

Background: The ΔE mutation in the AAA+ ATPase torsinA is associated with the neurological disease torsion dystonia.
Results: BiP and its co-factors maintain torsinA and torsinAΔE stability, glycosylation, and solubility.
Conclusion: torsinA/ΔE, a chaperone-like protein, requires the assistance of other chaperones to fold.
Significance: Therapeutics that modulate BiP may counteract torsinAΔE-associated physiological defects.
Keywords: Molecular Chaperone, Neurological Diseases, Protein Degradation, Protein Folding, Yeast, BiP, DYT1, GRP78, torsinA
Abstract
Early-onset torsion dystonia (EOTD) is a neurological disorder characterized by involuntary and sustained muscle contractions that can lead to paralysis and abnormal posture. EOTD is associated with the deletion of a glutamate (ΔE) in torsinA, an endoplasmic reticulum (ER) resident AAA+ ATPase. To date, the effect of ΔE on torsinA and the reason that this mutation results in EOTD are unclear. Moreover, there are no specific therapeutic options to treat EOTD. To define the underlying biochemical defects associated with torsinAΔE and to uncover factors that might be targeted to offset defects associated with torsinAΔE, we developed a yeast torsinA expression system and tested the roles of ER chaperones in mediating the folding and stability of torsinA and torsinAΔE. We discovered that the ER lumenal Hsp70, BiP, an associated Hsp40, Scj1, and a nucleotide exchange factor, Lhs1, stabilize torsinA and torsinAΔE. BiP also maintained torsinA and torsinAΔE solubility. Mutations predicted to compromise specific torsinA functional motifs showed a synthetic interaction with the ΔE mutation and destabilized torsinAΔE, suggesting that the ΔE mutation predisposes torsinA to defects in the presence of secondary insults. In this case, BiP was required for torsinAΔE degradation, consistent with data that specific chaperones exhibit either pro-degradative or pro-folding activities. Finally, using two independent approaches, we established that BiP stabilizes torsinA and torsinAΔE in mammalian cells. Together, these data define BiP as the first identified torsinA chaperone, and treatments that modulate BiP might improve symptoms associated with EOTD.
Introduction
In order to function proteins need to acquire proper secondary and tertiary structures. Although the amino acid sequence of a protein is a major determinant of its final fold, most proteins acquire intermediate folding states during translation that require the help of molecular chaperones and folding enzymes (1–5). Chaperones not only maintain protein solubility and facilitate folding but they also recognize misfolded proteins and target them for degradation via protein quality control pathways (6–11). Indeed, cell survival depends on the efficiency of these pathways, as defects in pathway function are lethal when cells are exposed to various stresses. Moreover, misfolded proteins that are not cleared from the cell can severely impact cellular physiology. These proteins can aggregate, they can gain a dominant negative function, and/or they can deplete chaperones, thereby exacerbating the accumulation of misfolded proteins and compromising protein homeostasis or “proteostasis” (12–15). Among all the proteins in the eukaryotic cell, approximately one-third enter the secretory pathway through the endoplasmic reticulum (ER)4 and either remain in the ER, are secreted from the cell, or are ultimately distributed among the plasma membrane, endosomes, vacuole/lysosomes, or Golgi apparatus. In the secretory pathway, misfolded proteins are cleared through diverse mechanisms, beginning with ER-associated degradation (ERAD), which represents a critical step in the protein quality control of secretory proteins (10, 11, 16).
A key chaperone that is required for protein folding in the ER is the lumenal Hsp70, BiP/GRP78 (also known as Kar2 in Saccharomyces cerevisiae) (17–19). Kar2/BiP is also a major contributor to general ER homeostasis, not only by participating in protein folding and protein translocation, assembly, and quality control, but also by regulating calcium homeostasis and ER stress signaling, including the unfolded protein response (20, 21). Kar2/BiP acts in conjunction with several co-factors that regulate its ATP hydrolytic activity and therefore its ability to bind substrates (21). These co-factors include the Hsp40s Erdj3 and Erdj4 (Scj1 and Jem1 in yeast) and the nucleotide exchange factors (NEFs) GRP170 and Sil1 (Lhs1 and Sil1 in yeast) (16, 22–30). Perhaps not surprisingly, BiP has also been associated with human diseases, including cancer, diabetes, and neurological disorders such as Parkinson disease and retinitis pigmentosa. These attributes suggest that BiP is a therapeutic target for the treatment of these ailments (31–33).
Many neurological disorders in humans are associated with defects in protein folding, in the function of the secretory pathway, or in protein quality control (34, 35), including amyotrophic lateral sclerosis, Parkinson, Huntington, and Alzheimer diseases, and prion diseases (36–40). Dystonia, the third most common movement disorder in humans, is also associated with defects in these pathways (41). Dystonia manifests as involuntary muscle contractions that can lead to paralysis and abnormal postures due to the simultaneous contraction of agonist and antagonist muscles (41, 42). A common inherited primary dystonia is early-onset generalized torsion dystonia (EOTD, also known as childhood onset torsion dystonia). EOTD is associated with an autosomal mutation in DYT1 (43, 44). Although dominant, the mutation in DYT1 has low penetrance, indicating the existence of other environmental and genetic factors that are critical for EOTD development (45). The average age of EOTD onset is ∼13 years, usually beginning in the lower limbs and spreading to other parts of the body. Brain biopsies from those afflicted with EOTD indicate the presence of inclusion bodies and neuronal cell enlargement, without associated neurodegeneration or neuronal cell death (46, 47). This lack of neurodegeneration suggests that therapeutic treatment for this chronic yet progressive movement disorder is possible (41).
DYT1 encodes torsinA, an ER- and nuclear envelope-localized ATPase that belongs to the AAA+ ATPases superfamily. AAA+ ATPases are a diverse group of enzymes that contain characteristic ATP-binding and hydrolysis domains defined by the Walker-A, Walker-B, Sensor-I, and Sensor-II motifs (Fig. 1A) (48). AAA+ ATPases can remodel protein complexes or alter protein conformation, and many of them play important roles in dissolving aggregates and in protein quality control, including ClpA, FtsH, Hsp104, p97/Cdc48, and one subassembly embedded within the 26 S proteasome (48–51). Indeed, torsinA appears to function as a chaperone and participates in ER protein quality control and the secretory pathway (48, 52–56). Until now it was unknown whether torsinA itself required the assistance of chaperones to aid in its maturation.
FIGURE 1.

TorsinA and torsinAΔE are ER-localized N-glycosylated proteins in yeast. A, SS, signal sequence; C, cysteines; N143 and N158, N-glycosylated Asn residues mutated in Fig. 8; K108 and E171, residues mutated in Fig. 8 that belong to the Walker motifs; D216, mutation that impacts ΔE penetrance; E302 or E303, glutamate residue deleted in patients with EOTD. Walker-A and -B, and Sensor-I and -II motifs (for ATP binding and hydrolysis) are indicated by black lines. B, ORFs of torsinA and torsinAΔE were cloned into the multicopy shuttle vector pRS426 and are under the control of the constitutive glyceraldehyde-3-phosphate dehydrogenase (GPD) promoter. C, protein samples were prepared from log phase wild-type yeast cells transformed with empty vector (V) or expression vectors for torsinA (T) or torsinAΔE (ΔE) grown at 28 °C and were treated with endoglycosidase H (EndoH) as indicated. The glycosylated species migrate at ∼33 kDa and the unglycosylated species resolve at ∼27 kDa. The * indicates a background band of the antibody. D, indirect immunofluorescence of log phase wild-type yeast cells transformed with expression vectors for torsinA-HA or torsinAΔE-HA or an empty vector control grown at 28 °C. The ER lumenal chaperone Kar2/BiP and the nuclear concentrated dye DAPI were used as controls.
The most common DYT1 mutation associated with EOTD is the deletion of a single glutamate residue from the Glu-302/Glu-303 pair near the C terminus of torsinA (“torsinAΔE”) (Fig. 1A). Deletion of Glu-302/Glu-303 causes a variety of alterations in torsinA's properties, including protein stability, degradation, subcellular localization, conformational change, and molecular interactions (57–68). How these alterations lead to EOTD is undetermined, which precludes the rational design of targeted therapies to control EOTD progression or prevent the disease.
To identify chaperones that aid in torsinA folding and that might be targeted to correct disease phenotypes associated with the ΔE allele, we co-opted the genetic tools available in the yeast S. cerevisiae and designed a new torsinA and torsinAΔE expression system. Yeasts have been used to define the molecular basis underlying several human diseases, including amyotrophic lateral sclerosis, antitrypsin deficiency, cancer, and Huntington, Alzheimer, and Parkinson diseases, among many others (69–72). Using this system, we found that Kar2/BiP and its Hsp40, Scj1, as well as the Kar2/BiP-associated NEF, Lhs1, contribute to torsinA and torsinAΔE stabilization in the ER. We also found that Kar2/BiP plays a dual role in controlling torsinA stability, depending on the presence of the ΔE mutation and/or of secondary mutations in unique functional motifs. Furthermore, Kar2/BiP affects torsinA and torsinAΔE solubility and N-linked glycosylation, consistent with a role in mediating protein folding. Finally, by depleting BiP levels in a mammalian cell model, we confirmed that BiP facilitates torsinA and torsinAΔE biogenesis. Together, these data represent the first demonstration of the cellular chaperones that orchestrate torsinA maturation.
EXPERIMENTAL PROCEDURES
Plasmid and Yeast Strain Construction
The pcDNA3.1-torsinA and pcDNA3.1-torsinAΔE expression plasmids were kindly provided by Dr. Xandra Breakefield (65). The torsinA and torsinAΔE open reading frames (ORF) were subcloned into the yeast expression vector pRS426GPD (73) by double restriction enzyme digestion with EcoRI/XhoI (Fermentas, Thermo Scientific) of pcDNA3.1-torsinA and pcDNA3.1-torsinAΔE and ligation into EcoRI/XhoI-linearized pRS426GPD (Fig. 1B). To construct HA-tagged torsinA and torsinAΔE vectors, a single HA tag was introduced at the C terminus of torsinA and torsinAΔE by in vivo recombination in S. cerevisiae following a previously published protocol (74). Briefly, primers LZJB12 and -13, encoding the HA tag sequence (Table 1), were annealed and co-transformed into S. cerevisiae together with NotI-digested pRS426GPD-torsinA or torsinAΔE. Recombined plasmids were extracted from S. cerevisiae and transformed into E. coli DH5α for amplification.
TABLE 1.
