

Background: Biochemical characterization of voltage-dependent anion channel 2 (VDAC2) is limited due to an inability to obtain functional protein.
Results: The crystal structure of VDAC2 suggests a dimer interface that is confirmed by double electron-electron resonance and cross-linking.
Conclusion: zfVDAC2 has a fractional dimeric population.
Significance: VDAC isoforms are structurally similar, but this study has identified a number of hot spots that require further exploration.
Keywords: Biophysics, Ion Channels, Mitochondria, X-ray Crystallography, Zebrafish, DEER, VDAC2
Abstract
In recent years, there has been a vast increase in structural and functional understanding of VDAC1, but VDAC2 and -3 have been understudied despite having many unique phenotypes. One reason for the paucity of structural and biochemical characterization of the VDAC2 and -3 isoforms stems from the inability of obtaining purified, functional protein. Here we demonstrate the expression, isolation, and basic characterization of zebrafish VDAC2 (zfVDAC2). Further, we resolved the structure of zfVDAC2 at 2.8 Å resolution, revealing a crystallographic dimer. The dimer orientation was confirmed in solution by double electron-electron resonance spectroscopy and by cross-linking experiments disclosing a dimer population of ∼20% in lauryldimethine amine oxide detergent micelles, whereas in lipidic bicelles a higher population of dimeric and higher order oligomers species were observed. The present study allows for a more accurate structural comparison between VDAC2 and its better-studied counterpart VDAC1.
Introduction
Mitochondria are often depicted as the cell's “powerhouse” because they generate ATP from ADP and inorganic phosphate through the oxidative phosphorylation process. It is now firmly established that mitochondria have a far more expansive role in cellular function, including signaling, cell death/survival, and differentiation (1–3). As such, mitochondria are central to normal human physiology as well as disease. The most abundant proteins in the outer mitochondrial membrane, the voltage-dependent anion channels (VDACs),5 are large pore-forming proteins that mediate the efficient exchange of metabolites and ions between the cytosol and the mitochondrial intermembrane space (IMS). In mammals, there are three VDAC isoforms (VDAC1–3), which have a high degree of sequence homology, yet their relative ratios and distribution between cell types are varied.
The sequence identity/similarity between human VDAC1 (hVDAC1), hVDAC2 and hVDAC3 is 75%/91% and 67%/85%, respectively (Fig. 1). Undoubtedly, VDAC1 is the best characterized isoform. After decades of struggle, three groups independently resolved the high resolution structure of VDAC1 (4–6). Using the lipidic bicelle crystallization technique (6–8), we solved a high resolution x-ray structure of mouse VDAC1 (mVDAC1) at a resolution of 2.3 Å. All reported high resolution structures of VDAC1 revealed a novel fold of a 19-stranded β-barrel with an N-terminal α-helix lying inside the pore. This helix is aligned nearly parallel to the membrane plane, causing a partial narrowing of the pore. Numerous studies have established the functional importance of the N-terminal α-helix in regulating the flux of metabolites through the channel (9, 10), and the observation that it is localized within the pore makes it ideally suited to regulate the passage of ions and metabolites through the pore. Based on molecular dynamics simulations and a series of cross-linking studies, the structure is believed to reside in the “open” conformation, facilitating the passage of ATP and other nucleotides (11, 12).
FIGURE 1.

Sequence alignment between VDAC1 and -2. A select set of VDAC1 and -2 sequences are aligned. For reference, the secondary structure elements are represented as two-dimensional schematics above sequences.
In contrast to VDAC1, structural and biochemical characterization of VDAC2 and -3 is rather limited, yet these isoforms have distinct physiological functions. In particular, VDAC2 null mice are embryonic lethal (13), whereas null mice of VDAC1 and -3 isoforms display mild phenotypes (14, 15). Furthermore, VDAC2 has been shown to interact and inhibit the proapoptotic protein BAK, thereby hampering mitochondrial apoptosis (13). In cardiomyocytes, VDAC2 specifically interacts with the ryanodine receptor and mediates Ca2+ transfer from the sarcoplasmic reticulum into mitochondria (16), implicating a role for VDAC2 in cardiac Ca2+ homeostasis. Additionally, the small quinazolinone agent erastin, an anti-tumor compound, interacts with VDAC2 to induce cell death in tumor cells expressing mutant RAS (17, 18). In total, there is significant evidence that despite the high sequence identity in this family, VDAC2 has a specific phenotype, which is distinct from those of the other two VDAC isoforms.
To gain insight into the structure and function of VDAC2, we expressed and purified zfVDAC2. The purified protein forms active channels in lipid planar bilayers, and we were able to determine the structure at 2.8 Å resolution. The overall structure is very similar to VDAC1, revealing a dimer interface between β17, β18, β19, β1, and β3. Using double electron-electron resonance (DEER) measurements and cross-linking experiments, we confirmed the dimer topology in solution and found a fractional dimer content of ∼20% in detergent micelles and ∼40% in lipidic bicelles.
EXPERIMENTAL PROCEDURES
Cloning of pQE60-zfVDAC2C127A and Single-cysteine Mutants
The full-length zfVDAC2 ORF was purchased from Open Biosystems and PCR-amplified. PCR primers were used to add an NcoI site (5′) and a His6 tag followed by a HindIII site (3′) for subsequent NcoI/HindIII cloning into the pQE60 expression plasmid. For DEER experiments, we created construct pQE60-zfVDAC2C127A/S232C. Three individual PCRs were used to create three overlapping fragments of the zfVDAC2 ORF and to introduce the C127A and S232C mutations, respectively, by mutagenesis primers. Individual PCR fragments were fused by splicing by overlap extension-PCR, and the resulting product was ligated into the pQE60 backbone using NcoI/HindIII. All PCR-amplified products were fidelity-checked by sequencing (Laragen, Inc., Culver City, CA).
To create single-Cys mutants for cross-linking experiments, the Cys-less backbone pQE60-zfVDAC2C127A was created by fusion of base pairs 427–870 of the ORF from pQE60-zfVDAC2 into pQE60-zfVDAC2C127A/S232C using PstI and HindIII, thereby eliminating the S232C mutation. Single-cysteine mutants were subsequently generated in the pQE60-zfVDAC2C127A backbone by mutagenesis PCR. For pQE60-zfVDAC2C127A/V27C, pQE60-zfVDAC2C127A/D50C, pQE60-zfVDAC2C127A/S52C, and pQE60-zfVDAC2C127A/T83C mutation primers were used to introduce the respective mutations in two independent PCRs, which were then fused in a splice overlap extension PCR. Final PCR products were ligated into pQE60-zfVDAC2C127A using XhoI and PstI. For pQE60-zfVDAC2C127A/Q196C, pQE60-zfVDAC2C127A/L251C, pQE60-zfVDAC2C127A/I255C, and pQE60-zfVDAC2C127A/I257C mutagenesis PCR was performed as described above, but PCR products were ligated into pQE60-zfVDAC2C127A using PstI and NheI. All PCR-amplified products were fidelity-checked by sequencing (Eurofins MWG Operon, Ebersberg, Germany).
