

Background: Endoplasmic reticulum (ER) stress is involved in β-cell failure and apoptotic death.
Results: Upon endogenous TRα knockdown, ER stress significantly enhanced apoptosis in pancreatic β-cells.
Conclusion: TRα facilitates reduced apoptosis in pancreatic β-cells under ER stress.
Significance: TRα is coupled to stress response modulation and improved survival of pancreatic β-cells.
Keywords: Diabetes, Endoplasmic Reticulum Stress, Islet, Thyroid, Thyroid Hormone
Abstract
Thyroid hormone receptor α (TRα) is critical to postnatal pancreatic β-cell maintenance. To investigate the association between TRα and the survival of pancreatic β-cells under endoplasmic reticulum (ER) stress, the expression of endogenous TRα was inhibited by infection with an adenovirus expressing double-stranded short hairpin RNA against TRα (AdshTRα). In control adenovirus-infected pancreatic β-cells, palmitate enhanced the expression of activating transcription factor 4 (ATF4) and heme oxygenase 1, which facilitates adaptation to oxidative ER stress. However, in AdshTRα-infected pancreatic β-cells, palmitate did not induce ATF4-mediated integrated stress response, and oxidative stress-associated apoptotic cell death was significantly enhanced. TRα-deficient mice or wild-type mice (WT) were fed a high fat diet (HFD) for 30 weeks, and the effect of oxidative ER stress on pancreatic β-cells was analyzed. HFD-treated TRα-deficient mice had high blood glucose levels and low plasma insulin levels. In HFD-treated TRα-deficient mice, ATF4 was not induced, and apoptosis was enhanced compared with HFD-treated WT mice. Furthermore, the expression level of 8-hydroxydeoxyguanosine, an oxidative stress marker, was enhanced in the β-cells of HFD-treated TRα-deficient mice. These results indicate that endogenous TRα plays an important role for the expression of ATF4 and facilitates reduced apoptosis in pancreatic β-cells under ER stress.
Introduction
Maintaining appropriate size of pancreatic β-cell mass plays an essential role in determining the amount of insulin that is secreted to maintain blood glucose levels within a narrow range. Elucidation of the mechanisms that control β-cell mass size is essential for the development of regenerative therapies for both type 1 and type 2 diabetes, which are both characterized by an insufficient β-cell mass (1). The apoptosis of β-cells occurs in type 1 as well as type 2 diabetes. In type 1 diabetes, β-cells are selectively destroyed by a combination of autoimmune and inflammatory processes, leading to an absolute insulin deficiency, whereas in type 2 diabetes, resistance to insulin in peripheral tissues, accompanied by a reduction in β-cell mass, leads to relative insulin deficiency (2, 3).
Endoplasmic reticulum (ER)2 stress is a newly identified pathophysiological paradigm in many chronic metabolic diseases that determines whether cells survive or die (4, 5). To combat the deleterious effects of ER stress, cells have evolved protective strategies, collectively termed the unfolded protein response (UPR) (6). This concerted and complex cellular response is mediated through ER transmembrane receptors: pancreatic ER kinase-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1). In resting cells, these three ER stress receptors are maintained in an inactive state through their association with the ER chaperone. When unfolded proteins accumulate, the ER chaperone dissociates from the three receptors, which leads to their activation and triggers the UPR. Activated PERK blocks general protein synthesis by phosphorylating eukaryotic initiation factor 2α (eIF2α). This phosphorylation enables translation of activating transcription factor 4 (ATF4), which occurs through an alternative eIF2α-independent translation pathway. ATF4 stimulates the expression of cAMP response element-binding transcription factor (C/EBP) and promotes cell survival by inducing genes involved in amino acid metabolism, redox reactions, stress response, and protein secretion (7). Thus, the UPR is a prosurvival response to reduce the accumulation of unfolded proteins and restore normal ER functions (5). Several reports have indicated that activation of the eIF2α-ATF4 signaling pathway reduced oxidative ER stress, and the deficiency of ATF4 impaired the expression of genes involved in resistance to oxidative ER stress (7, 8).
We reported the role of ligand-bound thyroid hormone receptor (TR) α in β-cell replication and in expansion of the β-cell mass during postnatal development (9). Our recent reports indicated that ligand-bound TRα plays a critical role in postnatal pancreatic β-cell maintenance. Overexpression of ligand-bound TRα also induced pancreatic β-cell proliferation and could be considered a survival factor that protects β-cells from streptozotocin-induced apoptosis. In the present study, we explored the function of endogenous TRα in pancreatic β-cells under ER stress. We demonstrated that TRα was necessary for the ATF4-mediated antistress response for adaptation to the palmitate-induced oxidative stress. This finding clearly shows that TR plays important roles for maintenance of β-cell mass under excessive intake of saturated fatty acid.
EXPERIMENTAL PROCEDURES
Cell Culture
MIN6 cells (kindly provided by Dr. Jun Miyazaki, Osaka University Graduate School of Medicine, Osaka, Japan) were maintained in Dulbecco's modified Eagle's medium containing 25 mm glucose, 13% heat-inactivated fetal bovine serum, 0.1 mm 2-mercaptoethanol, 100 units/ml penicillin, and 0.05 mg/ml streptomycin in a 5% CO2 humidified atmosphere at 37 °C (10).
Construction of Recombinant Adenovirus Vectors
A double-stranded short hairpin RNA (shRNA) encoding sense and antisense siRNA sequences against mouse TRα (5′-CGCTCTTCCTGGAGGTCTT-3′) (11) separated by a loop sequence (TTCAAGAGA) was cloned into the pENTR/U6 vector. A recombinant adenovirus expressing this shRNA (AdshTRα) was generated by using a pAd/BLOCK-iT-DEST vector kit (Invitrogen) according to the manufacturer's protocol. Adenovirus vector expressing ATF4 gene (AdATF4) was generated by using a pAd/CMV/V5-DEST Gateway vector kit (Invitrogen). ATF4 cDNA was cloned by GeneArt system service (Life Technology Japan, Tokyo). AdLacZ, which contains the lacZ gene controlled by the cytomegalovirus promoter, was provided by Quantum Biotechnologies (Montréal, Canada) and used as a control. Recombinant adenoviruses were purified by using a plaque-forming assay, harvested 48 h after infection of 293 cells, and further purified by using double-cesium chloride gradient ultracentrifugation. Viral titers were determined as described previously (9).
Treatment of Cells with Fatty Acids
A stock solution of 50 mm palmitate (Sigma-Aldrich) was prepared in 50% ethanol by heating to 70 °C. Palmitate and methyl palmitate (Sigma-Aldrich) were prepared by mixing with 90% ethanol at room temperature to produce 90 mm stock solutions. The fatty acid preparations were then bound to 10% fatty acid-free BSA by incubation for 1 h at 37 °C. The mixture was added to RPMI 1640 medium (containing 11 mm glucose) lacking fetal calf serum. The final concentrations present in the cell environment were 1% BSA and 0.5% ethanol. Control cells received BSA and vehicle only. One day after plating, the cells were infected with 30 m.o.i. of adenovirus. After 24 h of incubation in adenovirus-containing medium, the cells were cultured with or without palmitate, which is associated with ER stress, for an additional 24 h. Cell numbers were determined using a nonradioactive cell proliferation assay (Cell Counting Kit-8; Dojindo, Kumamoto, Japan), according to the manufacturer's protocol.