Primers used in this study
| Name | Sequence 5′ to 3′ | Ref. |
|---|---|---|
| LZJB5 | CCTTCCTTTTCGGTTAGAGCGGA | This study |
| LZJB12 | ACGGTGTTCACCAAGTTAGATTATTACTACGATGATTACCCATACGATGTTCCAGATTACGCTTGAGCGGCCGCTCGAGTCATGTAATTAGTTATGTCA | This study |
| LZJB13 | TGACATAACTAATTACATGACTCGAGCGGCCGCTCAAGCGTAATCTGGAACATCGTATGGGTAATCATCGTAGTAATAATCTAACTTGGTGAACACCGT | This study |
| LZJB17 | GAGCAGAAAGGATCACACATGTGGCTTTGGATTTC | This study |
| LZJB18 | GAAATCCAAAGCCACATGTGTGATCCTTTCTGCTC | This study |
| LZJB21 | TTGCACTTTCCACATGCTTCACAAATCACCTTGTACAAGGATCAG | This study |
| LZJB22 | CTGATCCTTGTACAAGGTGATTTGTGAAGCATGTGGAAAGTGCAA | This study |
| LZJB23 | GTTGTGGATTCGAGGCCAAGTGAGTGCCTGTGCGA | This study |
| LZJB24 | TCGCACAGGCACTCACTTGGCCTCGAATCCACAAC | This study |
| LZJB34 | AGTAAAGAAGTTTGGGTAATTCGCT | This study |
| LZJB35 | AGTGTTCTATGTTTGCCTTGATTTC | This study |
| LZJB44 | TCATTTGCGGGTGTCGATGG | This study |
| LZJB45 | CGTTGATGGCCTCCTTAAGC | This study |
| LZJB69 | TTGAACGTAATCTGAGCAATACAAA | This study |
| LZJB70 | TCACACTAAATGCTGATGCCTATAA | This study |
| LZJB71 | TCCTACTTATGGTAATGTGC | This study |
| LZJB72 | TAACTATATCCATTGCGTCC | This study |
| CG_GPD prom | CCCTGAAATTATTCCCCTACTTG | This study |
| K108A-F | GGACAGGCACCGGCGCAAATTTCGTCAGCAAG | This study |
| K108A-R | CTTGCTGACGAAATTTGCGCCGGTGCCTGTCC | This study |
| E171Q-F | CCATCTTCATATTTGATCAAATGGATAAGATGCATGC | This study |
| E171Q-R | GCATGCATCTTATCCATTTGATCAAATATGAAGATGG | This study |
Vectors containing the torsinA genes with mutations in the N-linked glycosylation sites, pLuBr85 (torsinA-N143Q), pLuBr86 (torsinAΔE-N143Q), pLuBr87 (torsinAΔE-N158Q), pLuBr100 (torsinA-N158Q), and pLuBr106 (torsinA-N143Q,N158Q) or at Asp-216, pLuBr60 (torsinA-D216H), and pLuBr61 (torsinAΔE-D216H) (Fig. 1A and Table 2) were made using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies) using primers LZJB21–24 or LZJB17–18, respectively, that were designed using the QuikChange Primer design application available online (Table 1). pRS426GPD-torsinA, pRS426GPD-torsinAΔE, and pLuBr100 were then used as the template in a mutagenic PCR using primers LZJB21 and LZJB22 to introduce the N143Q single mutation and LZJB23 and LZJB24 to introduce the N158Q single mutation. Mutations in the Walker-A (K108A) and -B (E171Q) motifs in torsinA and torsinAΔE were generated as above, with primer pairs K108A-F and K108A-R and E171Q-F and E171Q-R (Table 1), respectively. The mammalian expression vectors pcDNA3.1-torsinA and pcDNA3.1-torsinAΔE were used as templates. These constructs were then subcloned into pRS426GPD through EcoRI/XhoI digestion and ligation to generate the yeast expression vectors containing a single mutation in the Walker-A motif (K108A; pLuBr20 and -23) or Walker-B motif (E171Q; pLuBr21 and -24) (Fig. 1A and Table 2). Similarly, the torsinA constructs lacking the N-terminal hydrophobic domain (Fig. 1A), torsinA-Δ24–40 and torsinAΔE-Δ24–40, were subcloned by EcoRI/XhoI digestion and ligation from pcDNA3.1-torsinA-Δ24–40 and pcDNA3.1-torsinAΔE-Δ24–40 (75) into pRS426GPD, to generate pLuBr18 and pLuBr19, respectively (Table 2). The sequence integrity of all the subcloned DNA fragments and mutagenized sequences was verified by DNA sequence analysis using primers CG_GPDprom and LZJB5 which anneal at the GPD promoter and CYC1 terminator regions in pRS426GPD, respectively (Table 1 and Fig. 1B).
TABLE 2.
Plasmids used in this study
| Details | Plasmid no. | Ref. |
|---|---|---|
| pcDNA3.1-torsinA | 65 | |
| pcDNA3.1-torsinAΔE | 65 | |
| pcDNA3.1-torsinA-HA | 63 | |
| pcDNA3.1-torsinAΔE-HA | 63 | |
| pRS426GPD-torsinA | pLuBr281 | This study |
| pRS426GPD-torsinAΔE | pLuBr280 | This study |
| pRS426GPD-torsinA-HA | pLuBr27 | This study |
| pRS426GPD-torsinAΔE-HA | pLuBr28 | This study |
| pRS426GPD-torsinA-N143Q | pLuBr85 | This study |
| pRS426GPD-torsinA-N158Q | pLuBr100 | This study |
| pRS426GPD-torsinAΔE-N143Q | pLuBr86 | This study |
| pRS426GPD-torsinAΔE-N158Q | pLuBr87 | This study |
| pRS426GPD-torsinA-N143,N158Q | pLuBr106 | This study |
| pRS426GPD-torsinA-D216H | pLuBr60 | This study |
| pRS426GPD-torsinAΔE-D216H | pLuBr61 | This study |
| pRS426GPD-torsinA-K108A | pLuBr20 | This study |
| pRS426GPD-torsinA-E171Q | pLuBr21 | This study |
| pRS426GPD-torsinAΔE-K108A | pLuBr23 | This study |
| pRS426GPD-torsinAΔE-E171Q | pLuBr24 | This study |
| pRS426GPD-torsinA-Δ24–40 | pLuBr18 | This study |
| pRS426GPD-torsinAΔE-Δ24–40 | pLuBr19 | This study |
All S. cerevisiae strains used in this study are described in Table 3. To construct the strains containing a deletion of the PDR5 or PEP4 ORFs in the kar2-1 strain background, the pdr5Δ::KanMX and pep4Δ::KanMX deletion cassettes containing >200 bp of homology region upstream and downstream of each ORF were PCR-amplified (Pfu Turbo, Agilent) from DNA extracted from the corresponding deletion mutant in the haploid BY4742 S. cerevisiae deletion strain collection (Open Biosystems, Thermo Scientific) using primer pairs LZJB69 and -70 (for PDR5) and LZJB34 and -35 (for PEP4) (Table 1), and transformation of these cassettes into the kar2-1 strain (Table 3). The correct genotype of the mutants was confirmed by PCR analysis of genomic DNA of strains resistant to the antibiotic geneticin sulfate G418 (Research Products International Corp.) using primer pairs LZJB71 and -72 (for PDR5) and LZJB44 and -45 (for PEP4), as well as the KanB primer (CTGCAGCGAGGAGCCGTAAT), which anneals to the KanMX gene (Table 1). The phenotypes of the constructed strains were verified as follows. Deletion of PEP4 was verified by a defect in the maturation of the vacuolar protease aminopeptidase 1 (Ape1) (76), and deletion of PDR5 was verified by a general increase in the level of ubiquitinated proteins in MG132 (Peptide Institute Inc., Japan)-treated cells (data not shown) (77, 78). All yeast transformations were performed using lithium acetate/PEG3350, and DNA extractions were performed following standard protocols (79, 80).
TABLE 3.
Strains used in this study
| Strain | Genotype | Reference |
|---|---|---|
| BY4742 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Open Biosystems |
| lhs1Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 lhs1::KanMX | Open Biosystems |
| KAR2 | MATa leu2-3,112 ura3-52 ade2-101 | 103 |
| kar2-1 | MATa leu2-3,112 ura3-52 ade2-101 kar2-1 | 103 |
| kar2-1 pep4Δ | MATa leu2-3,112 ura3-52 ade2-101 kar2-1 pep4::KanMX | This study |
| kar2-1 pdr5Δ | MATa leu2-3,112 ura3-52 ade2-101 kar2-1 pdr5::KanMX | This study |
| JEM1 SCJ1 | MATα prc1-1 leu2-3,112 ura3-52 trp1-Δ901 his3-Δ200 lys2-801 suc2-Δ9 GAL | 29 |
| jem1Δ | MATα prc1-1 leu2-3,112 ura3-52 trp1-Δ901 his3-Δ200 lys2-801 suc2-Δ9 GAL jem1::LEU2 | 29 |
| scj1Δ | MATα prc1-1 leu2-3,112 ura3-52 trp1-Δ901 his3-Δ200 lys2-801 suc2-Δ9 GAL scj1::TRP1 | 29 |
| jem1Δ scj1Δ | MATα prc1-1 leu2-3,112 ura3-52 trp1-Δ901 his3-Δ200 lys2-801 suc2-Δ9 GAL jem1::LEU2 scj1::TRP1 | 29 |
| CNE1 | MATa leu2-3,112 his3-11,15 trp1-1 ura3-1 ade2-1 can1-100 | 145 |
| cne1Δ | MATa leu2-3,112 his3-11,15 trp1-1 ura3-1 ade2-1 can1-100 cne1::LEU2 | 145 |
| CNE1 | MATα leu2-3,112 his3-11,15 trp1-1 ura3-1 ade2-1 can1-100 | 145 |
| cne1Δ | MATα leu2-3,112 his3-11,15 trp1-1 ura3-1 ade2-1 can1-100 cne1::LEU2 | 145 |
| yos9Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 yos9::KanMX | Open Biosystems |
Media and Growth Conditions
Yeast strains were grown at 26–28 °C on YPD medium (1% yeast extract, 2% peptone, 2% dextrose) or on synthetic complete (SC) medium lacking specific amino acids required for auxotrophic selection, as described previously (81). For selection of KanMX-expressing strains, YPD plates were supplemented with 250 μg/ml G418.
TorsinA Secretion Assays
Overnight cultures of yeast cells grown at 26–28 °C in selective medium were spotted onto a nitrocellulose membrane. The membrane was layered on top of solid selective medium, and plates were incubated ∼20 h at 30 °C. To detect secreted protein or total cell protein (which includes the secreted material), we followed a previously published protocol (82). The membranes were then probed for the presence of torsinA or G6PDH and developed as described below.
Biochemical Methods
To assess protein stability, cells were grown overnight in selective medium at 28 °C, diluted to an initial A600 of 0.2 in fresh medium, and incubated at the same temperature for ∼6 h to an A600 of ∼1. Cycloheximide (CHX) (Sigma) was added to a final concentration of ∼190 μg/ml, and cells were incubated at 37 °C for 90 min. A 1-ml aliquot was taken before adding CHX (time 0 min) and every 30 min after adding CHX. The cells were then pelleted by centrifugation at 18,000 × g for 1 min at 4 °C. The cell pellet was immediately frozen in liquid nitrogen. Samples were processed for Western blot analysis as described below. To inhibit the proteasome, cells were preincubated in the presence of MG132 for 30 min at 37 °C before addition of CHX.
Yeast protein extracts were prepared as described previously (83). Samples were resuspended in 50 μl of trichloroacetic acid (TCA) sample buffer (80 mm Tris, pH 8, 8 mm EDTA, pH 8, 3.5% SDS, 5% glycerol, 0.4% Tris base, 0.01% bromphenol blue, and 4% fresh 2-mercaptoethanol), disrupted for 20 s with a mechanical pestle, heated at 75 °C for 5 min, resolved by SDS-PAGE, and fast-transferred to nitrocellulose membranes (fast semi-dry blotter, Thermo Scientific Pierce).
Protein extractions from mammalian cells were performed as follows. Cells were washed with cold PBS, resuspended in RIPA buffer with protease inhibitors (Roche Applied Science), rocked at 4 °C for 40 min, and harvested. The cell suspension was then centrifuged at 18,000 × g for 10 min at 4 °C, and the supernatant was used for Western blotting.