Expression and Purification of zfVDAC2
Cloning, expression, and purification of zfVDAC2 followed the previously reported protocol for mVDAC1 (6). In short, M15 Escherichia coli cells carrying pQE60-zfVDAC2 were grown in Luria broth with agitation at 37 °C, induced at an OD of 1 with 0.4 mm isopropyl-1-thio-β-d-galactopyranoside and grown for an additional 4–5 h. Cells were harvested by centrifugation and resuspended in 50 mm Tris·HCl (pH 8.0), 2 mm EDTA, 20% sucrose. The cell slurry was lysed by consecutive incubation with 12.5 μg/ml lysozyme and 0.6% Triton X-100 followed by sonication. Inclusion bodies were obtained through centrifugation at 12,000 × g for 15 min, washed, and solubilized in 20 mm Tris·HCl (pH 8.0), 300 mm NaCl, 6 m guanidinium hydrochloride. zfVDAC2 protein was purified using a Talon metal affinity column and refolded in a three-step dialysis at 10 mg/ml. Dialysis buffers were as follows: 1) 20 mm Tris·HCl (pH 8.0), 300 mm NaCl, 1 mm DTT, 3 m guanidinium hydrochloride; 2) 20 mm Tris·HCl (pH 8.0), 300 mm NaCl, 1 mm DTT, 0.1% LDAO; and 3) 20 mm Tris·HCl (pH 8.0), 50 mm NaCl, 1 mm DTT, 0.1% LDAO. 2% LDAO was added to the protein solution prior to the second dialysis step. Refolded protein was concentrated, ultracentrifuged (355,000 × g, 30 min) to remove aggregated protein, and passed over a Superdex 200 column in 150 mm NaCl, 0.1% LDAO, 20 mm Tris·HCl (pH 8.0) to obtain a homogenous protein population (Fig. 2, A and B). For crystallization, fractions containing the non-aggregated zfVDAC2 peak were collected, concentrated, and buffer changed to 50 mm NaCl, 0.1% LDAO, 20 mm Tris·HCl (pH 8.0) using an Amicon Ultra-30k (Millipore) concentrator.
FIGURE 2.

Functional characterization of zfVDAC2. A, size exclusion chromatography of zfVDAC2. The dashed box represents a non-aggregated homogenous population of protein used for further characterization. B, 12% SDS-polyacrylamide gel stained with Coomassie Brilliant Blue showing a pure sample of zfVDAC2. C, electrophysiology recordings of zfVDAC2 in diphytanoyl phosphatidylcholine bilayers in 10 mm Tris·HCl (pH 7.4), 1 m KCl, 5 mm CaCl2. Total point histograms at each applied voltage were plotted to calculate the conductance (G) of the open and closed state. mAU, milliabsorbance units.
Crystallization
Crystals of ∼0.6 × 0.2 × 0.1 mm were grown by the hanging drop vapor diffusion method using the Mosquito crystallization robot. A 12 mg/ml protein solution was mixed (1:1) with a well solution of 100 mm Tris·HCl (pH 8.0), 100 mm KCl, and PEG 2000 ranging between 19 and 24%. Subsequent optimization was performed by the addition of n-undecyl-β-d-thiomaltopyranoside at its critical micelle concentration (0.011%). Crystals were cryoprotected by transferring them for 10 s into a solution containing 100 mm Tris·HCl (pH 8.0), 100 mm KCl, 50% PEG 2000, 0.011% n-undecyl-β-d-thiomaltopyranoside prior to flash freezing in liquid nitrogen.
Lipid Planar Bilayer
Lipid bilayer experiments were performed as described previously (6). In brief, recordings were performed in diphytanoyl phosphatidylcholine bilayers across a 0.1-mm hole in 10 mm Tris·HCl (pH 7.4), 1 m KCl, 5 mm CaCl2. After channel insertion, currents were recorded in response to test pulses between −50 and +50 mV. Total point histograms at each applied voltage were plotted to calculate the conductance (G) of open and closed states (Fig. 2C).
X-ray Data Collection and Processing
X-ray diffraction data were collected to 2.7 Å on flash-frozen crystals (−180 °C) at the Advanced Light Source (Berkeley, CA) (beam line 5.0.2). Indexing and integration were performed with XDS (19), using data up to 2.8 Å, and the space group was determined to be P3221 with the following unit cell dimensions: a = 72.47 Å, b = 72.47 Å, and c = 177.95 Å (α = β = 90° and γ = 120°). One molecule is present per asymmetric unit with an estimated solvent content of 70.4% (Vm = 4.15 Å3/Da) based on the Matthews probability calculator (20, 21). The structure was solved by molecular replacement using the murine VDAC1 coordinates (Protein Data Bank code 3EMN) as the search model in PHENIX (21). After generation of the initial model, it was manually rebuilt using Coot (22), followed by iterative rounds of refinement in PHENIX (21). The final model (Protein Data Bank code 4BUM) contained 283 amino acids, six water molecules, and one LDAO molecule with a final R-factor and R-free of 0.246 and 0.282, respectively. Data collection and refinement statistics are listed in Table 1. All figures containing structures were generated using PyMOL (23) or Chimera (24).
TABLE 1.
Crystallography statistics for zfVDAC2
| Data collection | |
| Space group | P3221 |
| Cell dimensions | |
| a, b, c (Å) | 72.47, 72.47, 177.95 |
| α, β, γ (degrees) | 90, 90, 120 |
| Resolution (Å) | 51.29–2.701 |
| Rsym or Rmerge | 0.08 (0.76)a |
| I/σI | 12.99 (1.95) |
| Completeness (%) | 97.98 (97.3) |
| Redundancy | 3.5 (3.4) |
| Refinement | |
| Resolution (Å) | 50–2.8 |
| No. of reflections | 53,149 |
| Rwork/Rfree | 24.55/28.22 (31.23/35.34) |
| No. of atoms | 2151 |
| Protein | 2133 |
| Ligand/ion | 12 |
| Water | 6 |
| B-Factors | |
| Protein | 51.0 |
| Ligand/ion | 58.3 |
| Water | 24.4 |
| Ramachandran favored (%) | 94 |
| Ramachandran outliers (%) | 0.71 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.004 |
| Bond angles (degrees) | 0.87 |
a Values in parentheses are for the highest resolution shell.
