

Background: AIRAP is a recently identified heat shock protein whose function and regulation are still not well defined.
Results: Proteasome inhibition causes abundant AIRAP expression in human primary cells.
Conclusion: The anticancer drug bortezomib regulates AIRAP expression through HSF1-HSF2 interplay.
Significance: This study reveals how persistent proteasomal inhibition by clinically relevant concentrations of bortezomib affects heat shock response regulation and HSF-mediated AIRAP induction.
Keywords: Endothelial Cell, Heat Shock Protein, Proteasome, Stress Response, Transcription Regulation
Abstract
The zinc finger AN1-type domain 2a gene, also known as arsenite-inducible RNA-associated protein (AIRAP), was recently identified as a novel human canonical heat shock gene strictly controlled by heat shock factor (HSF) 1. Little is known about AIRAP gene regulation in human cells. Here we report that bortezomib, a proteasome inhibitor with anticancer and antiangiogenic properties used in the clinic for treatment of multiple myeloma, is a potent inducer of AIRAP expression in human cells. Using endothelial cells as a model, we unraveled the molecular mechanism regulating AIRAP expression during proteasome inhibition. Bortezomib induces AIRAP expression at the transcriptional level early after treatment, concomitantly with polyubiquitinated protein accumulation and HSF activation. AIRAP protein is detected at high levels for at least 48 h after bortezomib exposure, together with the accumulation of HSF2, a factor implicated in differentiation and development regulation. Different from heat-mediated induction, in bortezomib-treated cells, HSF1 and HSF2 interact directly, forming HSF1-HSF2 heterotrimeric complexes recruited to a specific heat shock element in the AIRAP promoter. Interestingly, whereas HSF1 has been confirmed to be critical for AIRAP gene transcription, HSF2 was found to negatively regulate AIRAP expression after bortezomib treatment, further emphasizing an important modulatory role of this transcription factor under stress conditions. AIRAP function is still not defined. However, the fact that AIRAP is expressed abundantly in primary human cells at bortezomib concentrations comparable with plasma levels in treated patients suggests that AIRAP may participate in the regulatory network controlling proteotoxic stress during bortezomib treatment.
Introduction
Protein homeostasis is essential for life in eukaryotes. The 26 S proteasome, a multicatalytic protease complex consisting of a 20 S core particle with proteolytic activity and a 19 S regulatory particle (1, 2), represents the primary site for non-lysosomal protein degradation in mammalian cells and regulates the turnover and quality control of proteins, consequently affecting many aspects of cell physiology, including cell cycle progression (3), signal transduction (4), differentiation (5), apoptosis (6), and angiogenesis (7).
Because of its central role in protein homeostasis, over the past decades the proteasome complex has emerged as an important target in anticancer therapy, and a variety of heterogeneous reversible and irreversible inhibitors of the 26 S proteasome have been identified (8–10). Among these, the boronic acid dipeptide bortezomib (Velcade, formerly PS-341) is used in the clinic for treatment of multiple myeloma and several other malignancies (11) on the basis of its direct proapoptotic effects on cancer cells as well as its antiangiogenic action (12–14).
A critical consequence of ubiquitin-proteasome network down-regulation is the activation of the cellular heat shock response (15–18). The heat shock response is regulated by a family of heat shock (HS)3 transcription factors (HSFs) that are expressed and maintained in an inactive state under non-stress conditions. Mammalian genomes encode three homologues of HSF (HSF1, HSF2, and HSF4) regulating heat shock protein (HSP) expression (19). Among these, HSF1 is considered to be the paralog responsible for regulating the heat-induced transcriptional response (20). However, HSF2 has also been reported to contribute to inducible expression of HS genes through interplay with HSF1 (21, 22).
HSF1 is generally found in the cytoplasm as an inert monomer lacking transcriptional activity. Upon exposure to heat and other proteotoxic stresses, HSF1 is derepressed in a stepwise process that involves HSF1 oligomerization to a trimeric state, localization to the nucleus, inducible phosphorylation and sumoylation, and binding to DNA sequences known as heat shock elements (HSE). Functional HSE sequences are characterized by an array of inverted repeats of the pentameric motif nGAAn and are usually located in the proximal region of HSF1-responsive gene promoters. HSF1 binding to HSE sequences is followed by a rapid shift in the transcriptional program, resulting in high rates of expression of cytoprotective heat shock proteins, which include molecular chaperones of the HSP70 and HSP90 families, HSP27, and other proteins of the network (20).
We recently identified the zinc finger AN1-type domain 2a gene, also known as arsenite-inducible RNA-associated protein (AIRAP), as a novel human heat shock gene (23). Little is known about AIRAP gene regulation in human cells, and the functional role of AIRAP protein is still not well defined. Because it was found to associate with the proteasome 19 S cap (24), we hypothesized that AIRAP may be part of the response to proteotoxic stress induced by pharmacological proteasome inhibition. Therefore, we investigated the effect of the proteasome inhibitor (PI) bortezomib on AIRAP expression in primary human cells. Endothelial cells were selected as a model for this investigation on the basis of several in vitro and in vivo studies that identify the endothelium as an important component of the response to bortezomib treatment through angiogenesis control.
Here we report, for the first time, that bortezomib potently induces AIRAP expression in human primary cells. We demonstrate that, in endothelial cells, AIRAP expression is regulated at the transcriptional level by a mechanism involving both HSF1 and HSF2 transcription factors via the formation of HSF1-HSF2 heterotrimeric complexes. Furthermore, we show that, whereas HSF1 is critical for AIRAP gene transcription, HSF2 negatively regulates AIRAP expression after bortezomib treatment, further emphasizing an important modulatory role of this transcription factor during proteotoxic stress.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatment
Human umbilical vein endothelial cells (HUVECs) (Cambrex) were grown at 37 °C with 5% CO2 in EGM-2 (Endothelial Growth Medium-2) complete medium (Cambrex). All experiments were performed using HUVECs passages 2–5. Human peripheral blood monocytes and lymphocytes, isolated and purified from buffy coats of healthy blood donors as described previously (25) were grown in RPMI 1640 medium supplemented with 10% FCS and antibiotics for 24 h before treatments. The PI bortezomib (Millennium Pharmaceuticals) and MG132 (Calbiochem) were dissolved in dimethyl sulfoxide and diluted in culture medium immediately before use. Control media contained the same amount of dimethyl sulfoxide vehicle (<0.1%). For heating procedures, cells were subjected to HS at 43 °C for 40 min in a precision water bath (W14, Grant Instruments). Cell viability was determined in triplicate samples by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to MTT formazan conversion assay (Sigma-Aldrich), as described previously (26).
Cell Transfection and Reporter Assays
Transfections were performed using FuGENE HD transfection reagent (Roche). The different AIRAP constructs (23) were cotransfected with a control plasmid (pRL-TK encoding Renilla luciferase, Promega) to normalize transfection efficiency. Transfected cells were grown for 16 h before bortezomib and heat treatments, and the luciferase activity of quadruplicate samples was measured in a microplate luminometer (Wallac, PerkinElmer Life Sciences) using a Dual-Luciferase kit (Promega). AIRAP promoter Firefly luciferase activity was normalized to Renilla luciferase activity in the same sample.
Cell Extracts, Western Blot Analysis, and Immunoprecipitation
Whole-cell extracts were prepared by lysis in buffer B (50 mm Tris-HCl, pH 7.5, 400 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton, 0.5% Nonidet P-40, 2 mm dithiothreitol, 10% glycerol) (27) supplemented with 20 mm β-glycerolphosphate, 19 mm p-nitrophenyl phosphate, 500 mm Na3VO4, 1 mm PMSF, and protease inhibitor mixture (Roche), as described previously (27). Nuclear and cytoplasmic extracts were prepared as described previously (28). For Western blot analysis, equal amounts of protein (35 μg/sample) from whole-cell extracts were separated by SDS/PAGE and blotted to nitrocellulose. After blocking with 5% skim milk solution, membranes were incubated with rabbit polyclonal anti-HSF1 (catalog no. sc-9144, Santa Cruz Biotechnology), anti-HSF2 (catalog no. sc-13056, Santa Cruz Biotechnology), anti-ZFAND2A (Sigma) antibodies or monoclonal anti-ubiquitin antibodies (catalog no. sc-8017, Santa Cruz Biotechnology), anti-HSP70 antibodies (Stressgene), and anti-β-actin antibodies (Sigma), followed by decoration with peroxidase-labeled anti-rabbit or anti-mouse IgG, respectively (Super-Signal detection kit, Pierce). For HSF1 and HSF2 immunoprecipitation, whole-cell extracts (200 μg) were precleared at 4 °C for 1 h with 40 μl of protein A-agarose beads in 300 μl of buffer B and incubated for 4 h with rabbit polyclonal anti-HSF1 (catalog no. sc-9144, Santa Cruz Biotechnology) or anti-HSF2 (catalog no. sc-13056, Santa Cruz Biotechnology) antibodies, followed by a 2-h incubation at 4 °C with protein A-agarose beads. After extensive washing in buffer B, immunocomplexes were analyzed by Western blot analysis. Aliquots (25 μg) of extracts were used as input. Quantitative evaluation of proteins was determined by Versadoc 1000 (Bio-Rad) analysis.