The following antibodies were used for Western blot analysis: rabbit polyclonal anti-torsinA, produced by Cocalico Biologicals (Reamstown, PA), that was made against a soluble lumenal domain of human torsinA (residues 41–332), which was expressed in Drosophila S2 cells and purified (75); mouse monoclonal anti-torsinA D-M2A8 (Cell Signaling); rabbit polyclonal anti-G6PDH (Sigma); rabbit polyclonal anti-Kar2 (84); rabbit polyclonal anti-Pdi1 (a gift from V. Denic, Harvard University, Cambridge, MA); rabbit polyclonal anti-Sec61 (85); rabbit polyclonal anti-Bos1 (a gift from C. Barlowe, Geisel School of Medicine at Dartmouth University, Hanover, NH); mouse monoclonal horseradish peroxidase (HRP)-conjugated anti-HA (clone 3F10, Roche Applied Science); goat polyclonal anti-GRP78 (C20, Santa Cruz Biotechnology); mouse monoclonal anti-β actin ab6276 (Abcam); rat monoclonal anti-Grp94 (9G10, Enzo Life Sciences); and sheep or goat HRP-conjugated anti-mouse, anti-rat, anti-goat, or anti-rabbit IgG secondary antibodies (GE Healthcare and Cell Signaling). Western blots were developed with Supersignal West Pico or Supersignal West Femto Chemiluminescent Substrate (Pierce), and images were visualized using a Kodak 440CF Image Station. The signal was quantified using ImageJ Version 1.46r (National Institutes of Health).
To monitor the acquisition of N-linked glycosylation, whole cell protein extracts were digested with 50 milliunits of endoglycosidase H (Roche Applied Science) for 1 h at 37 °C in the presence of 1 mm PMSF and 100 mm KOAc. Samples were resolved by SDS-PAGE and used for Western blotting, as described above.
To measure protein extraction from microsomal membranes, ER-derived microsomes were prepared from cells grown overnight in selective medium at 28 °C, diluted to an A600 of 0.2, and incubated at the same temperature for ∼6 h to an A600 of ∼1.2. A total of 60 absorbance units of cells were harvested and frozen. The pellets were thawed on ice and resuspended in 600 μl of lysis buffer (20 mm Hepes, pH 7.4, 50 mm KOAc, 2 mm EDTA, pH 8, 100 mm sorbitol, 1 mm dithiothreitol (DTT)). A total of ∼500 μl of glass beads were added, and samples were agitated on a Vortex mixer seven times for 60 s, with a 60-s incubation on ice between each treatment. The lysate was collected in an Eppendorf tube and centrifuged twice at 3000 rpm in a tabletop centrifuge at 4 °C for 5 min to remove cell debris and unbroken cells. The supernatant was then centrifuged at 18,000 × g at 4 °C for 20 min to collect the microsomes, which were washed in 500 μl of Buffer 88 (20 mm Hepes, pH 6.8, 150 mm KOAc, 250 mm sorbitol, 5 mm MgOAc) and centrifuged as above. Membranes were resuspended in Buffer 88, and the protein concentration was adjusted spectrophotometrically by measuring the A280 in 2% SDS. Next, 20 μl of the isolated microsomes were resuspended in 1 ml of the following buffers: Buffer 88, pH 6.8, 1% Triton X-100 (Sigma) in Buffer 88, pH 6.8, 0.1 m freshly prepared sodium carbonate in water, 25 mm CHES buffer, pH 9.5, 25 mm CAPS buffer, pH 10.5, or 6 m urea in 37.5 mm Tris, pH 8, 2 mm EDTA. After the samples were incubated for 30 min on ice, the samples were centrifuged at 100,000 × g for 1 h at 4 °C in a Sorvall RC M120EX ultracentrifuge. The pellets were washed with 500 μl of Buffer 88, and re-centrifuged at 146,000 × g for 15 min at 4 °C. The supernatants were transferred to Eppendorf tubes, and total protein was precipitated by adding 110 μl of 100% TCA and incubating on ice for 15 min, followed by a centrifugation at 18,000 × g for 10 min at 4 °C. Pellets from samples containing Triton X-100 were washed with acetone. All pellets were resuspended in 35 μl of TCA sample buffer, and proteins were resolved by SDS-PAGE and examined by Western blotting. All buffers were supplemented with a proteinase inhibitor mixture.
For protease protection assays, protease inhibitors were removed from the microsomes by washing them twice with 500 μl of Buffer 88. The washed microsomes were incubated on ice for 30 min in the presence or absence of 1% Triton X-100, followed by a 1-h incubation on ice in the presence or absence of 50 μg/ml proteinase K (Sigma). Proteins were precipitated with 500 μl of ice-cold 10% TCA and centrifuged at 18,000 × g for 10 min at 4 °C. Pellets were resuspended in 30 μl of TCA sample buffer. Samples were resolved by SDS-PAGE and analyzed by Western blotting.
For protein co-immunoprecipitation studies, yeast cells were grown overnight in selective medium at 28 °C, diluted to an A600 of 0.2, and grown at the same temperature for ∼6 h to an A600 of ∼1.2. A total of ∼45 absorbance units of cells were harvested and frozen. The pellets were thawed on ice and resuspended in 600 μl of lysis buffer supplemented with 0.5% Triton X-100 and a protease inhibitor mixture and transferred to borosilicate tubes containing ∼500 μl of glass beads. Samples were agitated on a Vortex mixer 9 times for 60 s, with >60 s of incubation on ice between each treatment. The lysate was collected in an Eppendorf tube and centrifuged twice at 3000 rpm in a tabletop centrifuge at 4 °C for 5 min to remove cell debris and unbroken cells. The supernatant was further cleared by centrifuging twice at 18,000 × g at 4 °C for 20 min. The protein concentration was adjusted spectrophotometrically by measuring the A280 of an aliquot in 2% SDS. Extracts were precleared by incubation for 1.5 h at room temperature with 30 μl of protein G-agarose (Roche Applied Science) and then incubated overnight with 30 μl of protein G-agarose (Roche Applied Science) in the presence or absence of 1 μl of anti-Kar2 antiserum. Beads were washed four times with 500 μl of wash buffer (20 mm Hepes, pH 7.4 (buffered with KOH), 150 mm NaCl, and supplemented with a protease inhibitor mixture). 30 μl of TCA sample buffer was added to the beads, and proteins were extracted by heating the beads for 5–7 min at 75 °C. Samples were resolved by SDS-PAGE and analyzed by Western blot.
Indirect Immunofluorescence Microscopy
The preparation of yeast for indirect immunofluorescence was performed following a previously published protocol (86). Briefly, cells were grown overnight in selective medium at 28 °C, diluted to an A600 of 0.3 in fresh medium, and grown at 28 °C for ∼5 h to an A600 of ∼0.7. To fix the yeast, 37% formaldehyde was added (final concentration of 4%), and the cells were incubated for 10 min at 28 °C with shaking. The cell suspension was centrifuged, and the pelleted yeast were resuspended in 5 ml of KM solution (40 mm KPO4, pH 6.5, 0.5 mm MgCl2) with 4% formaldehyde and incubated for 1 h at 28 °C with shaking. The cells were washed twice with KM solution and once with KM solution supplemented with 1.2 m sorbitol (KM + sorbitol) and were resuspended in 500 μl of KM + sorbitol. Next, the cell walls were digested with 30 μl of 10 mg/ml Zymolyase (20T, MP Biomedicals LLC) for no more than 25 min at 37 °C, and the spheroplasts were washed with KM + sorbitol buffer, resuspended in 500 μl of the same buffer, and incubated overnight at 4 °C. A 20-μl aliquot of the cell suspension was spotted on polylysine-coated slides, and the cells were dehydrated with methanol/acetone, blocked with PBS, pH 7.4, supplemented with 0.5% BSA, 0.5% ovalbumin, and 0.1% Triton X-100, and incubated at 37 °C for 1 h. Primary antibodies (mouse anti-HA 12CA5 (Roche Applied Science) (1:100) or rabbit anti-Kar2 (1:250)) diluted in blocking solution were applied, and the cells were incubated overnight at 4 °C, before secondary antibodies (Alexa Fluor 488 anti-mouse and Alexa Fluor 569 anti-rabbit (1:250)) were added for 1 h at room temperature. The slides were mounted using Prolong Gold Antifade reagent with DAPI (Invitrogen). Pictures were taken using a Leica TCS SP5 confocal microscope, ×63 oil immersion objective, and analyzed with Adobe Photoshop (Version 7.0) software.
Analysis of TorsinA and TorsinAΔE Stability and Biogenesis in HeLa Cells
HeLa Tet-On cells (Clontech) were maintained in DMEM (Invitrogen) supplemented with antibiotics and 10% fetal bovine serum at 37 °C in a 5% CO2 humidified incubator. Vectors engineered for the transient expression of torsinA/ΔE or C-terminal HA-tagged versions of torsinA/ΔE (Table 2) were transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions (the medium was changed ∼3.5–4 h post-transfection). siRNA transfections were performed using RNAiMax (Invitrogen) (the medium was changed ∼7 h post-transfection). In these experiments, the torsinA expression vectors were transfected into siRNA-treated cells 24 h after the siRNAs had been introduced. The following Stealth siRNAs (Invitrogen) were used: HSS105077 and HSS179390 against BiP/GRP78, and Stealth RNAi siRNA Negative Control Medium GC (Invitrogen), at a concentration of 1.6 nm. Protein extracts were prepared from cells incubated for another 24 h (a total of 48 h after siRNA transfection). Although higher efficiency of BiP knockdown can be achieved, our protocol was adjusted to minimize cell toxicity (2). As an alternative means to deplete the levels of BiP, we employed the AB5 subtilase cytotoxin (87).
To monitor torsinA stability, pulse chase analyses were performed as previously published (63) and included a 30-min starvation period and a 15-min labeling period with 200 μCi/well of [35S]Met/Cys (ICN Biochemicals) in a 6-well plate. Protein levels at the indicated time points were analyzed after immunoprecipitation with mouse monoclonal anti-HA 16B12 (Covance) and protein G-agarose (Roche Applied Science) using a Typhoon FLA7000 and associated software (version 1.2) (General Electric). Where indicated, the active subtilase toxin or an inactive mutant (SubAA272B) was added to a final concentration of 0.5 μg/ml during amino acid starvation and was present throughout the labeling and chase steps.
Statistical Analysis
Statistical analyses were performed using Student's t test (Microsoft Excel Software), assuming equal variances. A p value < 0.05 was considered significant. All statistical analyses of protein degradation assays were performed only with the values at the end of the chase, because they reflect maximal differences between data sets.
RESULTS
A Yeast Model to Study TorsinA and TorsinAΔE Biogenesis
To express torsinA and torsinAΔE in S. cerevisiae, we subcloned the ORF of torsinA and torsinAΔE into the multicopy yeast expression vector pRS426 (73), placing torsinA under the control of the constitutive glyceraldehyde-3-phosphate dehydrogenase promoter (Fig. 1B). In this system, torsinA and torsinAΔE were expressed at similar levels and migrated by SDS-PAGE as an ∼33-kDa species, which corresponds to the monomeric forms of the proteins (Fig. 1C). Endoglycosidase H treatment of torsinA and torsinAΔE protein extracts produced a faster migrating band, indicating that both torsinA and torsinAΔE were N-glycosylated in yeast, as shown previously in mammalian cells (Fig. 1C) (65). Also, in accordance with results in mammalian cells, torsinA-HA and torsinAΔE-HA were localized in the ER/nuclear envelope compartments, as evidenced by co-localization with the ER chaperone, Kar2/BiP (Fig. 1D). Similar results were obtained using sucrose density gradients with untagged torsinA and torsinAΔE and through live cell fluorescence imaging of GFP-tagged forms of torsinA and torsinAΔE (data not shown).