Spin Labeling, Continuous Wave (CW) EPR Spectroscopy, and DEER Experiments
For CW and DEER experiments, DTT was removed during the size exclusion chromatography step, and the protein was incubated with a 5-fold molar excess of 1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl methanethiosulfonate for 16 h at 4 °C to generate the R1 side chain (25). Excess spin label was removed by repeated washes using an Amicon Ultra-30k (Millipore) concentrator in 20 mm Tris·HCl (pH 8.0), 50 mm NaCl, 0.1% LDAO, 20% glycerol. For zfVDAC2C127A/S232C, the fraction of spin-labeled protein was >98% as estimated from a 4,4′-dithiodipyridine titration for free thiol after the spin labeling reaction (26). A fraction of the spin-labeled zfVDAC2C127A/S232C was reconstituted in bicelles by mixing the protein with bicelles in a 4:1 ratio with a 35% (2.8:1) l-α-dimyristoylphosphatidylcholine/CHAPSO bicellar solution.
The CW EPR spectra of zfVDAC2 232R1 in LDAO and bicelles were recorded at room temperature on a Varian E-109 spectrometer fitted with a two-loop one-gap resonator; samples of 5 μl were contained in a borosilicate capillary tube (0.64-mm inner diameter × 0.84-mm outer diameter; VitroCom Inc.). For LDAO, a second spectrum was recorded with the sample containing 30% (w/w) sucrose solution. The spectra were recorded using 2-milliwatt microwave power and 1-G field modulation at 100 kHz.
Four-pulse DEER experiments (27) at Q band were conducted at 80 K on a Bruker ELEXSYS 580 spectrometer fitted with a Bruker EN5107D2 resonator. The protein concentration during the DEER experiments was maintained at or below 200 μm. 20 μl of spin-labeled protein reconstituted in LDAO or bicelles containing 20% (v/v) glycerol were loaded into a borosilicate capillary tube (1.4-mm inner diameter × 1.7-mm outer diameter; VitroCom) and subsequently flash-frozen in liquid nitrogen. A 36-ns π-pump pulse was set at the maximum absorption spectra, and the observer π/2 (16-ns) and π (32-ns) pulses were positioned 50 MHz (17.8 G) up-field. Distance distributions were obtained from the raw dipolar evolution time using the program LongDistances (available at the Hubbell Laboratory Web site). A dipolar evolution function (DEF) was obtained after applying an exponentially decaying background function (using a dimensionality of 3) to correct for random intermolecular dipolar interactions. The DEF was fit to obtain interspin distance distributions (Fig. 7, B and C).
FIGURE 7.

Interspin distance measurements of zfVDAC2 232R1. A, cartoon representation of zfVDAC2 232R1 showing the expected distance based on modeling of R1 on the zfVDAC2 structure. Models of the R1 side chain at site 232 are shown as stick representations. The black dashed line shows the expected interspin distance in the crystallographic dimer. B, left, raw data (black) and exponentially decaying background signal (magenta) arising from random intermolecular distances in LDAO. For analysis, the raw data were truncated beyond 2.6 μs (vertical dashed line) to suppress the artifact caused by pulse overlap. Right, background-corrected DEF of zfVDAC2 232R1 reconstituted in LDAO. The fit of the DEF is shown in red. Δ, “depth of modulation” see “Experimental Procedures.” C, left, raw data (cyan) and exponentially decaying background signal (magenta) of DEER data in bicelles. The raw data were truncated beyond 2.6 μs (vertical dashed line) to suppress artifact. Right, background-corrected DEF of zfVDAC2 232R1 reconstituted in bicelles. D, distance distributions of zfVDAC2 232R1 reconstituted in LDAO and bicelles are shown in black and cyan, respectively. The vertical black bar indicates the expected distance from modeling the R1 side chain at site 232 shown in A. E, top spectra, EPR signals of 232R1 in LDAO (black) and bicelles (cyan). Bottom spectra, EPR spectra of osmolyte perturbations for residue 232R1 in LDAO recorded in buffer (black) and in buffer containing 30% sucrose (red). Spectral intensities corresponding to relatively immobile (i) and mobile states (m) of R1 are indicated.
In addition to the distribution of distances between interacting spins, the average number of interacting spins in a molecular entity, nm, can be determined according to Equation 1 (28),
where Δ is the “depth of modulation” (shown in Fig. 7, B and C), and C is a constant that depends on the pump pulse profile, the resonator, and the spectral line shape of the nitroxide spin label (28). In the present experiments, C was determined using double cysteine mutants of T4 lysozyme (N68C/T109C and T109C/V131C) known to be quantitatively labeled with R1 as judged by determination of free thiols after the labeling reaction using a 4,4′-dithiodipyridine assay (26). From the depth of modulation and using nm = 2, C is obtained from Equation 1; the values obtained from the two T4 lysozyme mutants agreed to within 1%. From the experimentally determined depth of modulation and C, the value of nm for VDAC2 in LDAO or bicelles can be determined.
Cross-linking Experiments
Disulfide bonds between single-cysteine mutants were induced with the oxidizing reagent dichloro(1,10-phenanthroline)Cu(II). zfVDAC2 mutants were diluted to 15 μm in buffer containing 150 mm NaCl, 20 mm Tris·HCl (pH 8.0), 5 mm DTT, and 0.1% LDAO. The oxidizing reaction was performed with 5 mm dichloro(1,10-phenanthroline)Cu(II) at 4 °C for 2 min and subsequently quenched with 10 mm EDTA and 10 mm N-ethylmaleimide. The results were analyzed on 12% SDS-polyacrylamide gels stained with a Coomassie-based InstantBlue reagent (Expedeon).
RESULTS
Protein Purification and Lipid Bilayer Conductance
Native recombinant zfVDAC2 protein with a His tag fused to the C terminus was prepared using a two-step column purification that consisted of 1) solubilized inclusion bodies that were applied to a Talon metal affinity column and eluted with 150 mm imidazole and 2) concentrated protein that was refolded and applied to a Superdex 200 column to remove aggregation and obtain a homogenous protein population (Fig. 2, A and B). Purified zfVDAC2 protein was inserted in planar lipid bilayers, where it exhibited a single channel conductance of 4.2 ± 0.5 nanosiemens at 10 mV in 1 m KCl (open state; Fig. 2C). As the applied voltage was increased to ±30 mV, the channel presented distinct gating behavior between the open and closed states with a conductance of 1.7 ± 0.2 nanosiemens (Fig. 2C). This behavior is in good agreement with recordings from VDAC1 isolated from rats (29) and indicates that zfVDAC2, from our preparation, forms active channels. To further understand the functional aspects of ion and metabolite trafficking as well as the relation of zfVDAC2 to previously reported VDAC1, we pursued a high resolution structure of zfVDAC2.
Structural Overview
Optimized crystals of zfVDAC2 belong to the trigonal space group P3221 with one molecule per asymmetric unit. The structure was determined by molecular replacement using the original mVDAC1 coordinates (Protein Data Bank code 3EMN) as a search model. The only part of the model that required manual rebuilding following the molecular replacement procedure was the loop between β-strands 1 and 2. The model was ultimately refined from merged data to a resolution of 2.8 Å with an R-free of 24.6% and an R-work of 28.2% (Table 1). 282 of the 283 amino acids of zfVDAC2 were assigned along with six water molecules and one copy of LDAO. Further details regarding the crystallization and structure determination can be found under “Experimental Procedures.” A refined electron density map is shown in Fig. 3C.