EMSA
The 75-bp AIRAP-HSE DNA probe was prepared by PCR amplification of the AIRAP promoter region containing the HSF-binding site (from −164 to −238) using the following primers: sense, 5′-CAAGCGCGCCGCCTGC-3′; antisense, 5′-GCAGGCCCGCCCACTC-3′. The PCR-product was labeled at both ends using [γ-32P]ATP plus T4 nucleotide kinase. The 35-bp HSP70-HSE DNA probe has been described previously (29). Whole-cell extracts (15 μg) were incubated with 32P-labeled probes, followed by analysis of DNA-binding activity by EMSA. Binding reactions were performed as described previously (27). Complexes were analyzed by nondenaturing 3% or 4% polyacrylamide gel electrophoresis for AIRAP-HSE or HSP70-HSE probes, respectively. To determine the specificity of HSF-DNA complexes, whole-cell extracts were preincubated with different dilutions of anti-HSF1 or anti-HSF2 polyclonal antibodies for 15 min before electromobility supershift assay (30). Quantitative evaluation of HSF-HSE complex formation was determined by Typhoon 8600 imager (Molecular Dynamics) with the use of ImageQuant (Amersham Biosciences, Piscataway, NJ).
RNA Extraction, RT-PCR, and Real-time RT-PCR
Total RNA was prepared using TRIzol (Invitrogen) as described in the protocol of the manufacturer. For RT-PCR analysis, extracted RNA (1 μg) was digested with 2 units of DNase I (Invitrogen) for 30 min at 37 °C. Samples were reverse-transcribed to cDNA with 200 units of M-MLV RT (Moloney Murine Leukemia Virus Reverse Transcriptase) (Invitrogen) using 5 μg of random primers (Invitrogen) for 1 h at 45 °C in a total volume of 20 μl. RT was inactivated at 95 °C for 5 min. For each sample, an aliquot of DNase I-digested RNA without RT was used as negative control for PCR amplification. Real-time RT-PCR analysis was performed with ABI PRISM 7000 (Applied Biosystem), using RealMasterMix ROX (Eppendorf) to prepare the reaction mixes. The sequences of the HSF2 primers amplifying both α and β isoforms were as follows: sense, 5′-AAGCCAAGGGAGAGGATTTC-3′; antisense, 5′-GCTGGTTCATTCTGCTCTCC-3′. The AIRAP, AIRAPL, HSP70, and β-actin primers have been described previously (23).
ChIP Assay
Cells were fixed by adding formaldehyde (Sigma) to the medium to a final concentration of 1%. After 15 min, cells were washed with ice-cold PBS containing 1 mm phenylmethylsulfonyl fluoride and shaken for 20 min at 4 °C in lysis buffer 1 (50 mm HEPES-KOH, pH 7.5, 140 mm NaCl, 1 mm EDTA, 10% glycerol, 0.5% Nonidet P-40, 0.25% Triton X-100, and protease inhibitor mixture) (31). After centrifugation (3000 rpm, 10 min) the pellet was resuspended in lysis buffer 2 (10 mm Tris-HCl, pH 8.0, 200 mm NaCl, 1 mm EDTA, 0.5 mm EGTA, and protease inhibitor mixture) (31) and shaken at room temperature for 10 min. Nuclei were resuspended in lysis buffer 3 (50 mm Tris-HCl, pH 8.0, 1% SDS, 5 mm EDTA) (31), and chromatin was sheared by sonication. After removal of nuclear debris, lysates were diluted 10-fold with dilution buffer 3 (50 mm Tris-HCl, pH 8.0, 5 mm EDTA, 200 mm NaCl, 0.5% Nonidet P-40) (31) and then precleared for 3 h using 80 μl of 50% salmon sperm DNA-saturated protein A-agarose beads. Immunoprecipitation was carried out at 4 °C overnight, and immunocomplexes were collected with salmon sperm DNA-saturated protein A-agarose beads. Antibodies utilized included anti-HSF1 and anti-HSF2 (Santa Cruz Biotechnology) or preimmune rabbit serum as a control for nonspecific interactions. After washing three times with high-salt wash buffer 3 (20 mm Tris-HCl, pH 8.0, 0.1% SDS, 1% Nonidet P-40, 2 mm EDTA, 0.5 m NaCl) (31) and twice with TE buffer (10 mm Tris (pH 8.0) and 1 mm EDTA), immunocomplexes were eluted with 1% SDS-containing TE. Protein-DNA cross-links were reverted by incubating at 65 °C overnight. After proteinase K digestion, DNA was extracted with phenol-chloroform and precipitated with ethanol using 15 μg of tRNA as a carrier. Primers and PCR conditions have been described previously (23).
siRNA Interference
Chemically synthesized 19-nucleotide duplex siRNAs with 3′ overhanging UU dinucleotides and their scrambled control sequences were purchased from Qiagen (Hilden, Germany). Two siRNA target sequences were used for AIRAP: GGGAAGCATTGTTCAGAAA and CCTGGGAAGAAGAAAGAGA. The siRNA target sequences for HSF1 and HSF2 were CCCAAGTACTTCAAGCACA and GTAGGACTGAAGGTTTAAA, respectively. All siRNAs and their scrambled controls were transfected using HIPerFect transfection reagent (Qiagen). Briefly, for AIRAP down-regulation, confluent HUVECs growing on 6-well plates were cotransfected with 12.5 nmol/liter of each siRNA targeting sequence. For HSF1 and HSF2 down-regulation, HUVECs growing on 6-well plates were transfected with 25 nmol/liter of HSF1 or HSF2 siRNA targeting sequence or scrambled control sequences. After transfection, the HSF1 or HSF2 siRNAs duplexes were removed, and bortezomib treatment was started at the indicated times.
Confocal Microscopy
HUVECs grown on coverslips were fixed with 4% paraformaldehyde and permeabilized in 0.1% Triton X-100-PBS for 10 min. After blocking with 1% BSA, fixed cells were incubated with rabbit anti-AIRAP antibody for 1 h and incubated with Cy3-conjugated anti-rabbit IgG for 30 min at room temperature. Images were acquired on a Leica confocal microscope TCS-4D system.
Statistical Analysis
Statistical analysis was performed using Student's t test for unpaired data. Data are expressed as the mean ± S.D. of at least duplicate samples. p < 0.05 was considered significant.
RESULTS
Proteasome Inhibition Leads to AIRAP Induction in Human Endothelial Cells
To investigate the effect of proteasome inhibition on HSF activation and AIRAP expression in human cells, quiescent HUVECs were treated with different doses of the reversible peptide-aldehyde PI MG132 or slow-reversible peptide-boronate inhibitor bortezomib (8, 9). After 14 h, whole-cell extracts were analyzed for HSF DNA-binding activity by EMSA and for AIRAP levels by Western blot analysis. As shown in Fig. 1, both PI were found to strongly induce HSF activation, with bortezomib being a much more potent inducer, starting to promote HSF DNA-binding activity at concentrations as low as 10 nm. Consistent with HSF activation, a dose-dependent increase in AIRAP protein levels was detected starting at 10 nm for bortezomib and 100 nm for MG132, demonstrating that AIRAP expression is highly induced in human cells during proteasome inhibition. Because of its clinical relevance and its potency in activating AIRAP expression, bortezomib was selected for this study.
FIGURE 1.
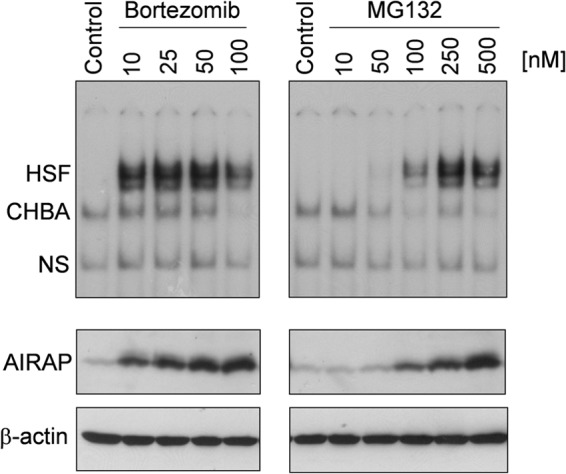
Dose-dependent induction of HSF DNA-binding activity and AIRAP expression by proteasome inhibitors in human endothelial cells. HUVECs were treated with different concentrations of the proteasome inhibitors bortezomib or MG132, and, at 14 h after treatment, whole-cell extracts were analyzed for HSF DNA-binding activity by EMSA (top panels). The positions of the HSF DNA-binding complex (HSF), constitutive HSE-binding activity (CHBA), and nonspecific protein-DNA interaction (NS) are shown. The levels of AIRAP protein and β-actin as a control were determined in the same samples by Western blot analysis (bottom panels). Data from one representative experiment of two with similar results are shown.
To characterize AIRAP intracellular localization, HUVECs were treated with bortezomib, and, after 14 h, nuclear and cytoplasmic fractions were isolated and analyzed by Western blot for AIRAP and HSP70 levels and for TATA-binding protein and GAPDH as a loading control for nuclear and cytoplasmic fractions, respectively. In parallel, HUVECs growing on coverslips were examined by immunofluorescence. As shown in Fig. 2, an intense AIRAP immunofluorescent signal was found to be distributed throughout the nucleus and cytoplasm in bortezomib-treated cells. Consistent with the immunofluorescence, AIRAP was detected in both nuclear and cytoplasmic fractions when analyzed by Western blot analysis.