TorsinA is a monotopic membrane protein (88) and behaves like a peripherally associated protein in mammalian cells (89). To determine whether this was also the case in yeast, we performed alkaline extractions using isolated ER-derived microsomal membranes from yeast expressing torsinA or torsinAΔE, following a previously published method for torsinA (89). All proteins remained associated with the microsomal pellet fraction at pH 6.8, as expected (Fig. 2A, lanes 1 and 2 and 9 and 10). At pH 9.5, both Pdi1, a soluble lumenal protein, and Kar2/BiP, a peripherally associated protein (90), were partially released into the supernatant. However, at pH 9.5, torsinA and torsinAΔE remained associated with the pellet (Fig. 2A, compare lanes 3 and 4 and 11 and 12). At pH 10.5, Pdi1 was almost completely released into the supernatant, whereas a considerable fraction of torsinA and torsinAΔE remained associated with the pellet fraction, similar to Kar2/BiP (Fig. 2A, compare lanes 5 and 6 and 13 and 14). At pH ∼11.5, Pdi1 was completely solubilized, while both torsinA and torsinAΔE were largely extracted, similar to Kar2/BiP (Figs. 2A, compare lanes 7 and 8 and 15 and 16, and 7C). The transmembrane protein Sec61 remained associated with the pellet at each pH, as expected. Treatment with 6 m urea efficiently released Pdi1, Kar2/BiP, and both torsinA variants but not Sec61, as expected (Fig. 2A, lanes 17–20). Overall, torsinA and torsinAΔE behaved more similarly to Kar2/BiP than to Pdi1, indicating that torsinA and torsinAΔE are peripherally membrane-associated proteins in yeast.
FIGURE 2.
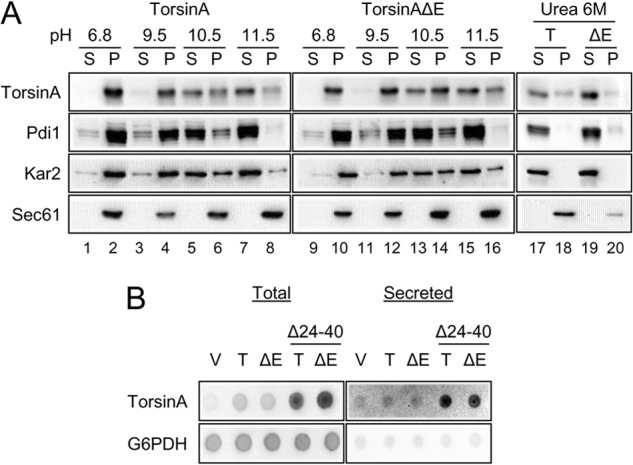
TorsinA and torsinAΔE peripherally associate with the yeast ER membrane through an N-terminal hydrophobic domain. A, ER-derived microsomes were prepared from log phase wild-type yeast cells grown at 28 °C that expressed torsinA or torsinAΔE and were incubated in a pH 6.8 buffer, in 25 mm CHES, pH 9.5 buffer, 25 mm CAPS, pH 10.5 buffer, 0.1 m Na2CO3, pH ∼11.5, or in 6 m urea for 30 min in ice. After centrifugation at 100,000 × g, aliquots from the supernatant (S) or the pellet (P) fractions were analyzed by SDS-PAGE, and the presence of torsinA and torsinAΔE was detected by Western blot. Pdi1 is a lumenal ER protein; Kar2 is a peripherally associated ER lumenal protein, and Sec61 is an integral membrane ER protein. B, nitrocellulose membranes spotted with overnight yeast cultures transformed with vector alone (V) or expressing torsinA (T) or torsinAΔE (ΔE), were layered on top of solid selective medium and incubated for ∼20 h at 30 °C. To detect secreted proteins, cells were washed off the nitrocellulose membrane with water. To detect total protein (intracellular and secreted material), cells were lysed in situ and then washed. TorsinA and G6PDH were detected by Western (“dot”) blot. Please note that secreted proteins yield a higher signal than their nonsecreted counterparts in the total protein blot (82).
FIGURE 7.

Kar2/BiP maintains torsinA and torsinAΔE membrane association and solubility. Microsomes prepared from log phase wild-type (KAR2) or kar2-1 yeast cells grown at 28 °C and that express torsinA (T) or torsinAΔE (ΔE) were incubated for 30 min in ice in pH 6.8 buffer or pH ∼11.5 carbonate buffer (A) or in a pH 6.8 buffer containing 1% Triton X-100 (B). After centrifugation at 100,000 × g, aliquots of proteins from the supernatant (S) or the pellet (P) fractions were separated by SDS-PAGE, and the presence of torsinA and torsinAΔE was detected by Western blot analysis and quantified. Significant p values for the carbonate extraction are as follows: *, p < 0.001 for KAR2 torsinA versus kar2-1 torsinA; p < 0.001 for KAR2 torsinAΔE versus kar2-1 torsinAΔE; ¥, p < 0.02 for kar2-1 torsinA versus kar2-1 torsinAΔE; and for Triton extraction are as follows: *, p < 0.008 KAR2 torsinA versus kar2-1 torsinA; p < 0.002 for KAR2 torsinAΔE versus kar2-1 torsinAΔE. C, graph shows the means ± S.E. of the fraction of torsinA or torsinAΔE remaining in the pellet from at least three independent experiments.
An N-terminal hydrophobic domain (HD; Fig. 1A) anchors torsinA to the lumenal side of the ER membrane (75, 89, 91). To test whether the HD was responsible for maintaining torsinA in the yeast ER, as anticipated, we examined whether torsinA, torsinAΔE, and versions lacking the HD (torsinA-Δ24–40 and torsinAΔE-Δ24–40) were secreted from cells by using a dot blot technique (82, 92). Cells expressing each torsinA variant or harboring an empty vector control were spotted onto nitrocellulose membranes. After ∼20 h of incubation on medium, we either lysed the cells in situ (to assess total protein) or removed the cells with vigorous washing (to examine secreted protein, which adhered to the membrane). We then performed an immunoblot for torsinA and the nonsecreted cytosolic protein G6PDH (Fig. 2B). As expected, we observed that torsinA-Δ24–40 and torsinAΔE-Δ24–40 were secreted, while torsinA, torsinAΔE, and G6PDH were not. Therefore, similar to mammalian cells, the N-terminal HD of torsinA prevents torsinA secretion to the extracellular medium, suggesting that this domain is properly folded and helps maintain torsinA and torsinAΔE in the yeast ER (75, 88).
Together, these data indicate that torsinA and torsinAΔE are N-linked glycosylated ER resident proteins that are peripherally associated with the ER membrane in yeast, as in mammalian cells. Because these basic cellular properties of torsinA are conserved in yeast, we pursued subsequent studies to define how torsinA and torsinAΔE impact cellular homeostasis and which cellular factors regulate their protein stability.
TorsinA can modulate the ER stress response, and torsinA overexpression leads to defects in ERAD and protein secretion (53, 56, 93–95). To test whether torsinA or torsinAΔE expression in yeast induces a stress response or leads to any overt phenotype, we compared the behavior of cells expressing torsinA, torsinAΔE, or harboring an empty vector control. Cells expressing torsinA or torsinAΔE showed no growth defect in the absence or presence of several ER stressors (including heat shock, tunicamycin, and DTT) compared with a strain transformed with an empty vector control (data not shown). Expression of torsinA or torsinAΔE in yeast also did not alter the heat shock response of wild-type cells incubated at 39 °C nor did it alter the unfolded protein response in wild-type cells incubated in the presence of tunicamycin (data not shown). These data are in-line with a recently published study in which the phenotypes of yeast expressing torsinA were analyzed (96). In addition, the degradation of the ERAD substrates CPY* and CFTR was unaltered by the co-expression of torsinA or torsinAΔE. Finally, no morphological changes were evident in the nuclear envelope of strains expressing torsinA or torsinAΔE, as determined by electron microscopy (data not shown) (53, 97, 98). These combined data indicate that torsinA is not toxic when expressed in yeast.
Stability of TorsinA and TorsinAΔE Depends on the ER Chaperone Kar2/BiP
Most of the current knowledge on torsinA is concentrated on the function and biochemical properties of torsinA and on the effect the ΔE mutation has on these properties (59, 99). However, nothing is known about whether molecular chaperones facilitate the folding, stabilization, and/or degradation of torsinA and torsinAΔE. Using yeast as a model system, we set out to identify which chaperones impact torsinA and/or torsinAΔE biogenesis.
A critical ER resident chaperone that plays a role in both protein folding and degradation is Kar2/BiP (17–19, 100–102). To test the role of Kar2/BiP on torsinA and torsinAΔE stability, we performed CHX chase experiments in a wild-type (KAR2) strain and in a Kar2/BiP mutant (kar2-1) strain. The kar2-1 allele carries a temperature-sensitive mutation that compromises ERAD and protein folding due to a defect in the controlled release of bound peptide substrates (101, 103–105). We first noted that there was no significant difference in the degradation of torsinA and torsinAΔE in wild-type cells (Fig. 3A). These results are in accordance with experiments in mammalian cells showing identical torsinA and torsinAΔE turnover when measured during a short time course (63). However, in the kar2-1 strain both torsinA and torsinAΔE were significantly destabilized (55% versus 81% for torsinA and 59% versus 84% for torsinAΔE remained after 90 min in the mutant strain versus the wild-type strain, respectively) (Fig. 3A). The data suggest that Kar2/BiP helps maintain the stability of torsinA and torsinAΔE in yeast.
FIGURE 3.

TorsinA and torsinAΔE stability in yeast depends on the ER chaperone Kar2/BiP. A, log phase yeast cells expressing torsinA or torsinAΔE grown at 28 °C were treated with cycloheximide and then incubated at 37 °C for 90 min. Samples were taken every 30 min, and the amount of torsinA or torsinAΔE in each time point was quantified relative to that at the 0-min time point. Data represent the average ± S.E. of at least four experiments with ≥2 independent replicates/experiment. The % of torsinA or torsinAΔE remaining after CHX addition in wild-type KAR2 or kar2-1 yeast strains is graphed (*, p < 0.04 for KAR2 torsinA versus kar2-1 torsinA; p < 0.003 for KAR2 torsinAΔE versus kar2-1 torsinAΔE). Representative Western blots are shown. B, torsinA and torsinAΔE N-linked glycosylation is Kar2-dependent. Protein samples were prepared from log phase KAR2 and kar2-1 yeast strains as before, except that they were treated with endoglycosidase H (EndoH) as indicated before Western blot analysis. Pdi1 is an N-glycosylated protein, whereas G6PDH is a cytosolic unglycosylated protein. * indicates a background band in the blot. C, torsinA and torsinAΔE glycosylated species are ER-embedded. Microsomes from KAR2 and kar2-1 strains expressing torsinA and torsinAΔE were incubated on ice for 30 min in the presence or absence of Triton X-100, followed by a 1-h incubation on ice in the presence or absence of proteinase K. Proteins were TCA-precipitated, resolved by SDS-PAGE, and analyzed by Western blot. Note that the ER protein Kar2 is clipped by proteinase K in the presence of Triton X-100. Bos1 is an ER transmembrane protein with an epitope exposed to the cytosol that is proteinase K-sensitive even in the absence of Triton X-100. UC, uncleaved Kar2/BiP; C, cleaved Kar2/BiP. D, Kar2/BiP co-immunoprecipitates with torsinA and torsinAΔE. Whole cell extracts from KAR2 cells transformed with a vector control (V) or expression vectors for torsinA (T) or torsinAΔE (ΔE) were immunoprecipitated (IP) under native conditions (0.5% Triton X-100) using anti-Kar2 antiserum. Immunoprecipitated material was resolved by SDS-PAGE and analyzed by Western blot. Controls from immunoprecipitations in which primary antiserum was absent (−) were also included.