FIGURE 3.
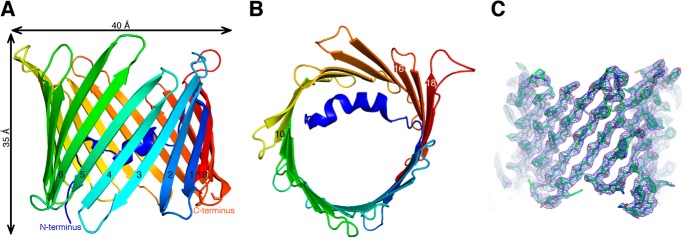
Overall structure of zfVDAC2. Shown are cartoon representations of zfVDAC2 viewed perpendicular to the membrane plane (A) and in the membrane plane (B). The protein backbone is rainbow-colored from the N terminus (blue) to the C terminus (red). A 2Fo − Fc map contoured at 1.5σ is displayed in a blue wire frame with the zfVDAC2 model shown in sticks (C).
The structure of zfVDAC2 is similar to that of mVDAC1 (Protein Data Bank code 3EMN), forming a β-barrel composed of 19 β-strands (Fig. 3). All of the strands exhibit an antiparallel pattern with the exception of strands 1 and 19, which associate in a parallel manner to seal the barrel. The maximal height and width of the β-barrel are 35 and 40 Å, respectively. The correct orientation of the protein within the outer membrane of the mitochondria is still debated, with multiple studies producing conflicting results on the endogenous position of the N and C termini (30–32); however, a recent study has shown the C terminus to reside in the IMS (33). The N terminus of the protein forms a distorted α-helix that is aligned nearly parallel to the plane of the membrane, causing a partial narrowing of the pore (Fig. 3). The pore forms a large pathway that traverses the entire length of the protein. There have now been a number of studies confirming that this conformation probably resides in the “open” state of the channel (11, 12).
A structural alignment between the crystal structure of zfVDAC2 and mVDAC1 (Protein Data Bank code 3EMN) showed a root mean square deviation of 0.98 Å (Fig. 4A). The well aligned structures, including the N-terminal segment, indicate that these proteins share many similar properties and functions. The sole exception is a large displacement (∼12 Å) of the loop between strands 1 and 2 (Fig. 4B). Further comparison using Columbic electrostatic calculations (Fig. 5) revealed a continuous electronegative patch on the cytosolic lip of mVDAC1 extending from the C terminus of the protein (β19) and within the loops connecting β1 to β2, β3 to β4, and β5 to β6. In zfVDAC2, the loop connecting β1 to β2 is electropositive, disrupting the electronegative continuity. In addition, there are a number of other subtle differences. Residues 61–65 in β-strand 3 contain four positive residues in zfVDAC2 (KYKRS) as opposed to two positive residues in mVDAC1 (KYRWT); residues 280–282, at the C terminus of the protein, contain two negative residues in zfVDAC2 (Glu-Leu-Glu) as opposed to one in mVDAC1 (Glu-Phe-Gln); and Glu-36, which resides in mVDAC1 in the mobile loop connecting β-strands 1 and 2, is substituted to an Ala in zfVDAC2. Additional hydrophobicity analysis did not show any significant difference between the two isoforms.
FIGURE 4.
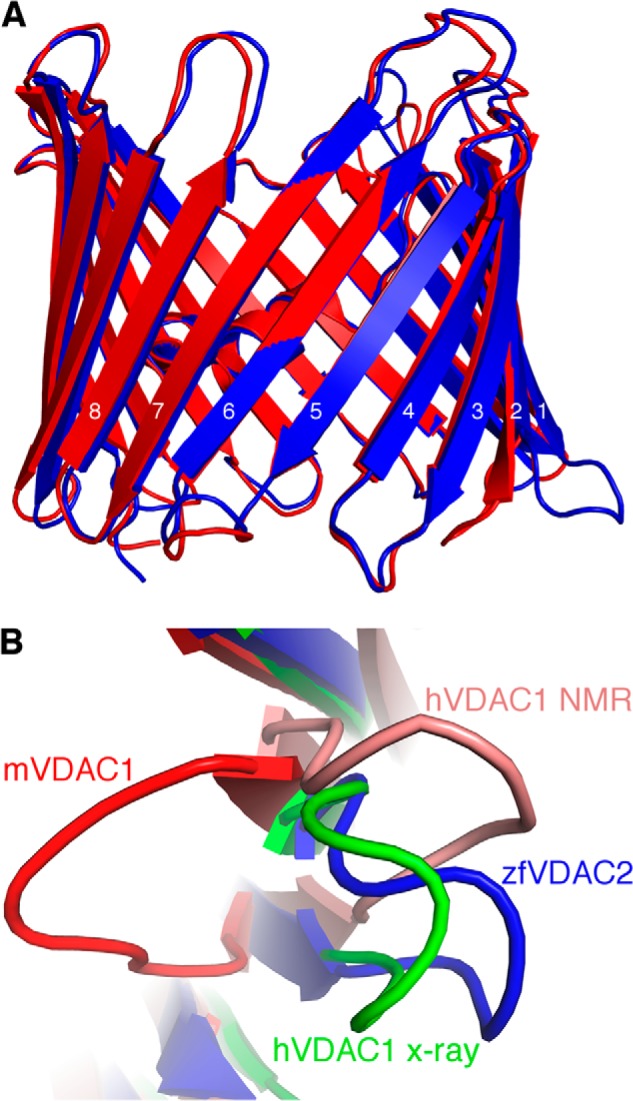
Superposition of zfVDAC2 and VDAC1. A, zfVDAC2 in blue and mVDAC1 (Protein Data Bank code 3EMN) in red were aligned with a Cα root mean square deviation of 0.98 Å. There is a large displacement (∼12 Å) in the loop between β-strands 1 and 2. B, an IMS view of the loop between β-strands 1 and 2, including zfVDAC2 in blue, mVDAC1 in red, hVDAC1 (Protein Data Bank code 2JK4) in green, and a representative NMR conformer of hVDAC1 (Protein Data Bank code 2K4T) in salmon.
FIGURE 5.

Electrostatic potentials of zfVDAC2 and mVDAC1. Electrostatic potentials for zfVDAC2 (top row) and mVDAC1 (bottom row) are shown. A, a view from the IMS. There is an elongated electronegative patch extending from the C terminus of mVDAC1 through the loops of β-strands 1–6, which is interrupted by positive charges in zfVDAC2 (dashed green circle). B, a side view. C, a view from the cytosolic side.