FIGURE 2.

Cytoplasmic and nuclear distribution of AIRAP in bortezomib-treated endothelial cells. A, HUVECs were treated with 10 nm bortezomib (Btz) (+) or vehicle (−) for 14 h, fractionated into nuclear (N) and cytoplasmic (C) extracts, and analyzed by Western blot for AIRAP and HSP70 protein levels. Equal amounts of protein extracts were loaded in each lane. Antibodies against TATA-binding protein (TBP) and GAPDH were used as a loading control for nuclear and cytoplasmic fractions, respectively. B, parallel samples were processed for immunofluorescence and labeled with anti-AIRAP antibodies (red). Nuclei were stained with DAPI (blue). The overlay of the two fluorochromes is shown (MERGE). Images were captured with a Leica confocal microscope TCS-4D system equipped with a ×40 oil immersion objective. Scale bar = 50 μm. Data from one representative experiment of two with similar results are shown.
Kinetics of HSF Activation and AIRAP Expression following Bortezomib-induced Proteotoxic Stress
To investigate the kinetics of HSF activation and AIRAP expression during proteasome inhibition, confluent HUVEC monolayers were treated with bortezomib at non-toxic (10 nm) concentrations (32, 33). At different times after treatment, whole-cell extracts were analyzed for HSF DNA-binding activity by EMSA (Fig. 3A, top panel) and for levels of polyubiquitinated proteins, AIRAP, HSP70, HSF1, HSF2, and β-actin by Western blot (Fig. 3A, bottom panels). In parallel samples, total RNA was analyzed for the levels of AIRAP, constitutively expressed AIRAP-like gene (AIRAPL), HSF2, and HSP70 mRNA by real-time PCR (Fig. 3B).
FIGURE 3.

Kinetics of AIRAP expression following bortezomib-induced proteotoxic stress in endothelial cells. A, HUVECs were treated with bortezomib (10 nm) or control diluent (C), and, at different times, whole-cell extracts were analyzed for HSF DNA-binding activity by EMSA (top panel). The positions of the HSF DNA-binding complex (HSF), constitutive HSE-binding activity (CHBA), and nonspecific protein-DNA interaction (NS) are shown. The levels of HSF1, HSF2, AIRAP, HSP70, polyubiquitinated proteins, and β-actin as a control were determined in the same samples by Western blot analysis (bottom panels). B, in parallel samples, total RNA was analyzed for AIRAP, constitutively expressed AIRAP-like gene AIRAPL, HSP70, HSF2, and β-actin expression by real-time PCR. Relative quantities of AIRAP, AIRAPL, HSP70, and HSF2 RNAs were normalized to β-actin. All reactions were made in duplicate using samples derived from at least three biological repeats. Error bars indicate mean ± S.D. The fold increase was calculated by comparing the induction of the indicated genes in the treated samples to the relative control, which was arbitrarily set to 1.
As shown in Fig. 3A, bortezomib treatment causes progressive accumulation of polyubiquitinated proteins starting at 3 h after treatment. In parallel with polyubiquitinated protein accumulation, HSF-HSE binding was detected between 3 and 6 h, increasing in the next hours up to 18–24 h, and then it started to decline (Fig. 3A). Minimal levels of HSF DNA binding activity were still detected at 48 h. In concomitance with HSF activation, AIRAP mRNA expression started to increase after 3 h of treatment, reaching maximal levels between 9 and 24 h, with kinetics similar to HSP70 mRNA (Fig. 3B). No change in AIRAPL mRNA levels was detected at any time (Fig. 3B).
As shown in Fig. 3A, a modest increase in the low-mobility phosphorylated HSF1 isoform started to be detected at 6 h after treatment. It should be emphasized that, as described previously for different PI (15, 16, 34), HSF2 levels were increased dramatically in bortezomib-treated cells starting 6 h after treatment and continuing up to 48 h. Interestingly, HSF2 mRNA levels were also found to increase up to 24 h after bortezomib treatment (Fig. 3B).
Following the increase in AIRAP mRNA, AIRAP protein was detected after 6 h of treatment. Similar to HSP70, AIRAP was expressed at high levels for the next 24 h. However, it should be noted that, different from HSP70, accumulation of AIRAP was found to decrease at 48 h after treatment. Hsp70 is known to be a highly stable protein under stress conditions (35, 36), whereas AIRAP protein stability has never been investigated. To determine whether the effect described above could reflect differences in protein stability, HUVECs were treated with 10 nm bortezomib for 14 h, at which time protein synthesis was arrested by adding 100 μg/ml cycloheximide. At different times after cycloheximide treatment, HSP70 and AIRAP levels were determined in whole-cell extracts by Western blot analysis, followed by quantitative determination by densitometric analysis. The results, shown in Fig. 4, indicate that, under the conditions described, HSP70 degradation rate is minimal, whereas AIRAP has a faster degradation rate with a half-life of 8 h.
FIGURE 4.
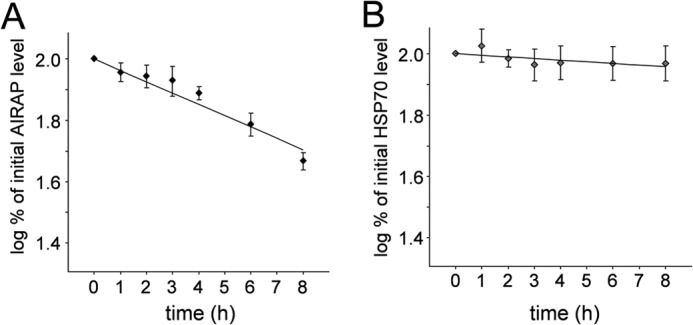
Estimation of AIRAP and HSP70 protein stability in bortezomib-treated HUVECs. HUVECs were treated with 10 nm bortezomib, and, after 14 h, the drug was removed, and cycloheximide was added at a concentration of 100 μg/ml. Samples were collected at various time points up to 8 h after cycloheximide treatment, and AIRAP and HSP70 levels were analyzed in whole-cell extracts by Western blot analysis and quantitated by scanning densitometry. The logarithmic plot of percentage of initial AIRAP (A) and HSP70 (B) protein levels versus time is shown. The degradation rate of AIRAP was calculated to be 8%/h, whereas the HSP70 degradation rate was found to be ∼1%/h. Data represent the mean ± S.D. of three independent experiments.
The effect of repeated bortezomib treatment on HSF activation and AIRAP expression was also investigated. HUVECs were treated with 10 nm bortezomib or vehicle for 24 h. At this time, the medium was removed and replaced with fresh medium containing the drug. Treatment was repeated every 24 h. For this study, both the classical HSE-containing sequence derived from the HSP70 promoter (HSP70-HSE) and a large, 75-bp DNA fragment (from −164 to −238) containing the HSE sequence of the human AIRAP promoter region (AIRAP-HSE) were used. As shown in Fig. 5A (top panel), under these conditions, HSF/HSP70-HSE complexes were detected up to 72 h after treatment. Similar results were obtained using the AIRAP-HSE sequence (Fig. 5A, bottom panel). In parallel with HSF activation, high levels of AIRAP and HSP70 proteins were detected up to 72 h. Interestingly, HSF2 was also found to accumulate at high levels up to 72 h of continuous exposure to bortezomib (Fig. 5B).
FIGURE 5.
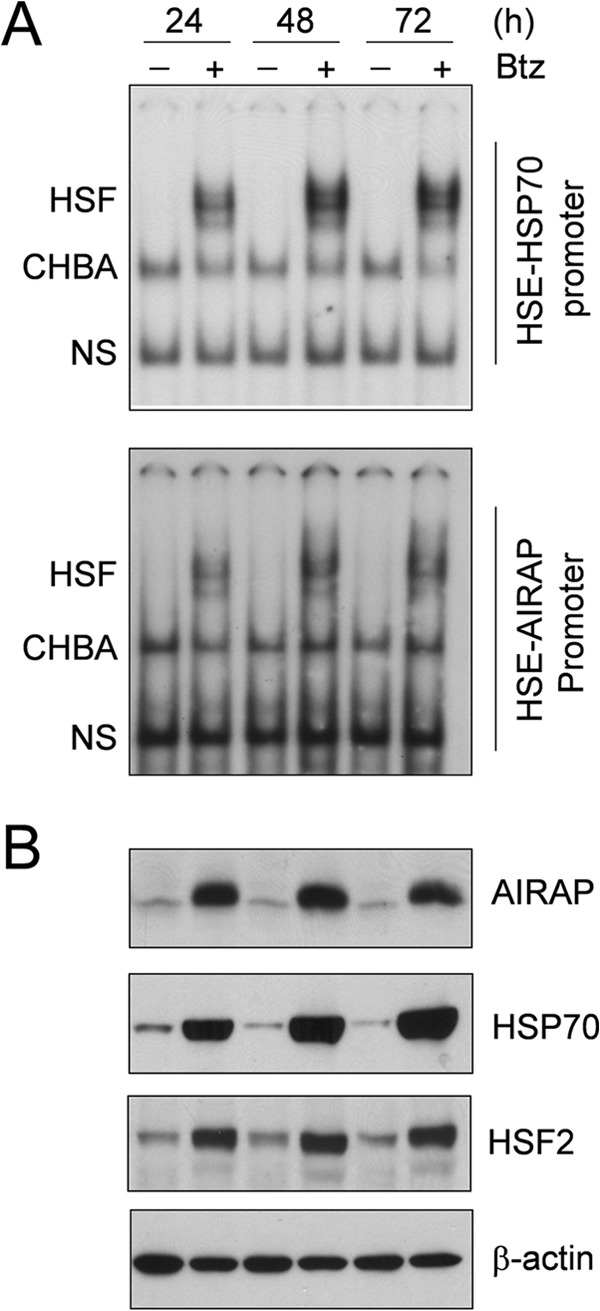
Analysis of HSF-AIRAP promoter complex formation and AIRAP expression during long-term bortezomib treatment. A, HUVECs were treated with 10 nm bortezomib (Btz) (+) or vehicle (−), and at 24, 48, and 72 h, whole-cell extracts were analyzed for HSF DNA-binding activity by EMSA in a 4% polyacrylamide gel using an HSP70-HSE ideal probe (29) (top panel) or a 3% polyacrylamide gel using a 75-bp (from −164 to −238) DNA fragment containing the HSE element of the AIRAP promoter region (23) (bottom panel). The positions of the HSF DNA-binding complex (HSF), constitutive HSE-binding activity (CHBA), and nonspecific protein-DNA interaction (NS) are shown. B, in parallel samples, whole-cell extracts were analyzed for levels of AIRAP, HSP70, HSF2, and β-actin proteins by Western blot analysis. Data from one representative experiment of two with similar results are shown.