We noticed that torsinA and torsinAΔE expressed in the kar2-1 strain migrated as three distinct bands, with the highest molecular weight species migrating at the position of torsinA expressed in the wild-type strain (Fig. 3, B and C). Because torsinA can be N-glycosylated at two asparagines, Asn-143 and Asn-158 (Fig. 1A, and see below) (65, 106), we hypothesized that the faster migrating bands corresponded to mono- and unglycosylated torsinA. Indeed, the mobility of the middle band coincided with the mobility of the torsinA-N143Q mutant, which can only be N-monoglycosylated at the Asn-158 residue (∼30 kDa) (Fig. 3B). Similar results were obtained for torsinAΔE (data not shown). This result indicates that, similar to mammalian cells, torsinA and torsinAΔE are N-glycosylated at both asparagines. Furthermore, the migration of the lowest molecular weight species coincided with the mobility of endoglycosidase H-treated samples of torsinA and torsinAΔE expressed in both the KAR2 and kar2-1 strains and with the mobility of a torsinA-N143Q,N158Q double mutant (∼27 kDa) (Fig. 3B). Therefore, a loss of Kar2/BiP function leads to inefficient N-linked glycosylation of torsinA and torsinAΔE. Importantly, the effect of the kar2-1 mutation on torsinA is specific because the N-linked glycosylation of other substrates, such as CPY*, is unaffected by this mutation (data not shown) (107).
Although the kar2-1 allele does not appear to impact protein translocation into the ER (101), it was formally possible that the mono- and unglycosylated torsinA species in the kar2-1 strain arise from defective translocation. To exclude this possibility, we performed a protease protection assay in the KAR2 and kar2-1 strains expressing torsinA and torsinAΔE. If indeed the mono- and unglycosylated torsinA species in the kar2-1 strain were caused by a translocation defect, they would be exposed on the surface of the microsomes and would be protease-accessible even in the absence of detergent. However, each of the torsinA and torsinAΔE species was stable after protease treatment (Fig. 3C, compare lanes 1 and 2 and 4 and 5, 7 and 8, and 10 and 11). In contrast, addition of 1% Triton X-100 initiated torsinA and torsinAΔE degradation (Fig. 3C, compare lanes 2 and 3, 5 and 6, 8 and 9, and 11 and 12). Kar2/BiP was also protected from protease in the absence of detergent but was clipped and migrated faster in the presence of detergent. The ER transmembrane protein Bos1, which exposes an epitope to the cytosol (108), was proteolyzed in the presence and absence of Triton X-100, as anticipated. These results indicate that the differentially N-glycosylated torsinA and torsinAΔE species are ER-encapsulated and support a role for Kar2/BiP in torsinA and torsinAΔE N-linked glycosylation and stability. Because a loss of protein stability is associated with misfolding (109), our data strongly suggest that Kar2/BiP functions as a torsinA chaperone.
If, as expected, Kar2/BiP is a torsinA chaperone, then both proteins should physically interact. To test this hypothesis, we obtained whole cell extracts from wild-type cells, immunoprecipitated Kar2/BiP under native conditions, and probed for torsinA and torsinAΔE by Western blot. Both torsinA and torsinAΔE co-precipitated with Kar2/BiP in the absence of cross-linking agents (Fig. 3D). Only a relatively small fraction of torsinA and torsinAΔE co-precipitated with Kar2/BiP, suggesting a transient interaction. Prolonged Hsp70-substrate interactions normally lead to substrate degradation (110, 111). However, transient interactions between chaperones and their substrates occur during substrate folding, as observed previously for Kar2/BiP (102, 112, 113). The combined data indicate that Kar2/BiP is a pro-folding chaperone for torsinA and torsinAΔE.
The ER Lumenal Hsp40 Scj1 and the Nucleotide Exchange Factor Lhs1 Also Maintain TorsinA and TorsinAΔE Stability
Kar2/BiP function in yeast is augmented by the action of co-chaperones, including the Hsp40s Jem1 and Scj1 and the NEFs Lhs1 and Sil1 (21, 114). Therefore, we compared torsinA and torsinAΔE stability by CHX chase analysis in a jem1Δ scj1Δ strain to the wild-type strain (JEM1 SCJ1) (Fig. 4A). Similar to the results observed in the kar2-1 strain (Fig. 3A), torsinA and torsinAΔE stability was significantly decreased in the jem1Δ scj1Δ strain compared with the wild-type strain (55% versus 84% torsinA, and 62% versus 80% torsinAΔE remained after 90 min in the mutant and wild-type strains, respectively) (Fig. 4A). To determine whether one of the two Hsp40s played a more important role in stabilizing torsinA and torsinAΔE, we performed chase analyses in the jem1Δ and scj1Δ single mutant strains. In agreement with previous reports showing a more prominent role of Scj1 than Jem1 during protein maturation (27, 29), torsinA and torsinAΔE stability was exclusively SCJ1-dependent (89% versus 48% torsinA and 83% versus 46% torsinAΔE remained after 90 min in the jem1Δ and scj1Δ strains, respectively) (Fig. 4B). Interestingly, we observed a glycosylation defect in the jem1Δ scj1Δ mutant but not in the jem1Δ or scj1Δ single mutants (Fig. 4, A and B). Therefore, although only Scj1 is required to stabilize torsinA and torsinAΔE, both Hsp40s support efficient torsinA and torsinAΔE glycosylation. These experiments suggest that torsinA instability is independent of the glycosylation defect caused by the absence of these chaperones.
FIGURE 4.

The ER lumenal Hsp40 Scj1 and the nucleotide exchange factor Lhs1 stabilize torsinA and torsinAΔE. CHX chases were performed as described before (see legend for Fig. 3). A, torsinA and torsinAΔE degradation in JEM1 SCJ1 and jem1Δ scj1Δ strains (*, p < 0.002 for JEM1 SCJ1 torsinA versus jem1Δ scj1Δ torsinA; p < 0.03 for JEM1 SCJ1 torsinAΔE versus jem1Δ scj1Δ torsinAΔE). B, torsinA and torsinAΔE degradation in jem1Δ and scj1Δ strains (*, p < 0.0005 for jem1Δ torsinA versus scj1Δ torsinA; p < 0.02 for jem1Δ torsinAΔE versus scj1Δ torsinAΔE). C, torsinA and torsinAΔE degradation in LHS1 and lhs1Δ strains (*, p < 0.04 for LHS1 torsinA versus lhs1Δ torsinA; p < 0.02 for LHS1 torsinAΔE versus lhs1Δ torsinAΔE). Representative Western blots are shown below. Experiments were performed at least twice, with≥2 independent replicates per experiment.
To test the role of the NEFs on torsinA and torsinAΔE stability, we performed CHX chases in the lhs1Δ and sil1Δ strains (Fig. 4C and data not shown). In accordance with a recent paper indicating a more prominent role for Lhs1 than Sil1 on ER protein biogenesis (115), the stability of torsinA and torsinAΔE was significantly decreased in the lhs1Δ strain compared with the wild-type strain (70% versus 91% torsinA and 73% versus 92% torsinAΔE remained after 90 min in the lhs1Δ and the wild-type strains, respectively) (Fig. 4C), whereas the sil1Δ mutation had no effect on torsinA or torsinAΔE stability (data not shown). In the absence of Lhs1, there was no effect on glycosylation (Fig. 4C and data not shown), which is consistent with the proposal, above, that the defect in glycosylation is uncoupled from torsinA stabilization. Together, these data strongly suggest that Kar2/BiP functions with a cognate Hsp40, Scj1, as well as a NEF, Lhs1, to facilitate the folding of torsinA and torsinAΔE in the ER.
Importantly, deletion of other ER chaperones had no impact on the stability or N-linked glycosylation of torsinA and torsinAΔE, indicating that these phenomena are not the result of a general ER stress response (Fig. 5 and data not shown). For example, deletion of the chaperone-like lectins Cne1 and Yos9 had no effect on these properties (Fig. 5). Preliminary data suggest that Mnl1 (the yeast EDEM1 homolog (116)) also had no effect on torsinA or torsinAΔE stability or glycosylation (data not shown). Mutations in other ER resident enzymes, such as select protein-disulfide isomerases, also showed no effect on torsinA or torsinAΔE (data not shown). Furthermore, similar to mammalian cells, the defects in N-linked glycosylation could also be induced by supplementing the growth medium with DTT (117), but this also occurred in an ire1Δ yeast mutant in which the unfolded protein response cannot be induced (data not shown) (118). These results strongly support our conclusion that compromised torsinA and torsinAΔE stability and folding are direct consequences of the lack of Kar2/BiP, Scj1, and Lhs1 activity.
FIGURE 5.

The lectin-like chaperones Cne1 and Yos9 do not affect torsinA and torsinAΔE stability. CHX chases were performed as described before (see legend for Fig. 3). A, torsinA and torsinAΔE degradation in CNE1 and cne1Δ strains. B, torsinA and torsinAΔE degradation in YOS9 and yos9Δ strains. Representative Western blots are shown below. Experiments were performed at least twice, with ≥2 independent replicates per experiment.
TorsinA and TorsinAΔE Are Degraded by the Proteasome and Vacuole When Kar2/BiP Function Is Disabled
In mammalian cells, torsinA and torsinAΔE are degraded in the lysosome via autophagy, but torsinAΔE is also degraded by ERAD (58, 62). To determine the role of the proteasome and the vacuole (the lysosome equivalent in yeast) during the turnover of torsinA and torsinAΔE when Kar2/BiP function is compromised in yeast, we deleted PDR5 or PEP4 in the kar2-1 background. The PDR5 deletion allows for efficient MG132-mediated inhibition of proteasome function (119, 120), and the PEP4 deletion inhibits nearly all vacuolar protease activity (121). We monitored torsinA and torsinAΔE degradation by CHX chase in the kar2-1 pdr5Δ strain treated with solvent (DMSO) or MG132 and found that both torsinA and torsinAΔE were partially but significantly stabilized by MG132 (73% versus 58% torsinA and 80% versus 61% torsinAΔE remained after 90 min in kar2-1 pdr5Δ cells treated with MG132 versus DMSO, respectively) (Fig. 6A).
FIGURE 6.

Loss of Kar2/BiP function causes torsinA and TorsinAΔE to be degraded by the proteasome and by the vacuole. CHX chases were performed as described before (see legend for Fig. 3). A, torsinA and torsinAΔE degradation in the kar2-1 pdr5Δ strain treated with DMSO or 100 μm MG132 for 30 min before addition of CHX (*, p < 0.02 for DMSO torsinA versus MG132 torsinA treatment; p < 0.02 for DMSO torsinAΔE versus MG132 torsinAΔE treatment). B, torsinA and torsinAΔE degradation in the kar2-1 pep4Δ strain. To facilitate the comparison, we included in the graph the data from the kar2-1 PEP4 strain from Fig. 3A (*, p < 0.002 for kar2-1 PEP4 torsinA versus kar2-1 pep4Δ torsinA; p < 0.02 for kar2-1 PEP4 torsinAΔE versus kar2-1 pep4Δ torsinAΔE). Experiments were performed at least twice, with ≥2 independent replicates per experiment. Representative Western blots are shown below.