Dimer Interface
The oligomeric organization of VDAC has been strongly correlated with its physiological function. Numerous studies have demonstrated that VDAC can achieve a number of oligomeric populations, depending on the cell's environment, including monomers, dimers, trimers, tetramers, hexamers, and even higher order oligomers (34). More recently, this arrangement has been shown to be dynamic and further influenced by the lipidic environment (35). As was done with hVDAC1 (5), a zfVDAC2 dimer was constructed using crystal symmetry operators. zfVDAC2 forms crystals of a space symmetry group P3221, which leads to three distinct zfVDAC2 symmetry-related interfaces. The most extensive surface, with a buried surface area of 554 Å2 and a solvation free energy gain of ΔG = −14.9 kcal/mol, is formed by buried hydrophobic residues in strands β17 (Arg-252 and Leu-251), β18 (Leu-259, Leu-257, and Ile-255), β19 (Leu-281, Leu-279, Leu-277, and Leu-275), β1 (Val-31, Leu-29, and Val-27), and β3 (Val-54, Ser-52, Asp-50, Thr-49, and Ser-47) (Fig. 6).
FIGURE 6.

zfVDAC2 dimer. A symmetry-related dimer of zfVDAC2 is presented in a transparent space-filling model on top of a cartoon representation (same coloring as in Fig. 3). The expanded view shows the interaction site composed of residues from strands 1, 3, and 17–19 presented in stick representations.
To confirm the dimer interface of zfVDAC2 in solution and the relative abundance of zfVDAC2 oligomers, we generated a cysteine-less construct by replacing the endogenous cysteine residue with alanine (C127A) and subsequently engineered a cysteine at position 232, which is the N-terminal residue of β16. This mutant was then spin-labeled with 1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl methanethiosulfonate reagent to generate an R1 side chain, and the interspin distance distribution was measured using DEER spectroscopy in LDAO and in lipidic bicelles. The DEER experiment measures the dipolar interaction between unpaired electrons in the range of 17–70 Å (27) and thus can be used to detect the presence of homodimers or higher oligomers in a solution of singly labeled proteins. Residue 232 was selected for this study because modeling of the R1 side chain at site 232 in the crystallographic dimer shown in Fig. 6 suggests a distance between R1 labels of ∼31 Å (Fig. 7A), which is well within the range of interspin distance that can be reliably measured with DEER spectroscopy.
In the DEER experiment, magnetic interactions between randomly distributed spins in different molecular entities give rise to an exponentially decaying “background” signal (red traces in the left-hand panels of Fig. 7, B and C). The experimental DEER signals (black and cyan traces in the left-hand panels of Fig. 7, B and C) deviate from the background at short evolution times, unequivocally demonstrating the presence of specific oligomers in solutions of both LDAO and bicelles. Removal of the background signal from the raw data gives the corresponding DEFs (right-hand panels of Fig. 7, B and C), which can be fit to obtain a probability distribution of interspin distances (Fig. 7D). In LDAO, the distance distribution is broad with a full width at half-height of nearly 20 Å, which exceeds the width anticipated from a distribution of R1 rotamers (34) and suggests flexibility in the region of β16. The broad distribution may also arise from heterogeneity regarding the relative orientation of each molecule in the dimer. However, some of the local maxima in the broad distribution could arise from distinct rotamers of R1. The median of the distribution is 30.5 Å, close to that determined from modeling of R1 in the crystallographic dimer.
In bicelles, the median of the distribution is 32.4 Å (the most probable distance is 30.3), again close to that expected from the crystallographic dimer. However, there is a small population near 55 Å (arrow in Fig. 7D). The shape of the distribution is not well determined due to the limited data collection time of 2.5 μs, but a detectable population at long distance is required to fit the data. A distance in this range cannot correspond to any possible homodimer but must originate from higher order oligomers.
The depth of modulation of the DEF (Δ; Fig. 7, B and C) is <4% for VDAC2 232R1 in either LDAO or bicelles, compared with ∼10% for the doubly labeled T4 lysozyme pairs used for determination of the constant C under identical conditions (see “Experimental Procedures”). The low depth of modulation could be due to incomplete labeling of the protein and/or due to a small fraction of protein in oligomeric form (28). Because labeling was found to be quantitative (see “Experimental Procedures”), it is concluded that only a small fraction of the protein exists in oligomeric form, consistent with recent spectroscopy studies on hVDAC1 (35).
The distance distribution for 232R1 in LDAO clearly supports the formation of a homodimer. Within the context of this model, the fraction of VDAC2 in dimer form, fd, can be estimated from the modulation depth of the DEER signal according to Refs. 24 and 25 (fd = n − 1 = ln(1 − Δ)/C, which gives ∼18%.
The depth of modulation for VDAC2 232R1 in bicelles is larger than for the protein in LDAO (compare delta values in panels B and C of Fig. 7), which indicates a higher fraction of total oligomeric species (∼40%). The larger depth of modulation arises due to the existence of higher order oligomers (indicated by the 55 Å population in the distance distribution) and possibly a larger value for fd. An increase in the oligomeric population in bicelles compared with LDAO micelles is in agreement with previous studies, which reported that the percentage of dimer is influenced by detergent or lipidic environments (35).
To obtain information on the relative mobility of the 232R1 side chain in LDAO and bicelles, CW EPR spectra were recorded for the protein in the two environments. In both cases, the spectra show a dominant component reflecting restricted mobility of the R1 side chain (Fig. 7E, top spectra, i), and a second component reflecting higher mobility (m). The two spectral components can arise from slow exchange (microsecond to millisecond time scale) between protein conformational substates or from rotameric equilibria of R1 (36). It has been shown that stabilizing osmolytes, such as sucrose, shift protein conformational equilibria but have little effect on rotameric equilibria of R1. For 232R1, the addition of sucrose shifts the relative population toward the more immobilized state, suggesting that the two components arise from slow (microsecond to millisecond) exchange between conformational substates differing in structure at, or in the vicinity of, the N-terminal region of β16 (Fig. 7E, bottom spectra).
To further validate the data obtained by DEER, we employed cross-linking experiments of single-cysteine mutants in an effort to better define the dimer interface in solution. We generated three cysteine mutants that were part of the putative dimer interface close to the cytoplasmic side of VDAC at positions Val-27, Asp-50, and Ser-52 and another set of three mutants close to the IMS at positions Leu-251, Ile-255, and Leu-257 (Figs. 6 and 8). These mutants were designed in loop regions and on β-strands at S–S distance ranges of 3.7–11.1 Å. In addition, two cysteine mutants were generated at sites that should reside outside of the putative dimer interface at positions Thr-83 and Gln-196 (Fig. 8A). Upon the addition of dichloro(1,10-phenanthroline)Cu(II), significant cross-linking was observed at all six sites that are part of the putative dimer interface (Fig. 8, B and C), indicative of disulfide formation between monomeric proteins as opposed to the control sites that did not display appreciable cross-linking (Fig. 8D).
FIGURE 8.

