008) Reaction mechanism of apocarotenoid oxygenase (ACO): a DFT study. Chemistry 14, 2264–2276 [DOI] [PubMed] [Google Scholar]
- 36. Harrison P. J., Bugg T. D. (2013) Enzymology of the carotenoid cleavage dioxygenases: reaction mechanisms, inhibition and biochemical roles. Arch. Biochem. Biophys. 544, 105–111 [DOI] [PubMed] [Google Scholar]
- 37. Wang X., Wang T., Jiao Y., von Lintig J., Montell C. (2010) Requirement for an enzymatic visual cycle in Drosophila. Curr. Biol. 20, 93–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Voolstra O., Oberhauser V., Sumser E., Meyer N. E., Maguire M. E., Huber A., von Lintig J. (2010) NinaB is essential for Drosophila vision but induces retinal degeneration in opsin-deficient photoreceptors. J. Biol. Chem. 285, 2130–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oberhauser V., Voolstra O., Bangert A., von Lintig J., Vogt K. (2008) NinaB combines carotenoid oxygenase and retinoid isomerase activity in a single polypeptide. Proc. Natl. Acad. Sci. U.S.A. 105, 19000–19005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maeda T., Perusek L., Amengual J., Babino D., Palczewski K., von Lintig J. (2011) Dietary 9-cis-β,β-carotene fails to rescue vision in mouse models of leber congenital amaurosis. Mol. Pharmacol. 80, 943–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nagao A., Olson J. A. (1994) Enzymatic formation of 9-cis, 13-cis, and all-trans retinals from isomers of β-carotene. FASEB J. 8, 968–973 [DOI] [PubMed] [Google Scholar]
- 42. Kloer D. P., Ruch S., Al-Babili S., Beyer P., Schulz G. E. (2005) The structure of a retinal-forming carotenoid oxygenase. Science 308, 267–269 [DOI] [PubMed] [Google Scholar]
- 43. Messing S. A., Gabelli S. B., Echeverria I., Vogel J. T., Guan J. C., Tan B. C., Klee H. J., McCarty D. R., Amzel L. M. (2010) Structural insights into maize viviparous14, a key enzyme in the biosynthesis of the phytohormone abscisic acid. Plant Cell 22, 2970–2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sharma R. V., Mathur S. N., Ganguly J. (1976) Studies on the relative biopotencies and intestinal absorption of different apo-β-carotenoids in rats and chickens. Biochem. J. 158, 377–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Montgomery J., J. A., Vreven T., Kudin K. N., Burant J. C., Millam J. M., Iyengar S. S., Tomasi J., Barone V., Mennucci B., Cossi M., Scalmani G., Rega N., Petersson G. A., Nakatsuji H., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Klene M., Li X., Knox J. E., Hratchian H. P., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Ayala P. Y., Morokuma K., Voth G. A., Salvador P., Dannenberg J. J., Zakrzewski V. G., Dapprich S., Daniels A. D., Strain M. C., Farkas O., Malick D. K., Rabuck A. D., Raghavachari K., Foresman J. B., Ortiz J. V., Cui Q., Baboul A. G., Clifford S., Cioslowski J., Stefanov B. B., Liu G., Liashenko A., Piskorz P., Komaromi I., Martin R. L., Fox D. J., Keith T., Al-Laham M. A., Peng C. Y., Nanayakkara A., Challacombe M., Gill P. M. W., Johnson B., Chen W., Wong M. W., Gonzalez C., Pople J. A. (2004) Gaussian 03, revision C.02, Gaussian Inc., Wallingford, CT [Google Scholar]
- 46. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 49. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Winn M. D., Murshudov G. N., Papiz M. Z. (2003) Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374, 300–321 [DOI] [PubMed] [Google Scholar]
- 52. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thorn A., Sheldrick G. M. (2011) ANODE: anomalous and heavy-atom density calculation. J. Appl. Crystallogr. 44, 1285–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schneider T. R. (2000) Objective comparison of protein structures: error-scaled difference distance matrices. Acta Crystallogr. D Biol. Crystallogr. 56, 714–721 [DOI] [PubMed] [Google Scholar]
- 55. Callender R. (1977) Resonance Raman studies of visual pigments. Annu. Rev. Biophys. Bioeng. 6, 33–55 [DOI] [PubMed] [Google Scholar]
- 56. Rimai L., Heyde M. E., Gill D. (1973) Vibrational spectra of some carotenoids and related linear polyenes: a Raman spectroscopic study. J. Am. Chem. Soc. 95, 4493–4501 [DOI] [PubMed] [Google Scholar]
- 57. Braiman M., Mathies R. (1980) Resonance Raman evidence for an all-trans to 13-cis isomerization in the proton-pumping cycle of bacteriorhodopsin. Biochemistry 19, 5421–5428 [DOI] [PubMed] [Google Scholar]
- 58. Moiseyev G., Takahashi Y., Chen Y., Gentleman S., Redmond T. M., Crouch R. K., Ma J. X. (2006) RPE65 is an iron(II)-dependent isomerohydrolase in the retinoid visual cycle. J. Biol. Chem. 281, 2835–2840 [DOI] [PubMed] [Google Scholar]
- 59. Redmond T. M., Poliakov E., Yu S., Tsai J. Y., Lu Z., Gentleman S. (2005) Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc. Natl. Acad. Sci. U.S.A. 102, 13658–13663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Solomon E. I., Decker A., Lehnert N. (2003) Non-heme iron enzymes: contrasts to heme catalysis. Proc. Natl. Acad. Sci. U.S.A. 100, 3589–3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Feig A. L., Lippard S. J. (1994) Reactions of nonheme iron(II) centers with dioxygen in biology and chemistry. Chem. Rev. 94, 759–805 [Google Scholar]
- 62. Matthews B. W., Liu L. (2009) A review about nothing: are apolar cavities in proteins really empty? Protein Sci. 18, 494–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Teeter M. M. (1991) Water-protein interactions: theory and experiment. Annu. Rev. Biophys. Biophys. Chem. 20, 577–600 [DOI] [PubMed] [Google Scholar]