To determine whether vacuolar proteases contribute to torsinA and torsinAΔE degradation, we performed a CHX chase in the kar2-1 pep4Δ strain (Fig. 6B) and found that torsinA was stabilized but torsinAΔE was only partially, yet still significantly stabilized (91% versus 55% torsinA and 76% versus 59% torsinAΔE remained after 90 min in the kar2-1 pep4Δ and the kar2-1 PEP4 strains, respectively) (Figs. 3A and 6B). Of note, torsinA degradation in the kar2-1 strain was significantly more dependent on PEP4 than torsinAΔE (p < 0.04), similar to what has been observed in mammalian cells (58, 62). No evidence of torsinA or torsinAΔE aggregates was observed during chases in the absence of proteasome or vacuolar function (data not shown).
Defect in Kar2/BiP Function Alters TorsinA and TorsinAΔE Membrane Extraction
The Kar2/BiP-dependent defects in torsinA and torsinAΔE N-linked glycosylation and the observed destabilization of the proteins when Kar2/BiP function was curtailed suggest that this chaperone facilitates the folding of torsinA and torsinAΔE. One method to monitor a change in the folding of peripheral, membrane-associated proteins is to determine the relative strength of membrane association. In other words, defects in folding might lead to greater insolubility, which would be evidenced by reduced membrane extraction in the presence of chaotropic agents (29, 101, 102, 122). To this end, we compared torsinA and torsinAΔE extraction from microsomes prepared from wild-type and kar2-1 strains after treatment with alkaline sodium carbonate buffer (pH ∼11.5) (Fig. 7A). As expected, torsinA and torsinAΔE associated with the pellet fraction after microsomes prepared from the kar2-1 strain were incubated at pH 6.8 (Fig. 7A, lanes 1, 2, 5, and 6). Although incubation with sodium carbonate released the majority of torsinA and torsinAΔE from the membrane in the wild-type strain (Figs. 2A and 7C), alkaline extraction was significantly less efficient in kar2-1 microsomes (64% versus 25% torsinA and 88% versus 31% torsinAΔE remained in the membrane fraction in the kar2-1 and wild-type strains, respectively; p < 0.001) (Fig. 7, A, lanes 3, 4, 7, and 8, and C). Interestingly, torsinAΔE associated more avidly with the membrane than torsinA in the kar2-1 strain (p < 0.02), suggesting that the presence of the ΔE mutation causes a subtle folding defect, which is exacerbated when Kar2/BiP is disabled.
Kar2/BiP helps maintain substrate solubility, and in its absence substrates such as pro-α-factor, CPY*, and CPY oligomerize or aggregate (29, 101, 102). To test if decreased Kar2/BiP function also triggers torsinA and torsinAΔE aggregation, we extracted torsinA and torsinAΔE from membranes collected from wild-type and kar2-1 cells using the nonionic detergent Triton X-100. In this experiment, torsinA and torsinAΔE were readily extractable by 1% Triton X-100 from microsomes prepared from wild-type cells (Fig. 7B, lanes 1–4). However, in microsomes prepared from the kar2-1 strain, there was a significant increase in the fraction of torsinA and torsinAΔE that remained associated with the pellet (51% versus 14% torsinA and 48% versus 18% torsinAΔE in the kar2-1 versus the wild-type strains, p < 0.008) (Fig. 7, B, lanes 5–8, and C). All of the differentially glycosylated species of torsinA and torsinAΔE in the kar2-1 strain were extracted to a similar degree both by detergent and alkali, indicating that the glycosylation state had no effect on torsinA and torsinAΔE solubility (Fig. 7). Together, these results suggest that compromised Kar2/BiP function leads to folding alterations that decrease torsinA and torsinAΔE solubility, which in turn initiates torsinA and torsinAΔE aggregation. The data also provide further support for our hypothesis that Kar2/BiP is a torsinA chaperone in yeast.
Mutations in TorsinA Functional Motifs Exhibit a Synergistic Interaction with the ΔE Mutation and Destabilize TorsinA
Mutation of functional domains in torsinA has provided valuable information on torsinA function and on the role of the ΔE allele on disease-associated phenotypes. For example, mutations in torsinA's C-terminal cysteines prevent a redox-dependent conformational change in torsinA that is critical for torsinA function (63). The ΔE mutation yields the same phenotype in torsinA as the cysteine mutant, suggesting that it also impacts local conformation (63). Furthermore, some intragenic mutations may mimic the effects of cellular or ER stresses in torsinAΔE-expressing cells, potentially replicating events that lead to the manifestation of the DYT1 mutation and EOTD development. Indeed, mutations in the N-linked glycosylation sites (Fig. 1A) can alter torsinAΔE subcellular localization and inclusion formation in mammalian cells (106). Thus, an examination of secondary mutations in the context of the ΔE allele in yeast may unveil previously undiscovered ΔE phenotypes.
In our yeast system and in some mammalian cell studies, no difference was observed in the stability of torsinA and torsinAΔE (Figs. 3–5) (63). However, torsinAΔE showed enhanced membrane association compared with torsinA in the kar2-1 strain (see above, Fig. 7). The data suggest that the lack of key chaperones can exacerbate subtle effects of the ΔE allele, which appear otherwise indistinguishable from the wild-type allele by our methods. We reasoned that torsinAΔE instability may also be magnified if the ΔE allele is combined with additional intragenic mutations. Therefore, we set out to explore the effects on torsinA and torsinAΔE stability when other mutations were engineered into distinct functional domains in torsinA.
TorsinA has two asparagines to which N-linked glycans are added (Asn-143 and Asn-158; Fig. 1A) (106, 123). To examine how the presence of the ΔE mutation affects torsinA stability in combination with mutations in the first (N143Q) or second (N158Q) N-linked glycosylation sites, we performed CHX chases in a wild-type strain (KAR2) transformed with the torsinA and torsinAΔE alleles carrying either of these secondary mutations (Fig. 8A). The N143Q mutation had no significant effect on the stability of torsinA or torsinAΔE (89% torsinA-N143Q versus 81% torsinA and 84% torsinAΔE-N143Q versus 84% torsinAΔE remained after 90 min) (Fig. 9B). The N158Q mutation also had no significant effect on torsinA stability (74% of torsinA-N158Q versus 81% torsinA remained after 90 min) (Figs. 8A and 9B). However, the N158Q mutation significantly decreased torsinAΔE levels (42% torsinAΔE-N158Q versus 84% torsinAΔE remained after 90 min, p < 0.0004) (Figs. 8A and 9B). These results are in accordance with data suggesting a more prominent role of glycosylation at position Asn-158 in torsinAΔE in mammalian cells (106) and indicate that glycosylation at position Asn-158 is critical to stabilize torsinAΔE but not torsinA.
FIGURE 8.

Synergism between mutations in functional motifs and the ΔE mutation and dual role of Kar2/BiP in torsinA and torsinAΔE degradation. A, defect in N-linked glycosylation triggers the Kar2/BiP-dependent degradation of torsinAΔE. CHX chases were performed as described before (see legend for Fig. 3). KAR2 (A and C) and kar2-1 (B and D) strains expressing torsinA or torsinAΔE containing (A and B) Asn-Gln mutations that prevent glycosylation at Asn-143 (N143Q) and Asn-158 (N158Q). (*, p < 0.0004 for torsinAΔE versus torsinAΔE-N158Q in the KAR2 strain); or mutations in the Walker-A motif (K108A) or Walker-B motif (E171Q) (C and D). (*, p < 0.0007 for torsinA versus torsinA-K108A in the KAR2 strain; p < 0.00001 for torsinAΔE versus torsinAΔE-K108A in the KAR2 strain; p < 0.003 for torsinAΔE versus torsinAΔE-E171Q in the KAR2 strain). Below are representative Western blots used for quantitation. Experiments were done at least twice with ≥2 independent replicates/experiment.
FIGURE 9.

The D216H mutation does not significantly affect torsinA or torsinAΔE stability. CHX chases were performed as described before (see legend for Fig. 3). A, torsinA-D216H and torsinAΔE-D216H degradation in KAR2 and kar2-1 strains. Representative Western blots are shown below. Experiments were performed at least three times, with ≥2 independent replicates per experiment. (*, p < 0.001 for torsinA-D216H in the KAR2 versus kar2-1 strains; p < 0.02 for torsinAΔE-D216H in the KAR2 versus kar2-1 strains.) B, comparative graph showing the effects of the mutations in Figs. 8 and 9 on torsinA and torsinAΔE levels at the 90 min time point of the CHX chases in the KAR2 and kar2-1 strains. Specific mutations in the N-glycosylation, Walker-A and B motifs, or D216H are indicated. *, other relevant p values not already described in Figs. 3 and 8 (p < 0.03 for torsinA-N143Q in the KAR2 versus kar2-1 strains; p < 0.003 for torsinAΔE-N158Q in the KAR2 versus the kar2-1 strains); p < 0.04 for torsinA-K108A in the KAR2 versus kar2-1 strains; p < 0.01 for torsinA-E171Q in the KAR2 versus kar2-1 strains; p < 0.0002 for torsinAΔE-K108A in the KAR2 versus kar2-1 strains; p < 0.03 for torsinAΔE-E171Q in the KAR2 versus kar2-1 strains.
The integrity of the Walker motifs is essential for the function of AAA+ ATPases (48, 124–126). Indeed, mutations in the Walker motifs that are predicted to disrupt ATP binding (K108A) or hydrolysis (E171Q) (Fig. 1A) (48, 63, 124, 125, 127) can alter torsinA and/or torsinAΔE subcellular localization, inclusion formation, and/or binding to interacting partners in mammalian cells (56, 60, 61, 63, 127–129). To test how the ΔE mutation affects torsinA stability in the presence of mutations in the Walker motifs, we performed CHX chase experiments in a wild-type strain (KAR2) transformed with torsinA and torsinAΔE alleles carrying either of these additional mutations (Fig. 8C). The K108A mutation led to a significant decrease in the stability of both torsinA and torsinAΔE (40% torsinA-K108A versus 81% torsinA and 20% torsinAΔE-K108A versus 84% torsinAΔE remained after 90 min, p < 0.0007) (Figs. 8C and 9B). On the contrary, although the E171Q mutation appeared to increase the stability of torsinA (94% torsinA-E171Q versus 81% torsinA remained after 90 min), it significantly destabilized torsinAΔE (55% torsinAΔE-E171Q versus 84% torsinAΔE remained after 90 min, p < 0.0007) (Figs. 8C and 9B). Therefore, mutations in the Walker-B motif led to opposite effects on torsinA stability, depending on the presence of the ΔE mutation. Importantly, both the K108A and E171Q mutations had a greater destabilizing effect on torsinAΔE than on torsinA (p < 0.03) (Fig. 9B). K108A-mediated destabilization in torsinA and torsinAΔE and the stabilizing effect of the E171Q mutation in torsinA are in agreement with results for other AAA+ ATPases (124, 126, 130). Overall, these experiments support our hypothesis that the ΔE mutation harbors a subtle folding defect and that this defect is amplified in the presence of additional mutations that affect torsinA conformation.
A naturally occurring polymorphism in ΔE carriers (Asp-216 to His-216) influences ΔE penetrance (Fig. 1A) (131, 132). The D216H mutation alters torsinAΔE inclusion formation in mammalian cells and counteracts torsinAΔE-associated ER stress in a Caenorhabditis elegans model (93, 94). To test if the D216H mutation influences torsinA and torsinAΔE stability, we performed CHX chases (Fig. 9A). We found that introduction of the D216H mutation did not change the stability or glycosylation status of torsinA or torsinAΔE in a wild-type strain (85% torsinA-D216H versus 81% torsinA and 78% torsinAΔE-D216H versus 84% torsinAΔE remained after 90 min) (Fig. 9). Decreased Kar2/BiP function significantly destabilized both torsinA-D216H and torsinAΔE-D216H (p < 0.02) but at similar levels as torsinA and torsinAΔE (66% torsinA-D216H versus 55% torsinA, and 60% torsinAΔE-D216H versus 59% torsinAΔE remained after 90 min in the kar2-1 strain) (Fig. 9). We observed that torsinA-D216H appeared somewhat more stable than torsinAΔE-D216H in the wild-type strain (Fig. 9), but this difference was not significant (p = 0.084). These results suggest that the synthetic effects of the D216H mutation observed in patients and C. elegans models are not caused by altered torsinA stability.