Both HSF1 and HSF2 Are Recruited to the AIRAP Promoter and Regulate Bortezomib-induced AIRAP Expression
Down-regulation of proteasome function is known to induce DNA-binding activity of both HSF1 and HSF2 (18, 34). However, the level of HSF1 contribution to the activity induced may vary among cells of different species and tissue origins (15). To determine the composition of the HSF complex binding to the AIRAP promoter, whole-cell extracts from HUVECs treated with 10 nm bortezomib for 18 h or exposed to HS at 43 °C for 40 min were subjected to supershift analysis using specific anti-HSF1 and HSF2 antibodies. HSF-DNA complexes were found to contain both HSF1 and HSF2, as detected by the appearance of slower-migrating HSF1 and HSF2 antibody-containing ternary complexes in native gel electrophoresis (Fig. 6A). It should be noted that the supershift of HSF-DNA complexes was complete at high concentrations of anti-HSF1 but not of anti-HSF2 antibodies, suggesting that the bortezomib-induced HSF-DNA complex may contain a mixture of HSF1-HSF2 heterotrimers and HSF1-HSF1 homotrimers. Similar results were obtained for the HSP70 promoter (Fig. 6A). As expected (15), different from bortezomib, a supershift analysis confirmed that heat induced HSF1 (but not HSF2) DNA-binding activity for both the AIRAP-HSE and HSP70-HSE sequence (Fig. 6B).
FIGURE 6.
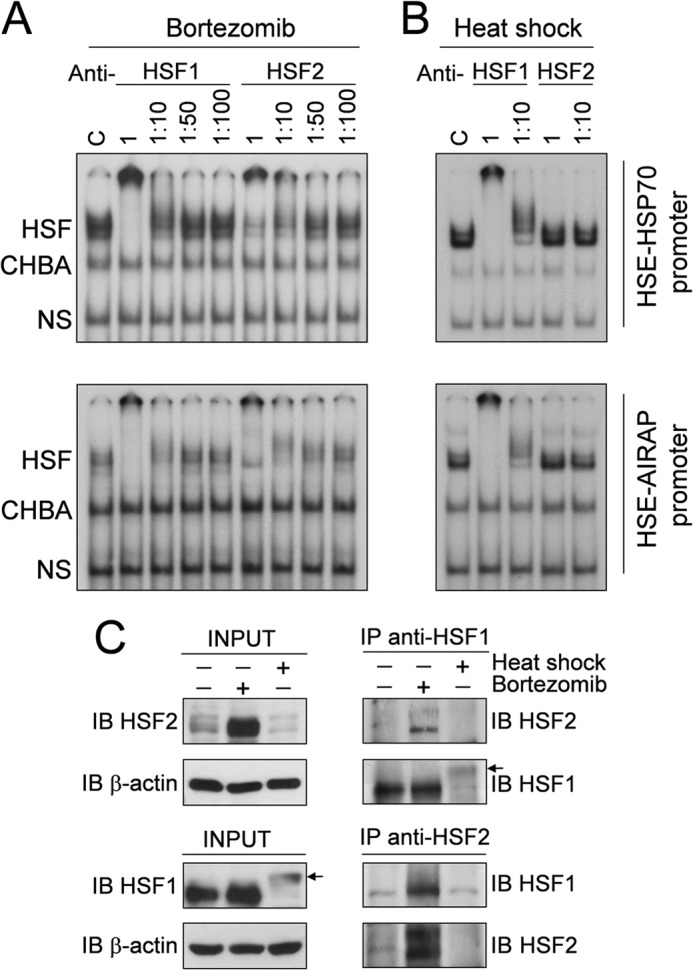
In vitro characterization of AIRAP promoter-HSF complexes in bortezomib-treated endothelial cells. HUVECs were treated with 10 nm bortezomib for 18 h (A) or subjected to heat shock (HS) at 43 °C for 40 min (B). Whole-cell extracts were preincubated with different dilutions of anti-HSF1 or anti-HSF2 polyclonal antibodies and analyzed for supershift assay using the same probes as in Fig. 5A. Positions of the HSF DNA-binding complex (HSF), constitutive HSE-binding activity (CHBA), and nonspecific protein-DNA interaction (NS) are shown. C, control. C, HUVECs treated as in A were used for HSF1 and HSF2 coimmunoprecipitations. Cell extracts from untreated cells or cells treated with bortezomib or heat shock were used to detect HSF1 or HSF2 proteins by immunoblot (IB) analysis before (INPUT) or after immunoprecipitation (IP) with anti-HSF1 or anti-HSF2 antibodies. Arrows indicate the low-mobility phosphorylated HSF1 isoform. Data from one representative experiment of two with similar results are shown.
The possibility that HSF1 and HSF2 interact physically to form heterocomplexes was verified by coimmunoprecipitation experiments. HUVECs treated with bortezomib (10 nm, 18 h) or exposed to HS (43 °C, 40 min) were subjected to coimmunoprecipitation with anti-HSF1 or HSF2 antibodies, followed by Western blot. As shown in Fig. 6C, coimmunoprecipitating HSF1 and HSF2 were detected after HSF2 or HSF1 immunoprecipitation in bortezomib-treated cells. The results confirmed that high levels of HSF2 accumulate in cells treated with the PI, whereas, as expected, in heat-shocked cells HSF2 levels are hardly detectable because of heat-induced HSF2 degradation (34, 37–39). It should be noted that, given the high levels of HSF2 in bortezomib-treated cells, we cannot exclude that the HSF2/HSF1 interaction detected in coimmunoprecipitation experiments could reflect a nonspecific, dose-dependent effect. It should also be noted that, under the conditions used, in heat-shocked cells HSF1 reached higher levels of phosphorylation compared with bortezomib-treated cells.
To investigate whether both HSF1 and HSF2 were recruited to the AIRAP-promoter in vivo, HUVECs were treated with bortezomib (10 nm, 18 h) or subjected to HS (43 °C, 40 min), and HSF1 and HSF2 recruitment to AIRAP and HSP70 promoters was analyzed by ChIP assay. HSF1- and HSF2-coprecipitating DNA was analyzed by PCR with promoter-specific primers amplifying the AIRAP and HSP70 promoters, respectively, and the rate of amplification was verified using cross-linked, non-immunoprecipitated chromatin. The specificity of chromatin immunoprecipitation was determined by using an unrelated control antibody (Fig. 7A). Consistent with the supershift analysis described above, both HSF1 and HSF2 were found to be recruited to the AIRAP and HSP70 promoters in bortezomib-treated cells, whereas mainly HSF1 was detected after HS. These data confirm that, upon proteasome inhibition, both factors bind to the AIRAP promoter. The fact that both HSF1 and HSF2 were also recruited to the HSP70 promoter suggests that HSF2 activation and recruitment to different HS genes may represent a general response to bortezomib treatment in endothelial cells.
FIGURE 7.