Cross-linking experiments validate the dimer interface. A, cartoon representation of the zfVDAC2 putative dimer with sites of cysteine mutations displayed in spheres, differently colored by position. B, 12% SDS-PAGE for zfVDAC2 single-cysteine mutants close to the cytosolic side. C, 12% SDS-PAGE for zfVDAC2 single-cysteine mutants close to the IMS. D, 12% SDS-PAGE of non-cross-linking single-cysteine mutants at positions outside of the putative dimer interface. Black box, monomeric zfVDAC2; red box, dimeric forms. MWM, molecular weight markers. Cu-oP, dichloro(1,10-phenanthroline)Cu(II) Groupings of images from different parts of the same gels are marked by vertical lines.
DISCUSSION
In recent years, there has been a vast increase in structural and functional understanding of VDAC1, but VDAC2 and -3 have been understudied. zfVDAC2 is a good structural model for mammalian VDAC2 due to a high degree of sequence homology (e.g. 83.0% identity and 93.0% similarity) between zfVDAC2 and hVDAC2 with no sequence gaps (Fig. 1 shows a zfVDAC2 alignment against selected mammalian VDAC sequences). Most of the minor sequence differences reside in the exposed loop regions where the transmembrane segments remain conserved (Fig. 9, top row). Interestingly, mammalian VDAC2s have a small 11-amino acid N-terminal extension that is not present in zfVDAC2. However, this N-terminal extension was shown not to be essential in mediating VDAC2-specific properties of the channel, such as tBid-induced cytochrome c release (37). Most of these minor sequence differences reside in the exposed loop regions where the transmembrane segments remain conserved (Fig. 9, top row). In total, zfVDAC is a good model for mammalian VDAC2.
FIGURE 9.

Mapping the sequence diversity in VDAC isoforms. The sequence diversity between zfVDAC2 and hVDAC2 (top row) is shown utilizing a cartoon representation of the zfVDAC2 structure and the sequence diversity between hVDAC1 and hVDAC2 (bottom row) is shown utilizing a schematic representation of the hVDAC1 structure. The views displayed from left to right are a side view (A), a 180° rotated side view (B), and a cytosolic view (C). Blue, sequence identity; green, sequence similarity; red, unconserved sequence substitutions.
VDAC2 has a number of unique and specific protein-interacting partners, including the proapoptotic protein BAK (13), the ryanodine receptor (16), and erastin and tubulin (38). A logical assumption is that these specific interactions would be governed by structural differences. However, a structural comparison between VDAC1 and -2 reveals few differences on a positional (Fig. 9, bottom row) or electrostatic basis (Fig. 5). The sole exceptions are large displacements of the loop between strands 1 and 2 (amino acid sequence 32–40) and the loop connecting β-strand 5 and β-strand 6 (amino acid sequence 88–95) as well as a change in the electrostatic properties on the cytosolic lip of VDAC. Indeed, differences between VDAC1 and VDAC2 accounting for the VDAC2-specific tBid-induced cytochrome c release were previously mapped to the N-terminal two-thirds, namely amino acids 13–188 (39). The loop connecting β-strand 1 and β-strand 2 is extended 12 Å away from the pore in the zfVDAC2 structure as compared with mVDAC1 (Protein Data Bank code 3EMN). In the hVDAC1 x-ray structure (Protein Data Bank code 2JK4), this loop occupies a position quite similar to that in the zfVDAC2, and the NMR structure of hVDAC1 has this loop even farther away from the mVDAC1 position (Fig. 4B). Interestingly, there is an amino acid divergence at position 36 located at the apex of this mobile loop. For VDAC1, there is a highly conserved Glu, but in VDAC2 the residue is primarily an Ala or a Cys. To date, this residue, as far as we know, has not been subjected to functional analysis.
The loop connecting β-strands 5 and 6 in the human x-ray structure is located ∼13 Å internally from the position in mVDAC1 and zfVDAC2 x-ray structures. In the NMR model, this loop resides in the middle of these two positions. In the case of zfVDAC2, this loop is pushed away due to the presence of a symmetry-related molecule.
It would seem that loop displacements between β-strands 1 and 2 and β-strands 5 and 6, as observed here, would not suffice to explain the vast phenotypic differences between isoforms. However, the dynamic and electrostatic properties of these loops could play a key role in establishing protein-protein interactions and generate protein complexes or modulate gating properties and thus alter the function of VDACs. Indeed, binding of partner proteins was shown to vary substantially between VDAC isoforms, with compelling evidence that VDAC2 has a greater repertoire of binding partners (40–42).
Another aspect of VDAC physiology that has major implications, in particular for apoptotic events, is the protein's oligomeric state. Current research has strongly implicated a dimeric organization of hVDAC1 by analysis of crystal packing (5, 7), chemical cross-linking (43), and fluorescence cross-correlation spectroscopy (35). However, these studies have been lacking for VDAC2.
Using crystallographic symmetry operators, we identified a parallel dimer interface for zfVDAC2 that is virtually identical to the one suggested for hVDAC1 with a buried surface area of 554 Å2. The interface is formed by strands β17, β18, β19, β1, and β3 (Figs. 6 and 8). Here we show that ∼18% of the protein exists in a dimeric form in solution, as demonstrated by DEER spectroscopy for a singly R1-labeled mutant of zfVDAC2 reconstituted in LDAO; similar dimers exist in bicelles, apparently along with higher order oligomers (Fig. 7B). Remarkably, previous fluorescence cross-correlation spectroscopy studies done by Betaneli et al. (35) on hVDAC1 reconstituted in giant unilamellar vesicles showed a ∼15% dimer population, comparable with that observed here for zfVDAC2 in LDAO.
The interspin distance distributions centered on ∼30 Å observed in DEER are consistent with the interspin distance expected from modeling of the R1 side chain in the x-ray structure presented here, but the broad distributions indicate significant protein flexibility. The main probability distributions for VDAC2 232R1 in LDAO and bicelles are similar but not identical, showing that the structure of the dimer in both environments is similar.
The 55 Å population for the protein in bicelles, but not in LDAO detergent micelles, cannot arise from a homodimer. A possible oligomeric configuration could be drawn from crystal packing analysis of mVDAC1 crystals originating in bicelles (7). This structure reveals a more elaborate hexameric arrangement mimicking the native oligomeric packing observed in EM (44) and atomic force microscopy (45) images of the outer mitochondria membrane. Intramolecular distances between side chain 232R1 in this hexamer identify a likely origin for both the ∼30 Å and 55 Å populations (Fig. 10).
FIGURE 10.