Kar2/BiP Plays a Dual Role during TorsinA and TorsinAΔE Biogenesis
Loss of Kar2/BiP led to the formation of incompletely glycosylated forms of torsinA and torsinAΔE and to a decrease in protein stability (Fig. 3). In our assays, all the differentially glycosylated species in the kar2-1 strain behaved similarly (Figs. 3 and 7). However, our results also indicate that robust glycosylation is required for torsinAΔE stability in a wild-type strain (Fig. 8A). These observations can be reconciled by previous results in yeast that demonstrate that chaperone requirements can vary depending on a protein's glycosylation state (133, 134). Thus, we hypothesized that although Kar2/BiP is required for stabilizing fully glycosylated torsinA and torsinAΔE (Fig. 3A), it is also required to degrade the mono- and unglycosylated torsinAΔE species. To test this model, we performed CHX chases of torsinA and torsinAΔE containing the N143Q or N158Q mutations in the kar2-1 strain (Figs. 8B and 9B). In agreement with our model, the lack of Kar2/BiP function significantly stabilized torsinAΔE-N158Q (69% torsinAΔE-N158Q versus 42% remained after 90 min in the kar2-1 or the wild-type strains, respectively, p < 0.002) (Figs. 8B and 9). We also found that torsinA-N143Q was significantly destabilized in the kar2-1 strain (75% versus 89% torsinA-N143Q remained after 90 min in the kar2-1 and the wild-type strains, respectively, p < 0.03) (Figs. 8B and 9B). This result suggests that a lack of N-linked glycans at N143 in torsinA does not impact the role of Kar2/BiP in maintaining torsinA stability. On the contrary, although there was a clear trend toward lower stability in the kar2-1 strain, neither torsinA-N158Q nor torsinAΔE-N143Q stability was significantly altered in the kar2-1 strain (65% versus 74% torsinA-N158Q and 76% versus 85% torsinAΔE-N143Q remained after 90 min in the kar2-1 or the wild-type strains, respectively). This result suggests that either lack of N-linked glycans at Asn-158 in torsinA and at Asn-143 in torsinAΔE abrogates Kar2/BiP dependence for stability or that these species are protected from degradation in the absence of Kar2/BiP (Figs. 8 and 9). Thus, combined with the data provided above (Figs. 3 and 8, A and C), Kar2/BiP can play either a pro-degradative or a pro-folding role depending on the glycosylation state of torsinA and torsinAΔE.
To determine whether Kar2/BiP's dual role was only associated with torsinA's glycosylation state or was more generally associated with protein folding, we measured the stability of torsinA and torsinAΔE Walker-A and Walker-B mutants in the kar2-1 strain (Fig. 8D). Consistent with the acquisition of an altered conformation, the degradation of torsinA-K108A, torsinAΔE-K108A, and torsinAΔE-E171Q was significantly attenuated in the kar2-1 strain (57% versus 40% torsinA-K108A, 66% versus 20% torsinAΔE-K108A, and 72% versus 55% torsinAΔE-E171Q remained after 90 min in the kar2-1 and wild-type strains, respectively, p < 0.04) (Figs. 8D and 9B). On the contrary, torsinA-E171Q was significantly destabilized in kar2-1 yeast compared with the wild-type strain (59% versus 94% torsinA-E171Q remained after 90 min in the kar2-1 and wild-type strains, respectively, p < 0.01) (Figs. 8D and 9B). Taken together, these results indicate that Kar2/BiP plays a dual role during torsinA degradation, which is linked to the folding or conformational states of torsinA.
BiP Also Promotes TorsinA and TorsinAΔE Stability in Mammalian Cells
To test whether the mammalian BiP homolog also contributes to torsinA and torsinAΔE biogenesis in mammalian cells, we first compared the levels of torsinA and torsinAΔE in HeLa cells in which BiP expression was silenced using siRNA (Fig. 10A). We designed a protocol using the BiP-specific siRNA 077 to obtain a reduction of BiP levels by ∼55% compared with a control siRNA, to prevent defects in cellular viability caused by greater BiP silencing (data not shown). These data are consistent with previous observations that BiP levels cannot be reduced to <40% of wild-type levels (2). Also consistent with previous data (135, 136), the depletion of BiP was accompanied by the induction of Grp94, which is another major ER chaperone (Fig. 10A). Under these conditions, the steady state levels of both torsinA and torsinAΔE were decreased by 70–75% compared with the negative control (Fig. 10). Similar results were observed using another BiP-targeted siRNA (data not shown). These results suggest that BiP affects the stability of both torsinA and torsinAΔE in mammalian cells.
FIGURE 10.
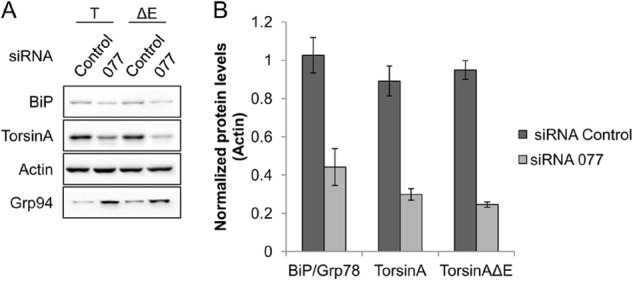
siRNA-mediated knockdown of BiP reduces the steady state levels of torsinA and torsinAΔE. HeLa cells were treated with siRNA against BiP for a total of 48 h and were transfected with torsinA (T) and torsinAΔE (ΔE) expression vectors 24 h before harvest. A, representative Western blot; B, graph showing the average ± S.E. of the normalized levels of BiP, torsinA, and torsinAΔE after treatment with a control siRNA or siRNA directed against BiP (077). Experiments were performed at least twice with three replicates per experiment. The graphed values were normalized as follows. The levels of BiP, torsinA, and torsinAΔE for each replicate in each experiment were normalized first to the corresponding actin levels. Then the actin-normalized value from one siRNA control replicate was used to normalize the values of all other replicates. This procedure was performed for each torsinA and torsinAΔE experiment independently. Actin was used as loading control. Grp94 levels were also monitored by Western blot.
To confirm these data, we implemented a second approach to deplete BiP. To this end, we performed a torsinA pulse-chase in HeLa cells transiently expressing torsinA or torsinAΔE in which BiP levels were acutely reduced by means of the AB5 subtilase cytotoxin, also known as the SubAB toxin (Fig. 11). SubAB is a bacterial serine protease that cleaves and inactivates BiP rapidly and specifically (87). Indeed, only 45 min after the toxin was added to the culture medium (t = “0” min of the chase), BiP levels dropped to ∼35% of the levels of BiP in cells treated with the catalytically inactive toxin, SubAA272B (Fig. 11A). Moreover, BiP could not be detected by Western blot in toxin-treated cells 1 h into the chase, whereas the catalytically inactive toxin had no effect on BiP (Fig. 11A). As anticipated, the reduction of BiP upon SubAB treatment also significantly destabilized torsinA (26% versus 44% of torsinA remained after a 4-h chase in cells treated with SubAB or SubAA272B, respectively, p < 0.02) (Fig. 11, A and B). Similar results were observed if only the fully glycosylated bands were quantified (data not shown). Although the effect was less pronounced, there was also a difference in torsinAΔE stability in the presence of active or inactive toxin (31% versus 39% torsinAΔE remained after a 4-h chase in cells treated with SubAB or SubAA272B, respectively) (Fig. 11, A and B). We also noticed that the levels of unglycosylated torsinA and torsinAΔE were higher at the beginning of the chase after toxin treatment compared with inactive toxin treatment or no treatment (72% versus 53% of unglycosylated torsinA and torsinAΔE in SubAB versus SubAA272B-treated cells or -untreated cells, respectively) (Fig. 11, A and C, data not shown and (63)). Because the unglycosylated species appears to mature into the fully glycosylated form of torsinA, the more rapid disappearance of torsinA is likely not due to the selective turnover of the unglycosylated protein (Fig. 11A). In fact, the unglycosylated form degraded similarly irrespective of whether the sample was treated with SubAB or SubAA272B (data not shown). Furthermore, because of the acute nature of the subtilase treatment, the effect on torsinA maturation and stability is unlikely to be caused by the up-regulation of other chaperones. In contrast to the knockdown experiments, GRP94 levels did not vary during the >4-h treatment with the toxin (Fig. 10 and data not shown). Taken together, the results using toxin-mediated BiP depletion are in accordance with the results using RNAi-mediated depletion of BiP in mammalian cells and with the experiments in the yeast genetic system, and establish BiP as a torsinA chaperone.
FIGURE 11.
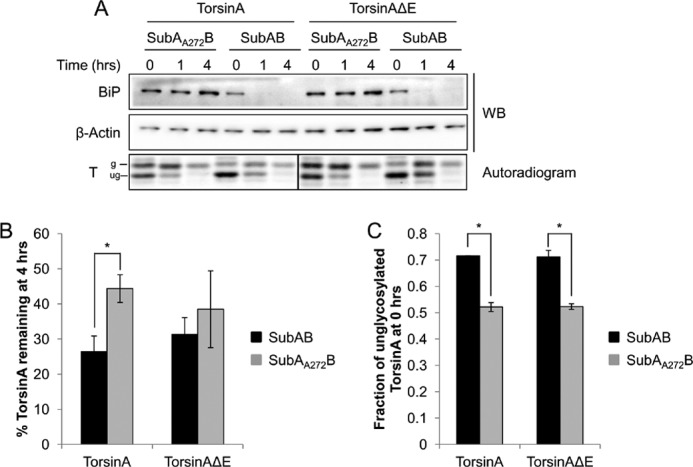
BiP is required for torsinA and torsinAΔE stability in mammalian cells. HeLa cells transiently transfected with torsinA-HA or torsinAΔE-HA expression vectors were treated with 0.5 μg/ml of an active SubAB or a catalytically inactive SubAA272B toxin for 45 min before a pulse-chase was performed (t = 0). A, BiP and torsinA levels were monitored at each time point by Western blot (WB) and autoradiography, respectively. B, graph showing the average ± S.E. of total torsinA or torsinAΔE remaining at the 4-h time point in the presence of SubAB or SubAA272B toxin quantified relative to that at the 0-h time point. (*, p < 0.02 for torsinA in cells treated with SubAB versus SubAA272B). C, graph showing the average ± S.E. of the relative levels of unglycosylated/total torsinA or torsinAΔE at the 0-h time point in the presence of SubAB or SubAA272B toxin (*, p < 0.002). Experiments were performed at least three times with at least one replicate/experiment.