Analyses of Reactive Oxygen Species (ROS) and Apoptosis
By Ficoll gradient centrifugation, the endocrine fraction was prepared from 4-week-old mice (12). Subsequently, the pancreatic β-cells were cultured for 6 h on 35-mm culture dishes with RPMI 1640 Gluta MAX-I medium supplemented with 10% resin-stripped FBS at 37 °C under 5% CO2 atmosphere. For cellular ROS measurements, cells were resuspended in prewarmed phosphate-buffered saline (PBS) supplemented with 5% fetal bovine serum (FBS) and incubated with 5 μm DCF (Invitrogen) for 30 min at room temperature, and then analyzed immediately by flow cytometry (BD FACSCalibur) (8).
In the apoptosis studies, cells were plated on glass coverslips (Fisher Scientific) at a density of 1 × 105 cells/coverslip. After 24 h, the cells were infected with 30 m.o.i. of adenovirus and then exposed to 250 μm palmitate for an additional 24 h. Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) staining was performed by using the DeadEnd Fluorometric TUNEL system (Promega) according to the manufacturer's instructions.
Real-time Reverse Transcriptase PCR
Total RNA was extracted by using an RNeasy mini kit (Qiagen) according to the manufacturer's instructions. After quantification by spectrophotometry, 5 μg of total RNA was reverse transcribed to obtain cDNA by using 160 μm deoxynucleotide triphosphate, 50 ng of random hexamer primers, and 200 units of SuperScript II according to the manufacturer's recommendations (Invitrogen). TaqMan probes for ATF4 (Mm00515325) and GAPDH were purchased from Applied Biosystems. PCR products were purified by PCR purification kit (Qiagen), and mRNA expressions were determined by loading to 2% agarose gel.
Plasmid Construction and Luciferase Assays
ATF4 translational control was analyzed using pTK-ATF4-Luc plasmid which was kindly provided by Dr. Ronald Wek (Indiana University School of Medicine) (13). pTK-ATF4-Luc plasmid contains the cDNA for the full-length 5′-leader of the ATF4 mRNA along with the ATF4 start codon, inserted between the thymidine kinase (TK) promoter and firefly luciferase gene in plasmid pGL3. Transient co-transfections were carried out in triplicate accompanied with the Renilla luciferase plasmid for normalization. Plasmid transfections were performed in AdshTRα or control virus-infected MIN6 cells using Lipofectamine 2000 (Invitrogen). Twenty-four hours after transfection, the cells were exposed to vehicle or palmitate for additional 12 h. Dual luciferase assays were carried out according to the manufacturer's instructions.
Western Blot Analysis
Protein lysate was prepared by using cell lysis buffer (Cell Signaling Technology) according to the manufacturer's instructions. Protein determination by Western blot analysis was performed as described previously (9) with the following primary antibodies: anti-eIF2α, anti-phosphorylated eIF2α, and anti-cleaved caspase3 antibody (Cell Signaling); anti-ATF4 and anti-tubulin antibodies (Santa Cruz Biotechnology).
Co-immunoprecipitation Analysis
To analyze protein expression of endogenous TRα in the cells, 100 μg of protein lysate from MIN6 cells was immunoprecipitated with 5 μg of C3 mouse monoclonal antibody against the C terminus of TRα (Santa Cruz Biotechnology) or with 5 μg of mouse IgG by using the Dynabeads protein G immunoprecipitation kit (Invitrogen) according to the manufacturer's protocol. Western blot analysis was performed with anti-TRα antibody (Santa Cruz Biotechnology) against an N-terminal TRα peptide.
Animals
The animal study protocol was approved by the Institutional Animal Care and Use Committee of the University of Yamanashi. TRα-deficient mice (TRα 0/0), which lack all known products of the TRα gene, were created in the laboratory of Dr. Samarut, as described elsewhere (14). The mice were maintained at the University of Chicago for several generations and back-crossed more than 10 times onto the C57BL/6 background before TRα 0/0 male and female mice were given to Dr. Suzuki's laboratory (Fukushima Medical University, F, Japan) and were then kindly given to the present authors by Dr. Suzuki with the permission of Dr. Samarut. We generated our colony by crossing TRα 0/0 with wild-type C57BL/6 mice. First-generation heterozygotes were inbred to generate future generations of wild-type mice (WT) and TRα 0/0. Experiments were performed on 1-month-old females ranging in weight from 20 to 30 g at the start of the experiment. For individual experiments, mice from each genotype were usually matched within 1 month of age and 5 g of body weight to minimize the confounding effects of age and size. Both types of mice were fed either a control regular diet (RD) or a high fat diet (HFD) consisting of 62.2% fat, 19.6% carbohydrate, and 18.2% protein content on an energy basis (Oriental Yeast, Tokyo, Japan).
Analyses of blood glucose levels and plasma insulin concentrations, and the glucose tolerance test, were performed as described previously (9). Mice were exsanguinated by bleeding from the retro-orbital plexus at 0, 30, 60, and 120 min after glucose injection. The pancreas was removed from mice, fixed in 10% buffered formalin, and subsequently embedded in paraffin. Three different parts of the pancreatic specimen from each mouse (n = 6–8 for WT and TRα 0/0 mice) were analyzed. All islets of Langerhans in each section were digitized, and the mean area of insulin-stained β-cells was calculated using hybrid cell count software (Keyence, Osaka, Japan). The number of β-cells/islet was quantified by counting the number of cell nuclei within the insulin immunoreactive area. The β-cell size was calculated by dividing β-cell area/islet with β-cell number/islet.
Statistics
Data are expressed as the mean ± S.D. Statistical analysis was performed by using one-way analysis of variance or the unpaired two-tailed Student's t test. Probability values of less than 0.05 were considered statistically significant.
RESULTS
To explore the mechanisms of TRα-associated antiapoptotic effects, we focused on the eIF2α-ATF4 pathway in pancreatic β-cells under ER stress. To explore the role of endogenous TRα, the expression of TRα was inhibited by infection with AdshTRα. MIN6 cells, a mouse pancreatic β-cell line, were infected with 30 m.o.i. of AdshTRα or control AdLacZ. After 24 h of incubation in adenovirus-containing medium, the cells were cultured with or without palmitate, which is associated with ER stress in pancreatic β-cells, for an additional 24 h. First, we analyzed the expression of endogenous TRα in AdLacZ-infected MIN6 cells by immunoprecipitation of a nuclear extract (100 μg) with the C3 mouse monoclonal antibody, which recognizes the TR C-terminal region, or with control mouse IgG. As shown in Fig. 1, TRα was clearly expressed in MIN6 cells with or without palmitate treatment; thus, palmitate treatment had no influence on TRα protein expression. In MIN6 cells infected with 30 m.o.i. of AdshTRα, the expression of TRα was completely inhibited after 48 h of incubation. The same amount of nuclear extract prepared from HepG2 cells, which express functional TR, was also immunoprecipitated as a positive control (15). There was no TRα signal in the protein lysates of HepG2 cells that were immunoprecipitated with mouse monoclonal antibody. We also used 10 μg of protein lysate to confirm the expression of tubulin in adenovirus-infected MIN6 cells with or without palmitate treatment as a loading control.