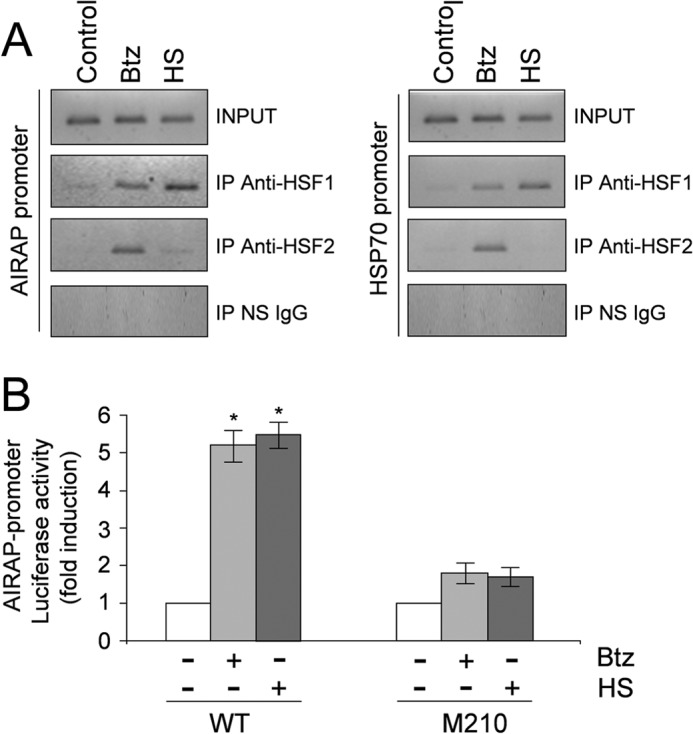
In vivo analysis of AIRAP promoter occupancy by HSF1 and HSF2 transcription factors after bortezomib treatment. A, HUVECs were treated with 10 nm bortezomib (Btz) for 18 h or subjected to heat shock (HS) at 43 °C for 40 min or left untreated (Control). Recruitment of HSF1 and HSF2 to the AIRAP and HSP70 promoters was analyzed by ChIP assay. ChIP-enriched DNAs using preimmune serum (IP NS IgG), anti-HSF1 (IP anti-HSF1), or anti-HSF2 (IP anti-HSF2) as well as input DNAs (INPUT) were prepared, and DNA fragments of the AIRAP gene (−208 to +45) and HSP70 gene (−262 to −70) were amplified by PCR. Data from one representative experiment of three with similar results are shown. B, HUVECs were transfected with the AIRAP-PGL3 construct (WT) and the AIRAP-PGL3-M210-HSE2 (M210) point-mutated (G to T) construct (23). After 16 h, cells were treated with bortezomib, subjected to heat shock (43 °C for 40 min), or left untreated. 9 h after bortezomib treatment and at 7 h of recovery at 37 °C after HS, whole-cell extracts were analyzed for luciferase activity. Data, expressed as fold induction of untreated control, represent the mean of quadruplicate samples from two independent experiments. Error bars indicate mean ± S.D. *, p < 0.05.
We have previously identified an HSE located at −211 from the transcription start site (HSE2) in the human AIRAP promoter as the critical element for heat-induced AIRAP transcription (23). To determine the functional role of HSFs in bortezomib-driven AIRAP expression using luciferase reporter activity analysis, HUVECs were transiently transfected with the AIRAP PGL3-M210-HSE2 (M210) construct, in which the functional HSE2 was mutated (23), and the wild-type WT-AIRAP-PGL3 (WT) construct, and, after 24 h cells were treated with bortezomib or subjected to HS (43 °C, 40 min). At 9 h after bortezomib treatment and at 7 h of recovery at 37 °C after HS, whole-cell extracts were analyzed for luciferase activity. As shown in Fig. 7B, under these conditions, both bortezomib and heat induced AIRAP promoter-driven transcription selectively in cells transfected with the wild-type construct.
Role of HSF1 and HSF2 on Bortezomib-induced AIRAP Expression
To better define how HSF1 and HSF2 contribute to bortezomib-induced AIRAP expression, both factors were down-regulated by siRNA. HUVECs were transiently transfected with HSF2 (si-HSF2), HSF1 (si-HSF1), or control (scrambled) siRNAs and, after 30 h, treated with 10 nm bortezomib. After 12 h, whole-cell extracts were analyzed for HSF2, HSF1, AIRAP, and HSP70 protein levels by Western blot analysis. As shown in Fig. 8A, transient transfection with si-HSF1 or si-HSF2 resulted in specific down-regulation of HSF1 or HSF2, respectively. It should be noted that transfection with si-HSF2 also prevented the bortezomib-induced increase in HSF2 levels.
FIGURE 8.
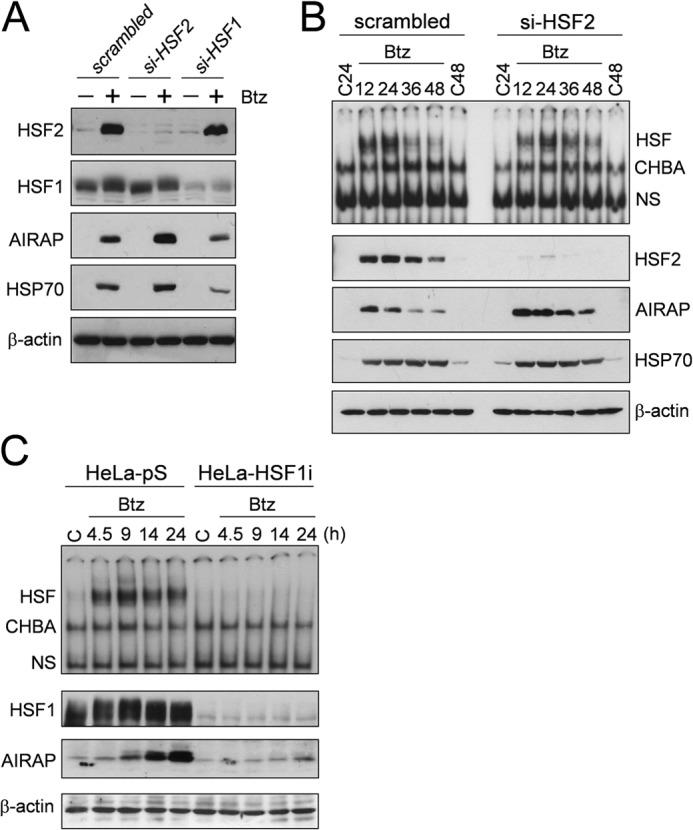
Effect of HSF1 and HSF2 silencing on bortezomib-induced AIRAP protein expression. A, HUVECs were transiently transfected with HSF2 (si-HSF2), HSF1 (si-HSF1), or control (scrambled) siRNAs and, after 30 h, cells were treated with 10 nm bortezomib (Btz) (+) or vehicle (−). After 12 h of bortezomib treatment, whole-cell extracts were analyzed for levels of HSF2, HSF1, AIRAP, HSP70, and β-actin proteins by Western blot analysis. Data from one representative experiment of two with similar results are shown. B, HUVECs were transiently transfected with HSF2 or scrambled siRNAs, and, after 10 h, cells were treated with 10 nm bortezomib or left untreated (C24 and C48). 12, 24, 36, and 48 h after bortezomib treatment, whole-cell extracts were analyzed for HSF DNA-binding activity by EMSA (top panel). The positions of the HSF DNA-binding complex (HSF), constitutive HSE-binding activity (CHBA), and nonspecific protein-DNA interaction (NS) are shown. The levels of HSF2, AIRAP, HSP70, and β-actin as a control were determined in the same samples by Western blot analysis (bottom panels). C, wild-type (HeLa-pS) and HSF1-silenced (HeLa-HSF1i) HeLa cells were treated with 25 nm bortezomib, and, at different times after treatment, whole-cell extracts were analyzed for HSF DNA-binding activity by EMSA using the AIRAP-HSE probe (top panel). The levels of HSF1, AIRAP, and β-actin as a control were determined in the same samples by Western blot analysis (bottom panels).
As shown in Fig. 8A, following bortezomib treatment, AIRAP and HSP70 proteins were down-regulated in HSF1-depleted cells compared with the control. Interestingly, HSF2 silencing, instead, resulted in an increase in AIRAP level, suggesting a regulatory role of HSF2 in AIRAP expression during proteasome inhibition.
On the basis of this observation, the kinetics of AIRAP expression was investigated in HSF2-depleted cells. HUVECs transiently transfected with HSF2 siRNAs were treated with 10 nm bortezomib. At different times after treatment, whole-cell extracts were analyzed for HSE-binding activity by EMSA and for HSF2, AIRAP, and HSP70 protein levels by Western blot analysis. The results shown in Fig. 8B confirm that si-HSF2 silencing results in an increase in AIRAP levels at all times analyzed. A modest increase in HSP70 levels was detected at early time points (12 h) after bortezomib treatment (Fig. 8, A and B).
Finally, the role of HSF1 in bortezomib-induced AIRAP expression was investigated using a stable human cervical carcinoma cell line (HeLa-HSF1i) with HSF1 loss of function, generated previously by using stable RNA-mediated interference (26). HeLa wild-type (HeLa-pS) and HeLa-HSF1i cells were treated with bortezomib (25 nm), and, at different time points after treatment, whole-cell extracts were analyzed for HSF DNA-binding activity by EMSA using the HSE-AIRAP probe and for AIRAP levels by Western blot analysis. HSF1 and β-actin levels were determined as a control. As shown in Fig. 8C, in the absence of HSF1, no DNA-binding activity was observed, and AIRAP expression was almost completely abrogated, confirming that HSF1 is critical for AIRAP gene transcription following proteasome inhibition.
Effect of Bortezomib on AIRAP Expression in Human Primary Blood-derived Cells
To investigate whether AIRAP induction is a general response of human cells to proteasome inhibition, the ability of bortezomib to trigger AIRAP expression was evaluated in human blood-derived primary cells. Peripheral blood monocytes and lymphocytes from healthy donors and HUVECs were treated with 10 nm bortezomib, and, after 14 h, whole-cell extracts were analyzed for the levels of polyubiquitinated proteins, AIRAP, HSF2, and β-actin by Western blot. As shown in Fig. 9, bortezomib increased AIRAP levels in all cells analyzed, indicating a general response of human cells to the drug. It should be noted that HSF2 levels were also increased in bortezomib-treated monocytes and lymphocytes.