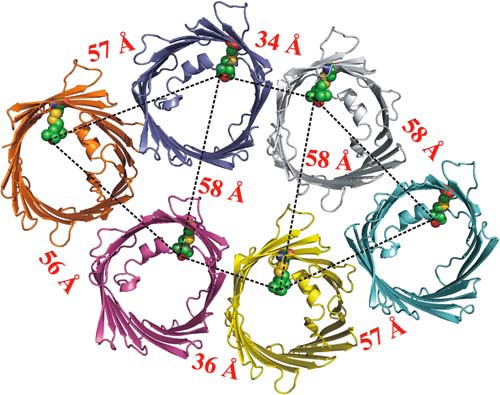
Interspin distances of mVDAC1 hexamer at position 232. A cartoon representation of mVDAC1 232R1 shows the expected distances based on modeling of R1 on the mVDAC1 structure. Models of the R1 side chain at site 232 are shown in spherical representations. The black dashed line shows the expected interspin distance in the crystallographic hexamer. Interspin distances of >70 Å are outside the range that can be reliably determined with the DEER data recorded in this study and thus are not shown.
Comparison of the CW EPR spectra of residue 232R1 in LDAO and bicelles shows an increase in the relative population of a more mobile state in LDAO (Fig. 7C), and osmolyte perturbation indicates that the two-component spectra probably arise from multiple conformational substates in equilibrium. Thus, the small differences that do exist in the distance distributions for LDAO and bicelles may be due to differences in conformational flexibility of the protein at or near residue 232R1.
The cross-linking experiments strongly support the validity of the suggested dimer interface in zfVDAC2. The two cysteine mutants designed outside of the dimer interface displayed no cross-linking, whereas mutants in the dimer interface displayed distinct levels of cross-linking. The highest levels of cross-linking were observed for mutants in long flexible loop regions (D50C with S–S located 11.1 Å apart and S52C with an S–S distance of 6.8 Å and for an internal mutant located in the cytosolic side of β1 (V27C), which is the closest cysteine mutant in this set of cross-linking experiments (S–S distance of only 3.7 Å). The other three mutants on the mitochondrial IMS side that displayed lower cross-linking efficiencies were designed in rigid β-strands or just at the interface between short loops and β17 and β18 situated at relatively long S-S distance ranges of 6.4–8.9 Å.
Although the present study advances our structural understanding in the VDAC field, in particular VDAC2, there are still many unanswered questions that will need to be addressed. Perhaps the varied phenotype is not purely structural but depends on different affinities to partner proteins, expression patterns, subcellular localization of the distinct isoforms, or differences in the intrinsic flexibility of functionally important structural elements between the different isoforms. There is plenty of evidence for different physiological roles of the three isoforms, but the search for the underlying mechanisms has to be further explored. The structural characterization of VDAC2, however, provides blueprints to test many hypotheses and identifies a loop between strands 1 and 2 as a plausible dynamic structural element, as judged by changes in its relative position in structures of different VDAC isoforms, and identifies an area with a change in the electrostatic surface potential between zfVDAC2 and mVDAC1.
Acknowledgments
We thank Armand S. Vartanian and Vinod Nayak for fruitful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 GM078844 (to J. A.) and 5R01 EY005216 (to C. J. L.). This work was also supported by Austrian Science Fund FWF Erwin-Schrödinger Fellowship J3065-B11 (to J. S.), Jules Stein Eye Institute Training Grant 2T32EY007026-36A1 (to C. J. L.), and the Jules Stein Professor Endowment (to W. L. H.).
This work is dedicated to the memory of Armand S. Vartanian, a colleague and friend.
The atomic coordinates and structure factors (code 4BUM) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- VDAC
- voltage-dependent anion channel
- hVDAC
- zfVDAC, and mVDAC, human, zebrafish, and mouse VDAC, respectively
- CHAPSO
- 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonic acid
- IMS
- intermembrane space
- DEER
- double electron-electron resonance
- LDAO
- lauryldimethyl amine oxide
- CW
- continuous wave
- DEF
- dipolar evolution function.
REFERENCES
- 1. Colombini M., Blachly-Dyson E., Forte M. (1996) VDAC, a channel in the outer mitochondrial membrane. Ion Channels 4, 169–202 [DOI] [PubMed] [Google Scholar]
- 2. Rostovtseva T. K., Sheldon K. L., Hassanzadeh E., Monge C., Saks V., Bezrukov S. M., Sackett D. L. (2008) Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. U.S.A. 105, 18746–18751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Colombini M. (2004) VDAC: the channel at the interface between mitochondria and the cytosol. Mol. Cell Biochem. 256, 107–115 [DOI] [PubMed] [Google Scholar]
- 4. Hiller S., Garces R. G., Malia T. J., Orekhov V. Y., Colombini M., Wagner G. (2008) Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science 321, 1206–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bayrhuber M., Meins T., Habeck M., Becker S., Giller K., Villinger S., Vonrhein C., Griesinger C., Zweckstetter M., Zeth K. (2008) Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. U.S.A. 105, 15370–15375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ujwal R., Cascio D., Colletier J. P., Faham S., Zhang J., Toro L., Ping P., Abramson J. (2008) The crystal structure of mouse VDAC1 at 2.3 Å resolution reveals mechanistic insights into metabolite gating. Proc. Natl. Acad. Sci. U.S.A. 105, 17742–17747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ujwal R., Cascio D., Chaptal V., Ping P., Abramson J. (2009) Crystal packing analysis of murine VDAC1 crystals in a lipidic environment reveals novel insights on oligomerization and orientation. Channels 3, 167–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ujwal R., Abramson J. (2012) High-throughput crystallization of membrane proteins using the lipidic bicelle method. J. Vis. Exp. 59, e3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guo X. W., Smith P. R., Cognon B., D'Arcangelis D., Dolginova E., Mannella C. A. (1995) Molecular design of the voltage-dependent, anion-selective channel in the mitochondrial outer membrane. J. Struct. Biol. 114, 41–59 [DOI] [PubMed] [Google Scholar]
- 10. Mannella C. A. (1998) Conformational changes in the mitochondrial channel protein, VDAC, and their functional implications. J. Struct. Biol. 121, 207–218 [DOI] [PubMed] [Google Scholar]
- 11. Choudhary O. P., Ujwal R., Kowallis W., Coalson R., Abramson J., Grabe M. (2010) The electrostatics of VDAC: implications for selectivity and gating. J. Mol. Biol. 396, 580–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Teijido O., Ujwal R., Hillerdal C. O., Kullman L., Rostovtseva T. K., Abramson J. (2012) Affixing N-terminal α-helix to the wall of the voltage-dependent anion channel does not prevent its voltage gating. J. Biol. Chem. 287, 11437–11445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng E. H., Sheiko T. V., Fisher J. K., Craigen W. J., Korsmeyer S. J. (2003) VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301, 513–517 [DOI] [PubMed] [Google Scholar]