FIGURE 1.
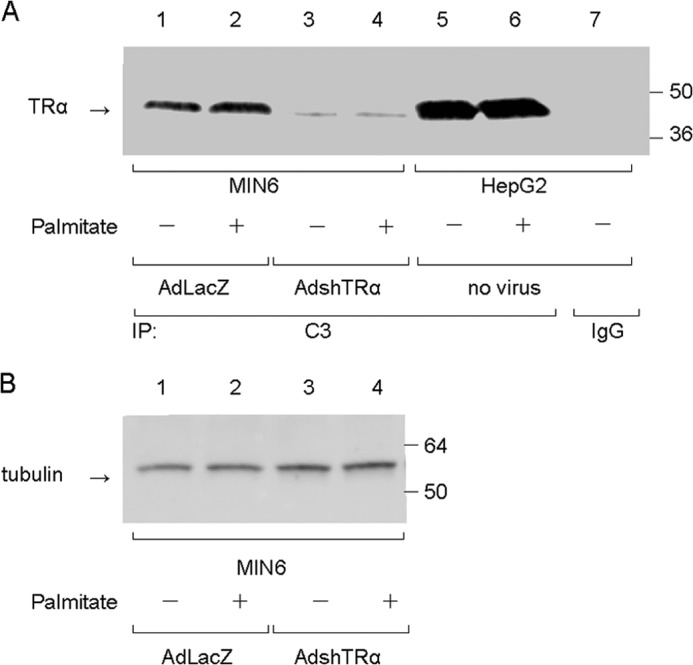
Effect of AdshTRα on the expression of endogenous TRα. A, cell lysates (100 μg) prepared from MIN6 cells were immunoprecipitated with 5 μg of mouse monoclonal anti-TRα antibody, which recognizes the TRα C terminus (C3), or 5 μg of normal mouse IgG. Western blot analysis of the precipitates was performed by using an antibody against an N-terminal TRα peptide. The same amount of protein extract prepared from HepG2 cells was used as a positive control. B, loading controls for tubulin are shown in the lower panel.
To analyze the function of endogenous TRα on free fatty acid-induced ER stress, MIN6 cells were infected with 30 m.o.i. of AdshTRα or AdLacZ and incubated with palmitate for an additional 24 h. Cell numbers did not differ between control virus- and AdshTRα-infected MIN6 cells. Twenty-four hours of palmitate treatment decreased the number of AdshTRα-infected MIN6 cells compared with that of control virus-infected cells (Fig. 2A). As shown in Fig. 2B, the TUNEL assay, an indicator of apoptosis, was positive in 1.7% of control cells, and palmitate treatment slightly enhanced apoptosis in AdLacZ-infected MIN6 cells by 1.8-fold. When endogenous TRα is knocked down, palmitate treatment significantly enhanced apoptosis by 10.9-fold. To further analyze the effect of endogenous TRα, primary cultured β-cells derived from WT or TRα 0/0 mice were incubated with palmitate. Twenty-four hours of palmitate treatment had no effect on cell proliferation of pancreatic β-cells from WT mice.
FIGURE 2.
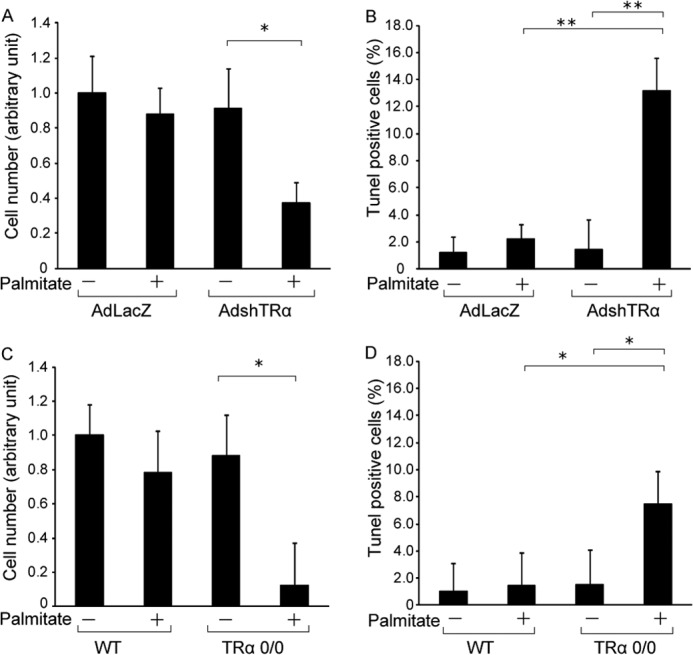
Influence of TRα on palmitate-induced apoptosis. A, MIN6 cells were infected with 30 m.o.i. of AdLacZ or AdshTRα and incubated in medium containing 250 μm palmitate for an additional 24 h. Relative cell numbers were determined by arbitrarily setting the value for control virus-infected cells in palmitate-free medium to 1. B, induction of apoptosis in MIN6 cells by palmitate treatment was evaluated using the TUNEL method. The ratio of TUNEL-positive to DAPI-stained cells is shown. C, primary cultured β-cells were treated with palmitate (250 μm) for 24 h and cell numbers analyzed. D, the ratio of TUNEL-positive to DAPI-stained β-cells is shown. Data are expressed as the mean ± S.D. (error bars; n = 6). *, p < 0.05; **, p < 0.01.
In contrast, palmitate treatment significantly decreased the cell numbers and enhanced apoptosis in pancreatic β-cells from TRα 0/0 mice (Fig. 2, C and D). These results indicate that palmitate-mediated apoptosis was significantly enhanced by TRα deletion, and the presence of TRα is a survival factor that protects β-cells from ER stress.
Harding et al. reported that eIF2α is phosphorylated and activated in stressed cells (16). Phosphorylated eIF2 α (p-eIF2α) selectively increases translation of ATF4, which contributes to adaptation to oxidative stress. In the present study, we focused on the eIF2α-ATF4 pathway to explore the mechanisms by which TRα deficiency enhanced palmitate-mediated apoptosis in pancreatic β-cells. Total eIF2α level did not differ with or without palmitate treatment. Expression of p-eIF2α and ATF4 was not observed in MIN6 cells without palmitate treatment (Fig. 3A). Palmitate treatment enhanced the expression of p-eIF2α and ATF4 in AdLacZ-infected MIN6 cells. In AdshTRα-infected MIN6 cells, the p-eIF2α level was enhanced by palmitate treatment. However, palmitate did not induce ATF4 protein expression in AdshTRα-infected MIN6 cells.