FIGURE 9.
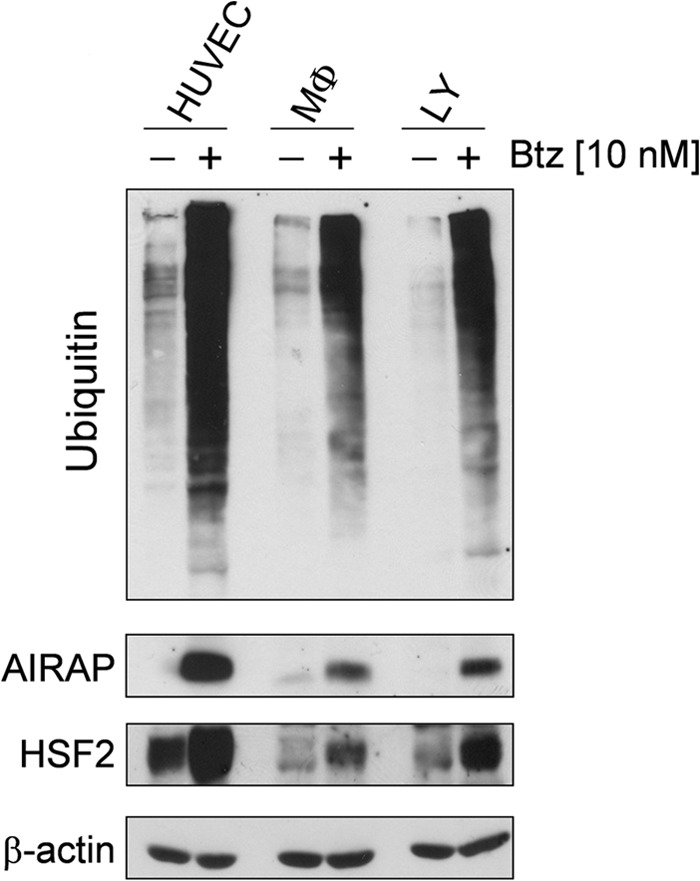
Induction of AIRAP expression by bortezomib in human primary blood-derived cells. HUVECs, peripheral blood monocytes (MΦ), and lymphocytes (LY) from healthy donors were treated with 10 nm bortezomib (Btz) (+) or vehicle (−) for 14 h. Levels of AIRAP, HSF2, polyubiquitinated proteins, and β-actin as a control were determined by Western blot analysis. Data from one representative experiment of three with similar results are shown.
DISCUSSION
AIRAP was originally identified as an arsenite-inducible, cysteine- and histidine-rich RNA-associated protein that is conserved among mammals, Drosophila, and Caenorhabditis elegans (40). We have shown recently that the human AIRAP gene behaves as a canonical heat shock gene whose expression is strictly controlled by HSF1 in a temperature-dependent fashion. Transcription is triggered at temperatures above 40 °C in different types of human primary and cancer cells (23). An HSE located at −211 from the transcription start site was identified in the human AIRAP promoter as the critical element for heat-induced AIRAP transcription (23).
We have shown now that the selective PI bortezomib, a drug in current clinical use for treatment of multiple myeloma and other chemotherapy-resistant cancers, is a potent inducer of AIRAP expression in human cells. In endothelial cells, bortezomib is 10 times more active than the peptide-aldehyde MG132, being effective at concentrations as low as 10 nm, which is comparable with plasma concentrations in treated patients (Millennium, Velcade prescribing information).
Pharmacokinetic/pharmacodynamic studies have shown that bortezomib is distributed rapidly into patient tissues after administration of a single dose, with proteasome inhibition occurring within 1 h and recovering within 72–96 h after administration (41). Analysis of AIRAP expression kinetics in vitro after a single bortezomib treatment has shown that AIRAP mRNA started to increase between 3–6 h of treatment, following polyubiquitinated protein accumulation and HSF activation, reaching maximal levels at 18 h and continuing up to 48 h with kinetics similar to HSP70 mRNA. Following the increase in AIRAP mRNA levels, AIRAP protein expression was detected after 6 h of treatment. The protein was expressed at high levels for the next 24 h both in the nucleus and cytoplasm of treated cells. Different from HSP70, accumulation of AIRAP was found to decrease at 48 h after treatment. This is likely a consequence of the lower stability of AIRAP compared with HSP70.
Because little is known about the effect of persistent proteasome inhibition on heat shock response regulation, we investigated the effect of repeated bortezomib treatments for a 72-h period. Under these conditions, sustained HSF DNA-binding activity and AIRAP expression were detected until 72 h. Interestingly, heat shock response attenuation was not observed, despite the high levels of HSP produced (42).
It is important to note that HSF2 protein levels were increased dramatically in bortezomib-treated cells starting at 6 h after treatment and continuing up to 48 h in single-treatment experiments and 72 h after repeated treatments. Inhibition of protein degradation coupled with an increase in HSF2 protein synthesis has been reported previously to be responsible for the accumulation of HSF2 protein following proteasome inhibition in different cell lines (15, 16). We now report, for the first time, an increase in HSF2 mRNA levels in bortezomib-treated human primary cells, suggesting that transcriptional/posttranscriptional regulation of HSF2 messages may also participate in increasing HSF2 protein levels during proteasome inhibition. Because of its regulatory role in differentiation and development (43), it should be emphasized that the dramatic increase in HSF2 detected in human primary endothelial cells, monocytes, and lymphocytes after bortezomib treatment may have relevant effects and should be considered in clinical regimens.
HSF1 and HSF2 differ in the nature of their transcriptional responses as well as in their pathways of activation. Whereas HSF1 is activated upon a wide variety of stress conditions (20, 34, 44), HSF2 is more selective, being induced during differentiation (43) and in early development (43) in addition to down-regulation of the ubiquitin-proteasome pathway (15). Unlike HSF1, which is a stable protein expressed evenly and constitutively, HSF2 is a labile protein, and its levels vary in different types of tissues and may fluctuate during developmental processes (43, 45). HSF2 expression levels are regulated both transcriptionally and by mRNA stabilization (46).
Upon activation, HSF1 undergoes a monomer-to-trimer transition, whereas HSF2 usually undergoes a dimer-to-trimer transition and, different from HSF1, does not appear to require phosphorylation (47, 48). Although both HSFs are able to recognize similar HSEs (49–51), different from HSF1, the mechanism of HSF2 recruitment to stress-inducible promoters has not yet been elucidated (21). Differences in HSE sequences and in the number of canonical and non-canonical nGAAn pentamers appear to be important for the type of HSF recruited. HSF1 binding has been shown to utilize a higher degree of cooperativity than HSF2, which appears to be more confined to short HSEs containing a small number of GAA blocks (49, 50).
It has been shown that, under specific conditions, including proteasome inhibition, HSF1 and HSF2 may interact because of the highly homologous trimerization domains and form heterocomplexes that could be recruited to specific promoters (21, 52). We now show that, after bortezomib treatment, both HSF1 and HSF2 are recruited to the AIRAP promoter. We also show that the two factors interact directly and form heterocomplexes. In addition, supershift analysis suggests that HSF-AIRAP promoter complexes may contain a mixture of HSF1-HSF2 heterotrimers and HSF1-HSF1 homotrimers. The possibility that different complexes may bind to the AIRAP promoter is supported by the presence of a contiguous array of seven units of the 5′-nGAAn-3′ consensus motif in the promoter that could adapt occupancy by at least two HSF trimers (23).
The fact that bortezomib is not able to induce AIRAP expression in human cells depleted of HSF1 indicates that HSF1 has a primary role in AIRAP transcription that cannot be compensated by HSF2 alone. Regarding HSF2, the potential impact of HSF2 on stress-induced heat shock gene expression and HSP accumulation is not yet well defined. At the transcriptional level, HSF2 has been reported to positively and/or negatively modulate the expression of specific heat shock genes (21, 22). On the other hand, a role of HSF2 in the regulation of proteasome activity with a consequent increase in HSP stability has also been described (53). In the case of AIRAP, we now report that HSF2 negatively regulates AIRAP expression after bortezomib treatment, further emphasizing an important modulatory role of this transcription factor during proteotoxic stress. A negative regulatory role of HSF2 on selected HSF1 target genes is of particular interest in the case of bortezomib because high levels of the factor can be reached with clinically relevant concentrations of the drug.
As indicated above, AIRAP biological function is still not well defined. Arsenite-induced AIRAP has been found to associate with the 19 S cap of the proteasome and has been suggested to alter the 19 S cap biochemical properties to facilitate substrate transit through the particle (24). It has been discovered recently that mammals have a second, constitutively expressed, AIRAP-like gene (AIRAPL) that also encodes a proteasome-interacting protein that shares with AIRAP the property of enhancing peptide accessibility to the proteasome active site (54). Bortezomib did not alter the expression of AIRAPL at concentrations that strongly induced AIRAP. On the other hand, inhibition of AIRAP expression by siRNA silencing only modestly (20%) decreased HUVEC viability at 48 h after 10 nm bortezomib treatment (Fig. 10). In addition, AIRAP silencing did not cause a significant change in proteasome function in the first 24 h after a single treatment with 10 nm bortezomib under conditions where chymotrypsin-like activity is inhibited by ∼80% (data not shown). It should be noted that, if AIRAP participates in accelerating polyubiquitinated protein transit through the particle, during bortezomib treatment, it could only act on the residual 20% proteasome activity, and only a modest proteasome regulatory effect could be expected under these conditions. It cannot be excluded that AIRAP may act at a different level during proteotoxic stress. AIRAP was discovered as an RNA-binding protein and, because of its nuclear and cytoplasmic localization in endothelial cells, the possibility that it may participate in mRNA and pre-mRNA protection/rescue and/or stabilization (55, 56) during proteotoxic stress should be considered.