- 14. Anflous K., Armstrong D. D., Craigen W. J. (2001) Altered mitochondrial sensitivity for ADP and maintenance of creatine-stimulated respiration in oxidative striated muscles from VDAC1-deficient mice. J. Biol. Chem. 276, 1954–1960 [DOI] [PubMed] [Google Scholar]
- 15. Sampson M. J., Decker W. K., Beaudet A. L., Ruitenbeek W., Armstrong D., Hicks M. J., Craigen W. J. (2001) Immotile sperm and infertility in mice lacking mitochondrial voltage-dependent anion channel type 3. J. Biol. Chem. 276, 39206–39212 [DOI] [PubMed] [Google Scholar]
- 16. Min C. K., Yeom D. R., Lee K. E., Kwon H. K., Kang M., Kim Y. S., Park Z. Y., Jeon H., Kim do H. (2012) Coupling of ryanodine receptor 2 and voltage-dependent anion channel 2 is essential for Ca2+ transfer from the sarcoplasmic reticulum to the mitochondria in the heart. Biochem. J. 447, 371–379 [DOI] [PubMed] [Google Scholar]
- 17. Yagoda N., von Rechenberg M., Zaganjor E., Bauer A. J., Yang W. S., Fridman D. J., Wolpaw A. J., Smukste I., Peltier J. M., Boniface J. J., Smith R., Lessnick S. L., Sahasrabudhe S., Stockwell B. R. (2007) RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bauer A. J., Gieschler S., Lemberg K. M., McDermott A. E., Stockwell B. R. (2011) Functional model of metabolite gating by human voltage-dependent anion channel 2. Biochemistry 50, 3408–3410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 20. Kantardjieff K. A., Rupp B. (2003) Matthews coefficient probabilities: improved estimates for unit cell contents of proteins, DNA, and protein-nucleic acid complex crystals. Protein Sci. 12, 1865–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 23. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrodinger, LLC, New York [Google Scholar]
- 24. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 25. Hubbell W. L., Mchaourab H. S., Altenbach C., Lietzow M. A. (1996) Watching proteins move using site-directed spin labeling. Structure 4, 779–783 [DOI] [PubMed] [Google Scholar]
- 26. Fleissner M. R., Bridges M. D., Brooks E. K., Cascio D., Kálai T., Hideg K., Hubbell W. L. (2011) Structure and dynamics of a conformationally constrained nitroxide side chain and applications in EPR spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 108, 16241–16246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jeschke G., Polyhach Y. (2007) Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys. Chem. Chem. Phys. 9, 1895–1910 [DOI] [PubMed] [Google Scholar]
- 28. Hilger D., Jung H., Padan E., Wegener C., Vogel K. P., Steinhoff H. J., Jeschke G. (2005) Assessing oligomerization of membrane proteins by four-pulse DEER: pH-dependent dimerization of NhaA Na+/H+ antiporter of E. coli. Biophys. J. 89, 1328–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Colombini M. (1989) Voltage gating in the mitochondrial channel, VDAC. J. Membr. Biol. 111, 103–111 [DOI] [PubMed] [Google Scholar]
- 30. De Pinto V., Prezioso G., Thinnes F., Link T. A., Palmieri F. (1991) Peptide-specific antibodies and proteases as probes of the transmembrane topology of the bovine heart mitochondrial porin. Biochemistry 30, 10191–10200 [DOI] [PubMed] [Google Scholar]
- 31. Engelhardt H., Meins T., Poynor M., Adams V., Nussberger S., Welte W., Zeth K. (2007) High-level expression, refolding and probing the natural fold of the human voltage-dependent anion channel isoforms I and II. J. Membr. Biol. 216, 93–105 [DOI] [PubMed] [Google Scholar]
- 32. Stanley S., Dias J. A., D'Arcangelis D., Mannella C. A. (1995) Peptide-specific antibodies as probes of the topography of the voltage-gated channel in the mitochondrial outer membrane of Neurospora crassa. J. Biol. Chem. 270, 16694–16700 [DOI] [PubMed] [Google Scholar]
- 33. Tomasello M. F., Guarino F., Reina S., Messina A., De Pinto V. (2013) The voltage-dependent anion selective channel 1 (VDAC1) topography in the mitochondrial outer membrane as detected in intact cell. PloS One 8, e81522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gonçalves R. P., Buzhynskyy N., Prima V., Sturgis J. N., Scheuring S. (2007) Supramolecular assembly of VDAC in native mitochondrial outer membranes. J. Mol. Biol. 369, 413–418 [DOI] [PubMed] [Google Scholar]
- 35. Betaneli V., Petrov E. P., Schwille P. (2012) The role of lipids in VDAC oligomerization. Biophys. J. 102, 523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. López C. J., Fleissner M. R., Guo Z., Kusnetzow A. K., Hubbell W. L. (2009) Osmolyte perturbation reveals conformational equilibria in spin-labeled proteins. Protein Sci. 18, 1637–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Naghdi S., Varnai P., Hunyady L., Hajnoczky G. (2012) The isoform specific N terminus of VDAC2 is dispensable for tBid induced cytochrome c release. Biophys. J. 102, 437A [Google Scholar]
- 38. Maldonado E. N., Sheldon K. L., DeHart D. N., Patnaik J., Manevich Y., Townsend D. M., Bezrukov S. M., Rostovtseva T. K., Lemasters J. J. (2013) Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: regulation by free tubulin and erastin. J. Biol. Chem. 288, 11920–11929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Naghdi S., Varnai P., Roy S. S., Hunyady L., Hajnoczky G. (2013) Which domain of VDAC2 is necessary for Bak insertion to the outer mitochondrial membrane and tBid-induced cytochrome c release? Biophys. J. 104, 656A [Google Scholar]
- 40. Alvira C. M., Umesh A., Husted C., Ying L., Hou Y., Lyu S. C., Nowak J., Cornfield D. N. (2012) Voltage-dependent anion channel-2 interaction with nitric oxide synthase enhances pulmonary artery endothelial cell nitric oxide production. Am. J. Respir. Cell Mol. Biol. 47, 669–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roy S. S., Ehrlich A. M., Craigen W. J., Hajnóczky G. (2009) VDAC2 is required for truncated BID-induced mitochondrial apoptosis by recruiting BAK to the mitochondria. EMBO Rep. 10, 1341–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Palaniappan K. K., Hangauer M. J., Smith T. J., Smart B. P., Pitcher A. A., Cheng E. H., Bertozzi C. R., Boyce M. (2013) A chemical glycoproteomics platform reveals O-GlcNAcylation of mitochondrial voltage-dependent anion channel 2. Cell Rep. 5, 546–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Geula S., Naveed H., Liang J., Shoshan-Barmatz V. (2012) Structure-based analysis of VDAC1 protein: defining oligomer contact sites. J. Biol. Chem. 287, 2179–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mannella C. A. (1982) Structure of the outer mitochondrial membrane: ordered arrays of porelike subunits in outer-membrane fractions from Neurospora crassa mitochondria. J. Cell Biol. 94, 680–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hoogenboom B. W., Suda K., Engel A., Fotiadis D. (2007) The supramolecular assemblies of voltage-dependent anion channels in the native membrane. J. Mol. Biol. 370, 246–255 [DOI] [PubMed] [Google Scholar]