FIGURE 3.

Impaired eIF2α-ATF4 signaling pathway caused by deficiency of TRα. MIN6 cells were infected with 30 m.o.i. of AdLacZ or AdshTRα and incubated in medium containing 250 μm palmitate for an additional 24 h. A, to analyze the activation of the eIF2α-ATF4 signaling pathway, 20 μg of cell lysates prepared from adenovirus-infected MIN6 cells was immunoblotted with anti-total eIF2α antibody, anti-p-eIF2α antibody, anti-ATF4 antibody, anti-XBP1 antibody, anti-ATF6 antibody, anti-Ho1 antibody, and anti-cleaved-caspase3 antibody. Loading controls for tubulin are shown in the bottom panel. B, wild-type 5′-leader sequences of the ATF4 mRNA, which mediate translational control, were inserted between the constitutive thymidine kinase promoter and the firefly luciferase reporter gene. AdLacZ- or AdshTRα-infected MIN6 cells were co-transfected with the pTK-ATF4-Luc plasmid and a control Renilla luciferase plasmid. The transfected cells were treated with 250 μm palmitate. C, AdLacZ- or AdshTRα-infected MIN6 cells were co-infected with 30 m.o.i. of AdATF4 and treated with cycloheximide (CHX) (50 μg/ml) to block protein synthesis. At the indicated time points, cells were harvested, and 20 μg of extracts was analyzed for the protein levels of ATF4 by Western blotting. D, the expression of ATF4 and GAPDH mRNA was determined by real-time RT-PCR with 100 ng of cDNA. E and F, cellular ROS levels were measured by DCF fluorescence.
Because the ATF4 protein expression was not induced by treatment with palmitate in AdshTRα-infected MIN6 cells despite phosphorylation of eIF2α, we analyzed whether translational control of ATF4 gene is involved in the palmitate-induced ATF4 protein expression when the ATF4 transcript is stably available. MIN6 cells were transfected with the pTK-ATF4-Luc plasmid, which contained the 5′-leader of the ATF4 mRNA expressed by the constitutive TK promoter and exposed to 250 μm palmitate for 12 h. As shown in Fig. 3B, palmitate enhanced translational efficiency of ATF4 gene not only in AdLacZ-infected cells but also AdshTR-infected cells. We also evaluated the effect of TRα on the degradation of ATF4 protein. AdshTRα-preinfected MIN6 cells were infected with an adenovirus vector expressing ATF4 (AdATF4) and were then incubated with 50 μg/ml cycloheximide. The pace of ATF4 protein degradation investigated at 0, 30, 60, or 90 min was not altered by knockdown of TRα (Fig. 3C). These results revealed that TRα did not involve in palmitate-induced preferential translation of ATF4, which was mediated by phosphorylation of eIF2α in response to oxidative stress. Therefore, we then analyzed the effect of endogenous TRα on the expression of ATF4 mRNA expression in MIN6 cells treated with or without palmitate. Knockdown of TRα expression inhibited the induction of the expression of ATF4 mRNA (Fig. 3D). This result suggested that palmitate-induced ATF4 expression was dependent on TRα-mediated transcriptional activation.
We also analyzed the effects of endogenous TRα on the expression of spliced X-box binding protein 1 (sXBP1) and ATF6, which are enhanced in pancreatic β-cells under ER stress (Fig. 3A). Palmitate treatment significantly enhanced the expression of sXBP1 or ATF6 in control virus- or AdshTRα-infected MIN6 cells.
Recent reports indicated that ER stress induces the expression of heme oxygenase 1 (Ho1), which is involved in resistance to oxidative ER stress via activation of the eIF2α-ATF4 pathway (8, 17). Moreover, ATF4-knock-out cells have impaired expression of genes involved in resistance to oxidative stress (7). Palmitate treatment enhanced the expression of Ho1 in AdLacZ-infected MIN6 cells. In contrast, palmitate did not induce Ho1 expression in AdshTRα-infected MIN6 cells. These results indicated that TRα is associated with the induction of Ho1 protein expression and is critical for the expression of genes involved in resistance to oxidative stress.
To explore the mechanisms of palmitate-induced apoptosis accompanied by ATF4 silencing in AdshTRα-infected MIN6 cells, we measured cellular ROS in AdLacZ- and AdshTRα-infected cells (Fig. 3E). ROS levels did not differ between AdLacZ- and AdshTRα-infected cells. ROS levels in AdshTRα-infected cells were significantly higher than those in control virus-infected cells 24 h after incubation with palmitate. These results suggest that endogenous TRα is required to mitigate oxidative stress induced by palmitate and that the increased oxidative stress of TRα-deficient cells upon palmitate exposure partly caused enhanced apoptosis, accompanied by lack of expression of genes involved in resistance to oxidative stress in β-cells under oxidative stress. To investigate the effect of re-expressed ATF4 in TRα-deficient β-cells, the purified pancreatic β-cells from TRα 0/0 mice were infected with AdATF4. AdATF4- or AdLacZ-infected β-cells from TRα 0/0 mice were cultured with or without palmitate. Whereas ROS levels in AdLacZ-infected β-cells from TRα 0/0 mice were significantly enhanced, there were no significant differences in ROS levels between AdATF4-infected β-cells treated with or without palmitate (Fig. 3F). These results indicated that the rescued ATF4 exhibited a protective effect on the palmitate-induced oxidative stress in the TRα-deficient β-cells.
To explore the function of endogenous TRα on the stress response process in pancreatic β-cells in vivo, 1-month-old female TRα 0/0 (n = 6) or WT (n = 6) mice were fed a HFD for 30 weeks, and the effects of ER stress on pancreatic β-cells were analyzed. Fat accounts for 60% of the energy content of the HFD, and mice on the HFD typically have increased weight and adiposity (18, 19). The body weight of WT mice increased linearly with time (Fig. 4, A and B). Also, weight gain was approximately 30% lower in the TRα 0/0 mice than in the WT control mice. Food intake did not differ between HFD-fed TRα 0/0 and WT mice (data not shown). At 20 and 30 weeks of observation, the serum insulin levels of HFD-fed TRα 0/0 were decreased compared with the HFD-fed WT mice (Fig. 4C). Consistent with these results, HFD-fed TRα 0/0 mice had high blood glucose levels compared with HFD-fed WT mice after 20 or 30 weeks (Fig. 4D).
FIGURE 4.