FIGURE 10.
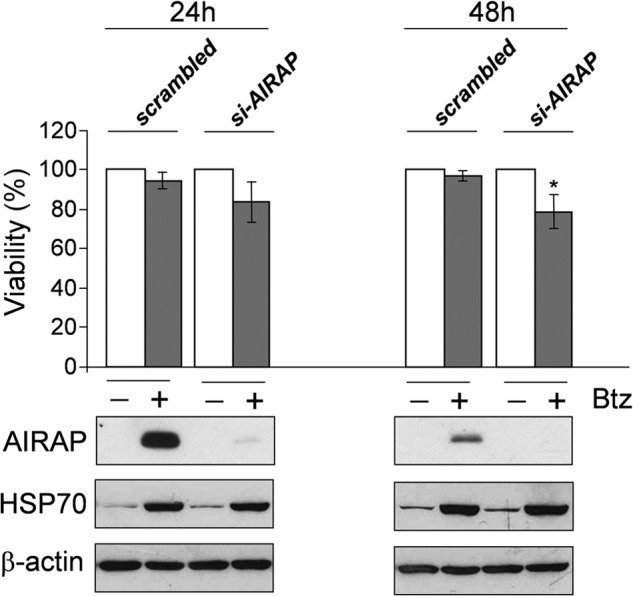
Effect of AIRAP silencing on HUVEC viability after bortezomib treatment. HUVECs were transiently transfected with AIRAP (si-AIRAP) or control (scrambled) siRNAs, and, after 10 h, cells were treated with 10 nm bortezomib (Btz) (+) or vehicle (−). 24 and 48 h after treatment, cell viability was determined by MTT assay. Data, expressed as a percentage of untreated control, represent the mean ± S.D. of three independent experiments (top panel). *, p < 0.05. The levels of AIRAP, HSP70, and β-actin as a control were determined by Western blot analysis in parallel samples (bottom panels).
Finally, in addition to endothelial cells, bortezomib was found to induce an abundant expression of AIRAP in human peripheral blood-derived cells and in multiple myeloma cells (data not shown). Because inhibition of proteasome activity in peripheral blood mononuclear cells and accumulation of proteasome-targeted polypeptides in tumor tissue are pharmacodynamic markers of bortezomib activity after intravenous administration (57, 58), the results described suggest that AIRAP may represent a new biomarker of bortezomib activity.
In conclusion, we have identified AIRAP as a novel bortezomib target gene in human cells. Differently from heat stress, bortezomib-induced AIRAP expression involves the interplay between HSF1 and HSF2, which should be taken into consideration when planning proteasome inhibitor-based therapies. AIRAP function in the endothelium remains to be elucidated, and its role in cancer is totally unexplored. However, the abundant presence of this recently identified heat shock protein in cells treated with clinically relevant concentrations of the drug suggests that AIRAP may be a novel component of the regulatory network controlling proteotoxic stress during proteasome inhibition and may, therefore, contribute to the outcome of bortezomib treatment.
Acknowledgments
We thank Prof. Adorno (Hematology Division, University of Rome Tor Vergata) and Prof. Girelli (Centro Trasfusionale Policlinico Umberto I, University of Rome La Sapienza, Rome, Italy) for providing the buffy coats. We also thank Dr. Ewa Krasnowska, Institute of Translational Pharmacology, Consiglio Nazionale delle Ricerche (IFT, CNR) for assistance with confocal microscopy.
This work was supported by Italian Ministry of University and Scientific Research PRIN project Grant N 2010PHT9NF-006.
- HS
- heat shock
- HSF
- heat shock factor
- HSE
- heat shock element
- HSP
- heat shock protein
- AIRAP
- arsenite-inducible RNA-associated protein
- PI
- proteasome inhibitor(s)
- HUVEC
- human umbilical vein endothelial cell
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
REFERENCES
- 1. Hershko A., Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 2. Tomko R. J., Jr., Hochstrasser M. (2013) Molecular architecture and assembly of the eukaryotic proteasome. Annu. Rev. Biochem. 82, 415–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bassermann F., Eichner R., Pagano M. (2014) The ubiquitin proteasome system: implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta 1843, 150–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kirkin V., Dikic I. (2007) Role of ubiquitin- and Ubl-binding proteins in cell signaling. Curr. Opin. Cell Biol. 19, 199–205 [DOI] [PubMed] [Google Scholar]
- 5. Moran-Crusio K., Reavie L. B., Aifantis I. (2012) Regulation of hematopoietic stem cell fate by the ubiquitin proteasome system. Trends Immunol. 33, 357–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vucic D., Dixit V. M., Wertz I. E. (2011) Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat. Rev. Mol. Cell Biol. 12, 439–452 [DOI] [PubMed] [Google Scholar]
- 7. Rahimi N. (2012) The ubiquitin-proteasome system meets angiogenesis. Mol. Cancer Ther. 11, 538–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beck P., Dubiella C., Groll M. (2012) Covalent and non-covalent reversible proteasome inhibition. Biol. Chem. 393, 1101–1120 [DOI] [PubMed] [Google Scholar]
- 9. Huang L., Chen C. H. (2009) Proteasome regulators: activators and inhibitors. Curr. Med. Chem. 16, 931–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goldberg A. L. (2012) Development of proteasome inhibitors as research tools and cancer drugs. J. Cell Biol. 199, 583–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen D., Frezza M., Schmitt S., Kanwar J., Dou Q. P. (2011) Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr. Cancer Drug Targets 11, 239–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kern J., Untergasser G., Zenzmaier C., Sarg B., Gastl G., Gunsilius E., Steurer M. (2009) GRP-78 secreted by tumor cells blocks the antiangiogenic activity of bortezomib. Blood 114, 3960–3967 [DOI] [PubMed] [Google Scholar]
- 13. Roccaro A. M., Hideshima T., Raje N., Kumar S., Ishitsuka K., Yasui H., Shiraishi N., Ribatti D., Nico B., Vacca A., Dammacco F., Richardson P. G., Anderson K. C. (2006) Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res. 66, 184–191 [DOI] [PubMed] [Google Scholar]
- 14. Ferrarini M., Ferrero E. (2011) Proteasome inhibitors and modulators of angiogenesis in multiple myeloma. Curr. Med. Chem. 18, 5185–5195 [DOI] [PubMed] [Google Scholar]
- 15. Mathew A., Mathur S. K., Morimoto R. I. (1998) Heat shock response and protein degradation: regulation of HSF2 by the ubiquitin-proteasome pathway. Mol. Cell. Biol. 18, 5091–5098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pirkkala L., Alastalo T. P., Zuo X., Benjamin I. J., Sistonen L. (2000) Disruption of heat shock factor 1 reveals an essential role in the ubiquitin proteolytic pathway. Mol. Cell. Biol. 20, 2670–2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bush K. T., Goldberg A. L., Nigam S. K. (1997) Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J. Biol. Chem. 272, 9086–9092 [DOI] [PubMed] [Google Scholar]
- 18. Kawazoe Y., Nakai A., Tanabe M., Nagata K. (1998) Proteasome inhibition leads to the activation of all members of the heat-shock-factor family. Eur. J. Biochem. 255, 356–362 [DOI] [PubMed] [Google Scholar]
- 19. Akerfelt M., Morimoto R. I., Sistonen L. (2010) Heat shock factors: integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 11, 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anckar J., Sistonen L. (2011) Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu. Rev. Biochem. 80, 1089–1115 [DOI] [PubMed] [Google Scholar]
- 21. Sandqvist A., Björk J. K., Akerfelt M., Chitikova Z., Grichine A., Vourc'h C., Jolly C., Salminen T. A., Nymalm Y., Sistonen L. (2009) Heterotrimerization of heat-shock factors 1 and 2 provides a transcriptional switch in response to distinct stimuli. Mol. Biol. Cell 20, 1340–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ostling P., Björk J. K., Roos-Mattjus P., Mezger V., Sistonen L. (2007) Heat shock factor 2 (HSF2) contributes to inducible expression of hsp genes through interplay with HSF1. J. Biol. Chem. 282, 7077–7086 [DOI] [PubMed] [Google Scholar]
- 23. Rossi A., Trotta E., Brandi R., Arisi I., Coccia M., Santoro M. G. (2010) AIRAP, a new human heat shock gene regulated by heat shock factor 1. J. Biol. Chem. 285, 13607–13615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stanhill A., Haynes C. M., Zhang Y., Min G., Steele M. C., Kalinina J., Martinez E., Pickart C. M., Kong X. P., Ron D. (2006) An arsenite-inducible 19S regulatory particle-associated protein adapts proteasomes to proteotoxicity. Mol. Cell 23, 875–885 [DOI] [PubMed] [Google Scholar]
- 25. Elia G., Polla B., Rossi A., Santoro M. G. (1999) Induction of ferritin and heat shock proteins by prostaglandin A1 in human monocytes. Evidence for transcriptional and post-transcriptional regulation. Eur. J. Biochem. 264, 736–745 [DOI] [PubMed] [Google Scholar]
- 26. Rossi A., Ciafrè S., Balsamo M., Pierimarchi P., Santoro M. G. (2006) Targeting the heat shock factor 1 by RNA interference: a potent tool to enhance hyperthermochemotherapy efficacy in cervical cancer. Cancer Res. 66, 7678–7685 [DOI] [PubMed] [Google Scholar]