Comparison of body weight, body composition, and glucose and insulin levels in mice A, 1-month-old female mice ranged in weight from 20 to 30 g at the start of the experiment. Mean weekly body weight gain in WT and TRα 0/0 mice fed RD or HFD is presented (n = 6–8). B, typical mice in the TRα 0/0 (left) or WT (right) groups after 30 weeks of HFD feeding are shown. C and D, nonfasting plasma levels of insulin (C) and glucose (D) are expressed as the mean ± S.D. (error bars; n = 6). *, p < 0.05.
We compared the glucose tolerance of TRα 0/0 and WT mice fed a HFD for 30 weeks. After a 6-h fast, no differences were observed in blood glucose levels of WT mice with or without HFD feeding. At 30 min after glucose injection, the blood glucose level was maximal (260 mg/dl) in mice, at which point the response to intraperitoneal injection of glucose solution (500 mg/kg body weight) was assessed. HFD-fed TRα 0/0 mice had significantly higher glucose levels after 30 min compared with HFD-fed WT mice (p < 0.05) (Fig. 5A). They showed peak glucose levels (407 mg/dl) after 30 min, and a high glucose level (>300 mg/dl) persisted after 60 min. In contrast, HFD-fed WT mice had the highest glucose value after 30 min and showed normal blood glucose levels from 60 min onward. Basal insulin levels in the HFD-fed WT mice were higher than those in the control mice (2.6-fold). HFD-fed WT mice demonstrated rapid secretion, which peaked at 30 min and subsequently dropped. HFD-fed TRα 0/0 mice did not exhibit a robust rise in levels of plasma insulin or an increase in plasma insulin levels during the glucose tolerance test despite glycemic excursion (Fig. 5B). To evaluate the early phase of insulin secretion, we calculated the insulinogenic index between 0 and 30 min (Δ insulin (IRI)0–30min/Δ blood glucose (BG)0–30min), which represents the early phase insulin response. The value of ΔIRI0–30min/ΔBG0–30min was significantly decreased in HFD-fed TRα 0/0 mice compared with the HFD-fed WT mice (0.09 ± 0.02 versus 0.69 ± 0.42, p < 0.05). Insulin area under the curve in HFD-fed TRα 0/0 mice was also lower than that in HFD-fed WT mice (2.60 ± 0.35 versus 4.80 ± 0.49, p < 0.05). These data suggested that HFD treatment reduces insulin secretion in the pancreas of TRα 0/0 mice and improves glucose levels.
FIGURE 5.
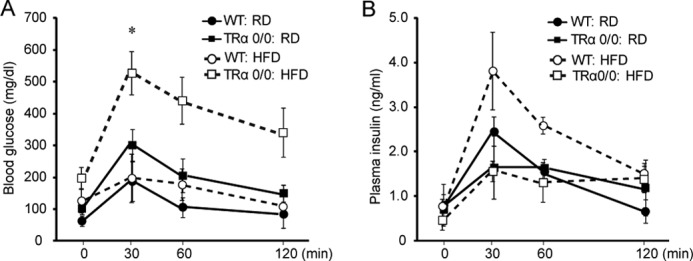
Intraperitoneal glucose tolerance test in WT or TRα 0/0 mice. Glucose tolerance test is shown in WT and TRα 0/0 mice after 30 weeks of RD or HFD feeding. Each group of mice (n = 6) was injected with glucose, and blood glucose (A) and plasma insulin (B) levels were measured after 0, 30, 60, and 120 min. Bars represent the mean ± S.D. (n = 6). *, p < 0.05.
Consistent with previous reports (20), pathological findings indicated that the numbers of β-cells were significantly enhanced in HFD-fed WT mice (Fig. 6A). In contrast, the HFD did not induce the proliferation of β-cells in TRα 0/0 mice. The individual β-cell size tended to increase in HFD-fed WT, whereas this tendency was not observed in HFD-fed TRα 0/0 mice (Fig. 6B). As shown in Fig. 6C, the TUNEL assay was positive in 0.7% of RD-fed TRα 0/0 mice, and the HFD slightly enhanced apoptosis in TR α0/0 mice by 9.6-fold. Cleaved caspase 3-positive β-cells were observed in HFD-fed TRα 0/0 mice but not in HFD-fed WT mice (Fig. 6D). These results indicate that the HFD enhanced apoptosis of β-cells in TRα 0/0 mice, thereby decreasing the β-cell population. These findings support the hypothesis that TRα plays a critical role in the protection of β-cells from HFD-induced apoptosis.
FIGURE 6.

Islet architecture in HFD-fed WT or TRα 0/0 mice. Islet architecture in WT and TRα 0/0 mice is shown after 30 weeks of RD or HFD feeding. The mice were sacrificed, and the morphology of pancreatic islets was analyzed immunohistochemically. A, β-cell number/islet was quantified by counting the number of cell nuclei within the insulin immunoreactive area. B, the mean size ± S.D. (error bars) of 30 β-cells in WT or TRα 0/0 mice is shown. C, islet apoptosis was histologically assessed in HFD-fed WT and TRα 0/0 mice using a TUNEL assay. The number of apoptotic cells in the islet area is shown. Approximately 30 islets were assessed, in which 10 slides were analyzed per mouse. D, islet apoptosis was histologically assessed in HFD-fed WT and TRα 0/0 mice using cleaved caspase3 staining. Pancreatic β-cells were visualized using anti-insulin antibody and Alexa Fluor 555-conjugated secondary antibody (red). Cleaved caspase3-positive cells were visualized with Alexa Fluor 488 (green) and are indicated by arrows.
Immunofluorescence studies revealed phosphorylated eIF2α in HFD-fed WT and TRα 0/0 mice (Fig. 7). Feeding of the HFD for 3 months induced the expression of ATF4 protein in the pancreatic β-cells of WT mice. However, ATF4 was not expressed in TRα 0/0 mice with or without the HFD. Recent reports indicate that ATF4 is involved in reducing oxidative ER stress (7) in the pancreatic β-cells of HFD-fed animals. These results indicate that TRα is important for the expression of ATF4 in pancreatic β-cells. To analyze the effects of attenuated Ho1, which is the target of ATF4 in pancreatic β-cells under ER stress, we analyzed the expression of 8-hydroxydeoxyguanosine (8-OHdG), an oxidative stress marker, in the TRα-deleted β-cells of mice after long term HFD (Fig. 8). 8-OHdG was not detected in the β-cells of WT mice fed the HFD for 3 months. The 8-OHdG-expressing β-cells were significantly enhanced in HFD-fed TRα 0/0 mice compared with WT mice. These results support the hypothesis that endogenous TRα in pancreatic β-cells is crucial for the response to oxidative stress via expression of Ho1.
FIGURE 7.
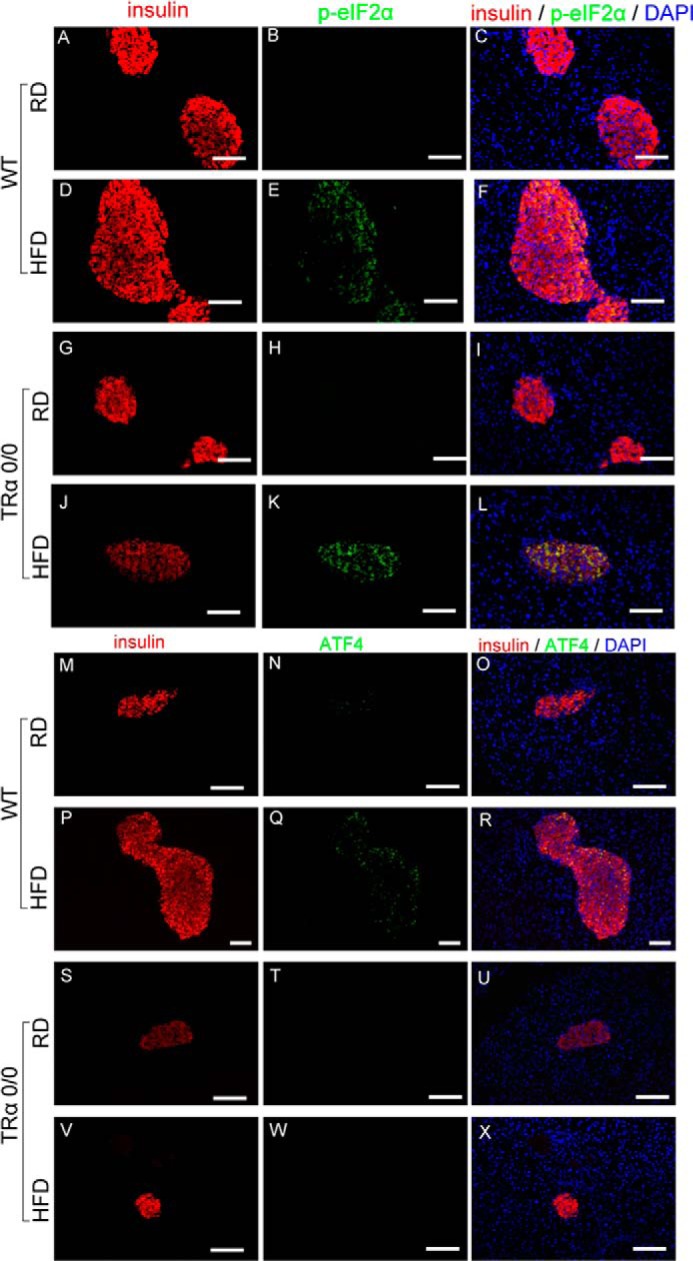
Impaired eIF2α-ATF4 signaling pathway in TRα 0/0 mice. WT and TRα0/0 mice were fed RD or HFD for 30 weeks. The expression of p-eIF2α or ATF4 was visualized with specific antibodies and Alexa Fluor 488-conjugated secondary antibody (green); β-cells were visualized with anti-insulin antibody and Alexa Fluor 555-conjugated secondary antibody (red). Scale bars, 100 μm.
FIGURE 8.
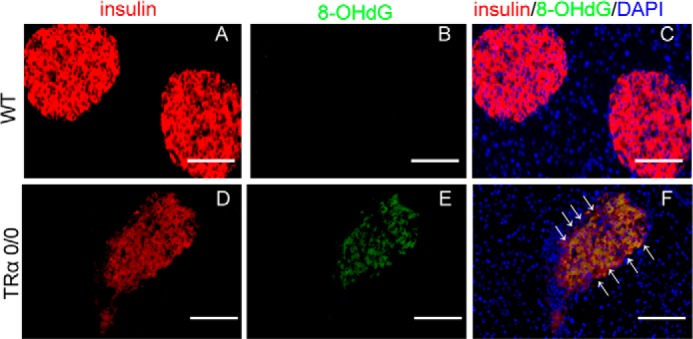
Accumulation of 8-OHdG in the islets of HFD-fed TRα 0/0 mice. WT and TRα 0/0 mice were fed HFD for 30 weeks. The expression of 8-OHdG, an oxidative stress marker, was visualized with anti-8-OHdG antibody and Alexa Fluor 488-conjugated secondary antibody (green); β-cells were visualized with anti-insulin antibody and Alexa Fluor 555-conjugated secondary antibody (red). Scale bars, 100 μm.
DISCUSSION
The marked increase in the worldwide incidence of type 2 diabetes in recent years is correlated with rising levels of obesity, implying that changing dietary habits and lifestyle may be closely associated with the development of type 2 diabetes. The loss of insulin-producing β-cells contributes to type 1 and type 2 diabetes. Normal postnatal β-cell homeostasis is maintained largely by self-replication, not by differentiation from progenitor cells. β-Cell proliferation is also essential for compensatory islet hyperplasia during metabolic stress and, therefore, for the prevention of diabetes. Proliferation is opposed by programmed β-cell elimination; the β-cell mass in the adult pancreas is dynamically regulated by constant adjustment of the balance between these two processes.
Thyroid hormones reduce glucose tolerance in humans and other animals (21). Experimental hyperthyroidism induced by thyroid hormone treatment leads to reduced glucose-induced insulin secretion from the isolated pancreas (22); this is not due to impaired insulin-secreting capacity of individual β-cells (23). It was also reported that stimulated insulin secretion is significantly increased in patients with hyperthyroidism, possibly reflecting increased β-cell sensitivity to glucose (24). This clinical evidence suggests that thyroid hormones and TR play a critical role in maintaining postnatal pancreatic β-cells.
Little is known about the molecular mechanisms of thyroid hormone regulation of the expression of ER-resident proteins. Recent reports indicate that a number of metabolic conditions closely associated with thyroid hormone signaling have been implicated in the pathogenesis of ER stress-aggravated obesity and insulin resistance (25), prompting our interest in further understanding ligand-bound TR function in pancreatic β-cells during ER stress. Our present study is the first report to demonstrate that ligand-bound TR is coupled to modulation of the stress response and improved survival of pancreatic β-cells.
In conjunction with its central role in insulin synthesis, protein folding, and transport, the ER serves as a major signal transduction organelle that integrates cellular responses to stress. The accumulation of misfolded proteins and other stresses activates an adaptive program by the ER, known as the UPR, to reestablish equilibrium. ER stress occurs when the ER exceeds its folding capacity with such disruptions, leading to an overall dysregulation of protein homeostasis, reflected as reduced maintenance of the quality and quantity of protein and an accumulation of unfolded/misfolded proteins that cannot be processed through the secretory pathway (26). This stress initiates or triggers an ER-to-nucleus signaling cascade, whereas UPR mitigates stress in three distinct manners: enhancing folding activity through increased chaperone/foldase expression; reducing ER workload through general translational attenuation; and clearance of protein aggregation and residual unfolded proteins through ER-associated protein degradation. When the UPR does not adequately reduce stress and return the cell to a state of protein homeostasis, the cell undergoes apoptosis.