- 27. Rossi A., Kapahi P., Natoli G., Takahashi T., Chen Y., Karin M., Santoro M. G. (2000) Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature 403, 103–108 [DOI] [PubMed] [Google Scholar]
- 28. Rossi A., Elia G., Santoro M. G. (1997) Inhibition of nuclear factor κ B by prostaglandin A1: an effect associated with heat shock transcription factor activation. Proc. Natl. Acad. Sci. U.S.A. 94, 746–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mosser D. D., Theodorakis N. G., Morimoto R. I. (1988) Coordinate changes in heat shock element-binding activity and HSP70 gene transcription rates in human cells. Mol. Cell. Biol. 8, 4736–4744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rossi A., Elia G., Santoro M. G. (1998) Activation of the heat shock factor 1 by serine protease inhibitors. An effect associated with nuclear factor-κB inhibition. J. Biol. Chem. 273, 16446–16452 [DOI] [PubMed] [Google Scholar]
- 31. Rossi A., Coccia M., Trotta E., Angelini M., Santoro M. G. (2012) Regulation of cyclooxygenase-2 expression by heat: a novel aspect of heat shock factor 1 function in human cells. PLoS ONE 7, e31304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Veschini L., Belloni D., Foglieni C., Cangi M. G., Ferrarini M., Caligaris-Cappio F., Ferrero E. (2007) Hypoxia-inducible transcription factor-1 α determines sensitivity of endothelial cells to the proteosome inhibitor bortezomib. Blood 109, 2565–2570 [DOI] [PubMed] [Google Scholar]
- 33. Belloni D., Veschini L., Foglieni C., Dell'Antonio G., Caligaris-Cappio F., Ferrarini M., Ferrero E. (2010) Bortezomib induces autophagic death in proliferating human endothelial cells. Exp. Cell Res. 316, 1010–1018 [DOI] [PubMed] [Google Scholar]
- 34. Mathew A., Mathur S. K., Jolly C., Fox S. G., Kim S., Morimoto R. I. (2001) Stress-specific activation and repression of heat shock factors 1 and 2. Mol. Cell. Biol. 21, 7163–7171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gerner C., Vejda S., Gelbmann D., Bayer E., Gotzmann J., Schulte-Hermann R., Mikulits W. (2002) Concomitant determination of absolute values of cellular protein amounts, synthesis rates, and turnover rates by quantitative proteome profiling. Mol. Cell. Proteomics 1, 528–537 [DOI] [PubMed] [Google Scholar]
- 36. Turturici G., Geraci F., Candela M. E., Cossu G., Giudice G., Sconzo G. (2009) Hsp70 is required for optimal cell proliferation in mouse A6 mesoangioblast stem cells. Biochem. J. 421, 193–200 [DOI] [PubMed] [Google Scholar]
- 37. Ahlskog J. K., Björk J. K., Elsing A. N., Aspelin C., Kallio M., Roos-Mattjus P., Sistonen L. (2010) Anaphase-promoting complex/cyclosome participates in the acute response to protein-damaging stress. Mol. Cell. Biol. 30, 5608–5620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sarge K. D., Murphy S. P., Morimoto R. I. (1993) Activation of heat-shock gene-transcription by heat-shock factor-I involves oligomerization, acquisition of DNA-binding activity, and nuclear-localization and can occur in the absence of stress. Mol. Cell. Biol. 13, 1392–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murphy S. P., Gorzowski J. J., Sarge K. D., Phillips B. (1994) Characterization of constitutive Hsf2 DNA-binding activity in mouse embryonal carcinoma-cells. Mol. Cell. Biol. 14, 5309–5317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sok J., Calfon M., Lu J., Lichtlen P., Clark S. G., Ron D. (2001) Arsenite-inducible RNA-associated protein (AIRAP) protects cells from arsenite toxicity. Cell Stress Chaperones 6, 6–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schwartz R., Davidson T. (2004) Pharmacology, pharmacokinetics, and practical applications of bortezomib. Oncology 18, 14–21 [PubMed] [Google Scholar]
- 42. Abravaya K., Phillips B., Morimoto R. I. (1991) Attenuation of the heat shock response in HeLa cells is mediated by the release of bound heat shock transcription factor and is modulated by changes in growth and in heat shock temperatures. Genes Dev. 5, 2117–2127 [DOI] [PubMed] [Google Scholar]
- 43. Pirkkala L., Nykänen P., Sistonen L. (2001) Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J. 15, 1118–1131 [DOI] [PubMed] [Google Scholar]
- 44. Morimoto R. I. (1998) Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 12, 3788–3796 [DOI] [PubMed] [Google Scholar]
- 45. Eriksson M., Jokinen E., Sistonen L., Leppä S. (2000) Heat shock factor 2 is activated during mouse heart development. Int. J. Dev. Biol. 44, 471–477 [PubMed] [Google Scholar]
- 46. Pirkkala L., Alastalo T. P., Nykanen P., Seppa L., Sistonen L. (1999) Differentiation lineage-specific expression of human heat shock transcription factor 2. FASEB J. 13, 1089–1098 [DOI] [PubMed] [Google Scholar]
- 47. Sistonen L., Sarge K. D., Morimoto R. I. (1994) Human heat shock factors 1 and 2 are differentially activated and can synergistically induce hsp70 gene transcription. Mol. Cell. Biol. 14, 2087–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu Y. M., Huang D. Y., Chiu J. F., Lau A. T. (2012) Post-translational modification of human heat shock factors and their functions: a recent update by proteomic approach. J. Proteome Res. 11, 2625–2634 [DOI] [PubMed] [Google Scholar]
- 49. Kroeger P. E., Morimoto R. I. (1994) Selection of new HSF1 and HSF2 DNA-binding sites reveals difference in trimer cooperativity. Mol. Cell. Biol. 14, 7592–7603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Manuel M., Rallu M., Loones M. T., Zimarino V., Mezger V., Morange M. (2002) Determination of the consensus binding sequence for the purified embryonic heat shock factor 2. Eur. J. Biochem. 269, 2527–2537 [DOI] [PubMed] [Google Scholar]
- 51. Yamamoto N., Takemori Y., Sakurai M., Sugiyama K., Sakurai H. (2009) Differential recognition of heat shock elements by members of the heat shock transcription factor family. FEBS J. 276, 1962–1974 [DOI] [PubMed] [Google Scholar]
- 52. Loison F., Debure L., Nizard P., le Goff P., Michel D., le Dréan Y. (2006) Up-regulation of the clusterin gene after proteotoxic stress: implication of HSF1-HSF2 heterocomplexes. Biochem. J. 395, 223–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lecomte S., Desmots F., Le Masson F., Le Goff P., Michel D., Christians E. S., Le Dréan Y. (2010) Roles of heat shock factor 1 and 2 in response to proteasome inhibition: consequence on p53 stability. Oncogene 29, 4216–4224 [DOI] [PubMed] [Google Scholar]
- 54. Yun C., Stanhill A., Yang Y., Zhang Y., Haynes C. M., Xu C. F., Neubert T. A., Mor A., Philips M. R., Ron D. (2008) Proteasomal adaptation to environmental stress links resistance to proteotoxicity with longevity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 105, 7094–7099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mazroui R., Di Marco S., Kaufman R. J., Gallouzi I. E. (2007) Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell 18, 2603–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gareau C., Fournier M. J., Filion C., Coudert L., Martel D., Labelle Y., Mazroui R. (2011) p21(WAF1/CIP1) upregulation through the stress granule-associated protein CUGBP1 confers resistance to bortezomib-mediated apoptosis. PLoS ONE 6, e20254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Orlowski R. Z., Stinchcombe T. E., Mitchell B. S., Shea T. C., Baldwin A. S., Stahl S., Adams J., Esseltine D. L., Elliott P. J., Pien C. S., Guerciolini R., Anderson J. K., Depcik-Smith N. D., Bhagat R., Lehman M. J., Novick S. C., O'Connor O. A., Soignet S. L. (2002) Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J. Clin. Oncol. 20, 4420–4427 [DOI] [PubMed] [Google Scholar]
- 58. Dy G. K., Thomas J. P., Wilding G., Bruzek L., Mandrekar S., Erlichman C., Alberti D., Binger K., Pitot H. C., Alberts S. R., Hanson L. J., Marnocha R., Tutsch K., Kaufmann S. H., Adjei A. A. (2005) A phase I and pharmacologic trial of two schedules of the proteasome inhibitor, PS-341 (bortezomib, Velcade), in patients with advanced cancer. Clin. Cancer Res. 11, 3410–3416 [DOI] [PubMed] [Google Scholar]