Human diabetic subjects exhibit an early defect in glucose-stimulated insulin secretion (27), and autopsy studies have demonstrated reduced β-cell mass in diabetic subjects (1); therefore, intervention strategies are needed to improve β-cell function and prevent β-cell death. Increasing experimental evidence implicates a role for ER stress in the progressive reductions in insulin secretion associated with β-cell failure and apoptotic β-cell death (26, 28). Our recent reports indicated that ligand-bound TRα maintains insulin biosynthesis and secretion while expanding islet mass through the direct activation of β-cell proliferation and inhibition of β-cell death in mice (9). In the present study, we demonstrate that endogenous TRα is coupled to the modulation of UPR and improved survival of ER-stressed pancreatic β-cells.
The signaling pathways engaged following ER stress involve three different arms of the UPR (29), which is activated by the ER stress luminal sensors ATF6, IRE1, and PERK. Activated PERK phosphorylates the translation initiation factor eIF2α, which transiently attenuates global protein synthesis, reducing the load on the ER. ER stress-mediated eIF2α phosphorylation and the ensuing repression of general translation allow the dysfunctional ER a window of opportunity to recover from cellular stress (16). Phosphorylated eIF2α leads to enhanced expression of ATF4, which in turn up-regulates the expression of key genes important for recovery from ER stress, including oxidative stress.
Our results indicate that ligand-bound TRα is important for the expression and induction of ATF4 in pancreatic β-cells. In TRα-depleted β-cells, the abundance of ATF4 protein was significantly decreased, and ER stress-induced expression of Ho1 was not observed. In AdshTRα-infected pancreatic β-cells, ER stress-mediated cell death was enhanced with oxidative stress. In contrast, an increase in PERK phosphorylation concomitant with the stimulation of eIF2α phosphorylation was observed in TRα-depleted β-cells. These results support the hypothesis that ligand-bound TRα is critical for the eIF2α-ATF4 pathway.
TRα 0/0 mice are viable and exhibit reduced linear growth, bone maturation delay, and moderate hypothermia (30). The skeletal phenotype of TRα 0/0 mice includes retarded ossification, failure of progression of hypertrophic chondrocyte differentiation, and disorganization of epiphyseal growth plate architecture. Basal thyroid hormone concentrations in TRα 0/0 mice were only mildly impaired, and growth hormone concentrations were unchanged compared with those of wild-type mice, indicating the notion that the deletion of the TRα gene fully accounts for this phenotype (30). Furthermore, TRα is also expressed in white adipose tissue, and TRα knock-in mutant mice had impairments in the adipogenesis of white adipose tissue due to the repression of the expression and transcription activity of the regulator genes of adipogenesis (31). TRα mediates growth retardation and reduction of white adipose tissue mass, contributing to the phenotype showing reduced body weight observed in HFD-treated TRα 0/0 mice.
Hypothyroidism is associated with deterioration of glucose metabolisms, and this effect is accompanied by reduced plasma insulin concentration (32). Several reports indicated that overt hypothyroidism is associated with profound changes in insulin sensitivity and insulin secretion, and these changes are reversible, at least in part, after recovery of thyroid function (33, 34). Thyroid hormone effects on glucose homeostasis and on endocrine function of the pancreas are the result of an interaction of the thyroid hormone with its receptors. Recently, we also reported that liganded TRα induced expansion of the β-cell mass after streptozotocin-induced β-cell loss by an increase in the β-cell replication and reprogramming of pancreatic acinar cells (12). In this study, long term exposure to oxidative stress enhanced cell death of pancreatic β-cells when the expression of endogenous TRα was extinguished. Nevertheless, the effect of hypothyroidism on the oxidative stress response is still unclear. Further study is needed to figure out the whole relationships between thyroid hormone and glucose homeostasis.
Pancreatic β-cells represent one of the tissues most susceptible to oxidative stress, due in part to low levels of antioxidant gene expression alongside an environment that produces ROS through the respiratory chain and during disulfide linkage creation in insulin biosynthesis. The generation of free radicals, which can be a consequence of persistent hyperglycemia and excessive intracellular levels of triglyceride and nonesterified fatty acids that attack cellular organelles, including the mitochondria, leads to dysfunctional electron transport and triggers cell death of pancreatic β-cells. The results of our study demonstrated why defective TRα leads to β-cell failure. Furthermore, we indicated that both p-eIF2α- and TRα-induced ATF4 were critical for successful adaptation to cellular stress. Our findings led to the hypothesis that TRα might be involved in a positive regulatory mechanism that controls the maintenance of pancreatic β-cell mass under ER stress.
Acknowledgments
We are grateful to Dr. Satoru Suzuki, Fukushima Medical University, for expert assistance with animal experiments; the staff of Life Technology Japan (Tokyo) for preparing the adenoviruses; and Dr. Ronald C. Wek, Indian University School of Medicine, for pTK-ATF4-Luc plasmid, which contained the 5′-leader of the ATF4 mRNA expressed from a constitutive TK promoter.
This work was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI Grant 24591359.
- ER
- endoplasmic reticulum
- AdATF4
- adenovirus vector expressing ATF4 gene
- AdLacZ
- adenovirus expressing lacZ
- AdshTRα
- adenovirus expressing double-stranded short hairpin RNA against TRα
- ATF
- activating transcription factor
- DCF
- 2′,7′-dichlorodihydrofluorescein diacetate
- HFD
- high fat diet
- Ho1
- heme oxygenase 1
- m.o.i.
- multiplicity of infection
- 8-OHdG
- 8-hydroxydeoxyguanosine
- PERK
- pancreatic ER kinase-like kinase
- RD
- control regular diet
- ROS
- reactive oxygen species
- TK
- thymidine kinase
- TR
- thyroid hormone receptor
- UPR
- unfolded protein response.
REFERENCES
- 1. Butler A. E., Janson J., Soeller W. C., Butler P. C. (2003) Increased β-cell apoptosis prevents adaptive increase in β-cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes 52, 2304–2314 [DOI] [PubMed] [Google Scholar]
- 2. Mathis D., Vence L., Benoist C. (2001) β-Cell death during progression to diabetes. Nature 414, 792–798 [DOI] [PubMed] [Google Scholar]
- 3. Eizirik D. L., Darville M. I. (2001) β-Cell apoptosis and defense mechanisms: lessons from type 1 diabetes. Diabetes 50, S64–69 [DOI] [PubMed] [Google Scholar]
- 4. Fonseca S. G., Urano F., Burcin M., Gromada J. (2010) Stress hyperactivation in the β-cell. Islets 2, 1–9 [DOI] [PubMed] [Google Scholar]
- 5. Szegezdi E., Logue S. E., Gorman A. M., Samali A. (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ellgaard L., Molinari M., Helenius A. (1999) Setting the standards: quality control in the secretory pathway. Science 286, 1882–1888 [DOI] [PubMed] [Google Scholar]
- 7. Harding H. P., Zhang Y., Zeng H., Novoa I., Lu P. D., Calfon M., Sadri N., Yun C., Popko B., Paules R., Stojdl D. F., Bell J. C., Hettmann T., Leiden J. M., Ron D. (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633 [DOI] [PubMed] [Google Scholar]
- 8. Suragani R. N., Zachariah R. S., Velazquez J. G., Liu S., Sun C. W., Townes T. M., Chen J. J. (2012) Heme-regulated eIF2α kinase-activated ATF4 signaling pathway in oxidative stress and erythropoiesis. Blood 119, 5276–5284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Furuya F., Shimura H., Yamashita S., Endo T., Kobayashi T. (2010) Liganded thyroid hormone receptor-α enhances proliferation of pancreatic β-cells. J. Biol. Chem. 285, 24477–24486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miyazaki J., Araki K., Yamato E., Ikegami H., Asano T., Shibasaki Y., Oka Y., Yamamura K. (1990) Establishment of a pancreatic β-cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 127, 126–132 [DOI] [PubMed] [Google Scholar]
- 11. Hassani Z., François J. C., Alfama G., Dubois G. M., Paris M., Giovannangeli C., Demeneix B. A. (2007) A hybrid CMV-H1 construct improves efficiency of PEI-delivered shRNA in the mouse brain. Nucleic Acids Res. 35, e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Furuya F., Shimura H., Asami K., Ichijo S., Takahashi K., Kaneshige M., Oikawa Y., Aida K., Endo T., Kobayashi T. (2013) Ligand-bound thyroid hormone receptor contributes to reprogramming of pancreatic acinar cells into insulin-producing cells. J. Biol. Chem. 288, 16155–16166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dey S., Baird T. D., Zhou D., Palam L. R., Spandau D. F., Wek R. C. (2010) Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. J. Biol. Chem. 285, 33165–33174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Macchia P. E., Takeuchi Y., Kawai T., Cua K., Gauthier K., Chassande O., Seo H., Hayashi Y., Samarut J., Murata Y., Weiss R. E., Refetoff S. (2001) Increased sensitivity to thyroid hormone in mice with complete deficiency of thyroid hormone receptor α. Proc. Natl. Acad. Sci. U.S.A. 98, 349–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin K., Chen S., Zhu X. G., Shieh H., McPhie P., Cheng S. (1997) The gene regulating activity of thyroid hormone nuclear receptors is modulated by cell type-specific factors. Biochem. Biophys. Res. Commun. 238, 280–284 [DOI] [PubMed] [Google Scholar]
- 16. Harding H. P., Zhang Y., Bertolotti A., Zeng H., Ron D. (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 5, 897–904 [DOI] [PubMed] [Google Scholar]
- 17. Liu X. M., Peyton K. J., Ensenat D., Wang H., Schafer A. I., Alam J., Durante W. (2005) Endoplasmic reticulum stress stimulates heme oxygenase-1 gene expression in vascular smooth muscle: role in cell survival. J. Biol. Chem. 280, 872–877 [DOI] [PubMed] [Google Scholar]
- 18. Woods S. C. (2004) Lessons in the interactions of hormones and ingestive behavior. Physiol. Behav. 82, 187–190 [DOI] [PubMed] [Google Scholar]
- 19. Hill J. O., Melanson E. L., Wyatt H. T. (2000) Dietary fat intake and regulation of energy balance: implications for obesity. J. Nutr. 130, 284S–288S [PubMed] [Google Scholar]
- 20. Ahrén J., Ahrén B., Wierup N. (2010) Increased β-cell volume in mice fed a high-fat diet: a dynamic study over 12 months. Islets 2, 353–356 [DOI] [PubMed] [Google Scholar]
- 21. Lenzen S., Bailey C. J. (1984) Thyroid hormones, gonadal and adrenocortical steroids and the function of the islets of Langerhans. Endocr. Rev. 5, 411–434 [DOI] [PubMed] [Google Scholar]
- 22. Lenzen S., Joost H. G., Hasselblatt A. (1976) Thyroid function and insulin secretion from the perfused pancreas in the rat. Endocrinology 99, 125–129 [DOI] [PubMed] [Google Scholar]
- 23. Lenzen S. (1978) Dose-response studies on the inhibitory effect of thyroid hormones on insulin secretion in the rat. Metabolism 27, 81–88 [DOI] [PubMed] [Google Scholar]
- 24. The Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 329, 977–986 [DOI] [PubMed] [Google Scholar]
- 25. Ozcan U., Cao Q., Yilmaz E., Lee A. H., Iwakoshi N. N., Ozdelen E., Tuncman G., Görgün C., Glimcher L. H., Hotamisligil G. S. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 [DOI] [PubMed] [Google Scholar]
- 26. Schröder M., Kaufman R. J. (2005) The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739–789 [DOI] [PubMed] [Google Scholar]
- 27. Bagdade J. D., Bierman E. L., Porte D., Jr. (1967) The significance of basal insulin levels in the evaluation of the insulin response to glucose in diabetic and nondiabetic subjects. J. Clin. Invest. 46, 1549–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang H., Kouri G., Wollheim C. B. (2005) ER stress and SREBP-1 activation are implicated in β-cell glucolipotoxicity. J. Cell Sci. 118, 3905–3915 [DOI] [PubMed] [Google Scholar]
- 29. Calfon M., Zeng H., Urano F., Till J. H., Hubbard S. R., Harding H. P., Clark S. G., Ron D. (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 [DOI] [PubMed] [Google Scholar]
- 30. Gauthier K., Plateroti M., Harvey C. B., Williams G. R., Weiss R. E., Refetoff S., Willott J. F., Sundin V., Roux J. P., Malaval L., Hara M., Samarut J., Chassande O. (2001) Genetic analysis reveals different functions for the products of the thyroid hormone receptor α locus. Mol. Cell. Biol. 21, 4748–4760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ying H., Araki O., Furuya F., Kato Y., Cheng S. Y. (2007) Impaired adipogenesis caused by a mutated thyroid hormone α1 receptor. Mol. Cell. Biol. 27, 2359–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Handisurya A., Pacini G., Tura A., Gessl A., Kautzky-Willer A. (2008) Effects of T4 replacement therapy on glucose metabolism in subjects with subclinical (SH) and overt hypothyroidism (OH). Clin. Endocrinol. 69, 963–969 [DOI] [PubMed] [Google Scholar]
- 33. Rochon C., Tauveron I., Dejax C., Benoit P., Capitan P., Fabricio A., Berry C., Champredon C., Thieblot P., Grizard J. (2003) Response of glucose disposal to hyperinsulinaemia in human hypothyroidism and hyperthyroidism. Clin. Sci. 104, 7–15 [DOI] [PubMed] [Google Scholar]
- 34. Stanicka S., Vondra K., Pelikánová T., Vlcek P., Hill M., Zamrazil V. (2005) Insulin sensitivity and counter-regulatory hormones in hypothyroidism and during thyroid hormone replacement therapy. Clin. Chem. Lab. Med. 43, 715–720 [DOI] [PubMed] [Google Scholar]