DISCUSSION
EOTD, one of the most common forms of inherited primary dystonias, is associated with a dominant ΔE302/303 mutation in the gene DYT1, which encodes the ER/nuclear envelope localized AAA+ ATPase torsinA (43, 44). However, the variable phenotypic manifestation and low penetrance of the ΔE mutation (45) indicate that additional environmental or genetic factors must play a role in EOTD onset. One hypothesis is that ΔE impacts torsinA folding, and thus modifies torsinA's half-life, solubility, and/or interaction with chaperones, as evident in other genetic “conformational” diseases (34). Cellular chaperone levels are sensitive to genetic and environmental insults. Thus, altering chaperone levels could augment the phenotypes associated with the ΔE mutation. By understanding the chaperone microenvironment required for the folding, stabilization, and degradation of wild-type torsinA and of the disease-associated torsinAΔE variant, we hope to identify cellular factors that can be targeted with therapeutics. In this work, we report for the first time on ER chaperones required for torsinA and torsinAΔE biogenesis.
We discovered that Kar2/BiP is required to maintain the N-glycosylation and solubility of torsinA and torsinAΔE in a yeast model (Figs. 3 and 7) and that in the absence of Kar2/BiP, torsinA and torsinAΔE are destabilized and degraded (Figs. 3 and 6). In addition, we obtained preliminary evidence that the proteins are ubiquitinated (data not shown), which is in agreement with a recent study showing that the E3 ubiquitin ligase, FBG1, is involved in the degradation of torsinA and torsinAΔE (137). We also found that Kar2/BiP plays a pro-degradative role when the stability of torsinA and torsinAΔE is compromised (Figs. 8 and 9). Finally, we provide evidence that BiP supports torsinA biogenesis in human cells (Figs. 10 and 11). Therefore, to our knowledge, BiP is the first ER molecular chaperone that has been reported to facilitate the maturation of torsinA and torsinAΔE.
The ΔE mutation affects torsinA's redox-sensitive conformational changes, potentially by altering the local structure of an α-helix in the C-terminal subdomain (63, 94). Nevertheless, several studies have failed to uncover significant structural differences between torsinA and torsinAΔE, suggesting that the ΔE mutation confers a more subtle effect on torsinA's structure (91, 94). This idea is supported by our observations that the ΔE mutation increases torsinA membrane association in the absence of functional Kar2/BiP (Fig. 7) and that the ΔE mutation and mutations in several functional motifs exhibit a synergistic interaction that alters torsinA stability (Figs. 8 and 9). These data also suggest that the effect of the ΔE mutation could be amplified through secondary genetic or environmental insults, potentially mimicking the sequence of events that is required to manifest the dominant but poorly penetrant ΔE mutation (45, 91, 94). Indeed, the secondary mutations we incorporated might mimic alterations in the availability or function of chaperones, enzymes, or interacting partners, which in turn could play a role in EOTD onset and/or progression.
N-Linked glycosylation is a post-translational modification that is intimately involved in ER protein quality control (4, 138). Defects in N-glycosylation are associated with several human diseases (139–141). Interestingly, we found that defects in Kar2/BiP or its associated Hsp40s (Scj1 and Jem1) altered torsinA and torsinAΔE N-glycosylation, but this defect was uncoupled from destabilization (Figs. 3 and 4). How do these chaperones impact N-linked glycosylation? The kar2-1 (BiP mutant) allele has not been shown to affect the glycosylation of other secreted substrates; therefore, a general effect on the glycosylation machinery can be ruled out. The efficiency of N-linked glycosylation is not only determined by the amino acid sequence to which the modification is appended (the NX(S/T) tripeptide motif) but is also affected by secondary and tertiary structure, hydrophobic collapse, and disulfide bond formation (142). Notably, torsinA has two asparagines that can be glycosylated, Asn-143 and Asn-158 (Figs. 1 and 3) (65, 123). It is possible that the presence of the cysteine at position 162 (Fig. 1A) may affect glycosylation at the second acceptor site, N158VS, by altering the local conformation through the formation of an intramolecular disulfide bond (143) or by forming a covalent bond with the oligosaccharyltransferase during translocation (144). Any effect on glycosylation may be potentiated if the polypeptide is improperly folded due to compromised chaperone function, e.g. when torsinA is expressed in the kar2-1 yeast. This hypothesis is supported by the observations that treatment with the reducing agent, DTT, or deletion of the N-terminal hydrophobic domain in torsinA (Fig. 1A) leads to a similar glycosylation defect observed in the kar2-1 strain.5 Furthermore, torsinA interacts with the chaperone and lectin calnexin in mammalian cells (60). S. cerevisiae lacks a true calnexin (145). Calnexin and calreticulin have been shown to compete with BiP for binding to specific glycoproteins (112), but calnexin also binds nonglycosylated substrates (146). This may explain why compromised Kar2/BiP function in yeast leads to a co-translational folding defect (evidenced by the appearance of different glycosylated species) that is not observed in mammalian cells (Figs. 3 and 9). Overall, our results indicate that torsinA and torsinAΔE glycosylation is sensitive to alterations in folding caused not only by the absence of specific chaperones but also by environmental insults and by intragenic mutations.
Protein aggregation can lead to profound effects on cell viability (147). A critical role played by molecular chaperones, including Kar2/BiP, is to maintain the solubility of nascent polypeptides until their final conformations are achieved (29, 148). The membrane extraction of torsinA and torsinAΔE with carbonate or a nonionic detergent was severely impaired in the absence of Kar2/BiP (Fig. 7). In principle, this phenotype could be due to a translocation defect, but translocation of the proteins into the ER was unaffected in the kar2-1 strain (Fig. 3C). More likely, enhanced membrane association arose from conformational changes that expose hydrophobic regions, which would enhance membrane association or which could lead to aggregation (122). Alternatively, torsinA and torsinAΔE might associate with other proteins that aggregate in the kar2-1 strain (29). This model would be consistent with the proposed chaperone function of torsinA (53–55, 149), although no effect of torsinA or torsinAΔE on the ERAD of select substrates was observed in yeast (data not shown), and only terminally misfolded ERAD substrates have been reported to aggregate in the kar2-1 yeast (29).
The introduction of mutations in torsinA and torsinAΔE generally led to a more profound loss of stability in torsinAΔE than in torsinA (Figs. 8 and 9). Lack of N-linked glycosylation at Asn-158 specifically affects torsinAΔE, both in mammalian cells and in our yeast model (Fig. 8) (106). Defects in glycosylation can impact local folding, alter the interaction with lectins and chaperones, and reduce protein solubility (138). Thus, it is possible that the glycan at Asn-158 is required for the interaction of torsinA and torsinAΔE with chaperones or lectins, which torsinAΔE may require for folding. This effect, in turn, would lead to increased degradation of torsinAΔE but not torsinA. Alternatively, altered folding caused by the lack of a glycan at Asn-158 may impact the ability of torsinAΔE to form hexamers, which is the typical oligomeric state assumed by AAA+ ATPases, including torsinA (48, 61, 150). These unassembled torsinAΔE monomers may be targeted for degradation via the ERAD pathway (10, 151). This model may also explain why mutating the Walker-A motif triggers torsinA and torsinAΔE degradation, whereas mutating the Walker-B motif stabilizes torsinA (Figs. 8 and 9). The K108A mutation in the Walker-A motif of AAA+ ATPases not only prevents ATP binding, it also impairs substrate interaction and hexamer formation (48, 152). Conversely, the E171Q mutation in the Walker-B motif locks the protein in an ATP- and substrate-bound conformation and promotes hexamer stabilization (48, 152). In the case of torsinAΔE, the C-terminal conformational defect and the lack of binding to interacting partners required for ATPase activity (63, 127) may prevent efficient hexamerization, a phenotype that could be aggravated by the E171Q mutation. This phenomenon would explain why the E171Q mutation destabilizes torsinAΔE (Fig. 9B).
Besides protecting nascent polypeptides and aiding in their folding, chaperones can also identify and target terminally misfolded intermediates for degradation (4, 9). Thus, depending on the substrate, chaperones can have a pro-folding and/or a pro-degradative role (109). For example, mammalian Hsp/c70 is a key pro-folding factor for CFTR that can also act as a pro-degradative chaperone, targeting CFTR for ERAD (153). The loss of stability of torsinA and torsinAΔE containing additional mutations in functional motifs also uncovered a dual role of Kar2/BiP on the folding and stabilization of these proteins (Figs. 8 and 9). Kar2/BiP normally aids in folding torsinA and torsinAΔE (Figs. 3 and 8–11) but is instead a pro-degradative factor when the stability and folding of torsinA and torsinAΔE are impaired by mutations in specific functional motifs (Figs. 8 and 9). This dual role of Kar2/BiP is not unprecedented. Kar2/BiP functions as a pro-folding chaperone for the vacuolar protease CPY but as a pro-degradative factor of the ERAD substrate CPY* (100, 102). Moreover, BiP aids in the folding and assembly of monomers into oligomers, while simultaneously contributing to the degradation of the unassembled, potentially misfolded monomeric subunits (154–157). Thus, Kar2/BiP may help torsinA and torsinAΔE to fold (Fig. 3) and hexamerize, while also facilitating the degradation of the torsinA and torsinAΔE monomers that fail to assemble into hexamers due to, for instance, the presence of the additional mutations mentioned above (Figs. 8 and 9). Alternatively, Kar2/BiP may simply recognize misfolded features of the mutated torsinA and torsinAΔE and aid in their degradation. These results illustrate the complex relationship between chaperones and their substrate clients, and underscore the delicate equilibrium between folding and degradation that ultimately dictates the fate of folding intermediates.
Because BiP is associated with the development of diseases as diverse as cancer and Parkinson disease (20, 31–33, 158, 159), there is growing interest in developing small molecule modulators of BiP. Chemical modulators of BiP activity have been developed and tested in vitro (87, 160), and small molecules that enhance the activity of cytosolic Hsp70 exist (161), which may be adapted in the future to act on BiP. Alternatively, the expression levels of BiP can be modified, a procedure that has shown therapeutic potential both in vitro and in mouse models (162–166). Because our results demonstrate that BiP stabilizes torsinA and torsinAΔE, and because patients afflicted with EOTD are heterozygous for the ΔE mutation, the design of effective therapies may need to combine the modulation of BiP activity/expression levels with the modulation of secondary targets that selectively affect torsinAΔE folding such as the glycosylation machinery (Fig. 8). A deeper understanding of the molecular and environmental factors that alter the biochemical properties of torsinA and of the factors that participate in torsinA folding and degradation will provide new clues to define the etiology of EOTD and may identify new therapeutic targets to prevent or treat the disease.
Acknowledgments
We are indebted to Laura Diaz-Martinez, Tom Harper, Nicole Kotchey, Ali Vetter, Allyson O'Donnell, and all the members of the Brodsky, Zolkiewski, and Thomas labs for their technical assistance and/or their insightful comments and suggestions. We are also grateful to Pedro Gonzalez-Alegre for reagents.
This work was supported, in whole or in part, by National Institutes of Health Grant GM75061 (to J. L. B.).
L. F. Zacchi and J. L. Brodsky, manuscript in preparation.
- ER
- endoplasmic reticulum
- EOTD
- early-onset torsion dystonia
- ERAD
- ER-associated degradation
- NEF
- nucleotide exchange factor
- HD
- hydrophobic domain
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid
- CHX
- cycloheximide
- G6PDH
- glucose-6-phosphate dehydrogenase
- CPY*
- mutant version of vacuolar carboxypeptidase Y
- CFTR
- cystic fibrosis transmembrane conductance regulator.
REFERENCES
- 1. Kister A. E., Potapov V. (2013) Amino acid distribution rules predict protein fold. Biochem. Soc. Trans. 41, 616–619 [DOI] [PubMed] [Google Scholar]
- 2. Gidalevitz T., Stevens F., Argon Y. (2013) Orchestration of secretory protein folding by ER chaperones. Biochim. Biophys. Acta 1833, 2410–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vendruscolo M. (2012) Proteome folding and aggregation. Curr. Opin. Struct. Biol. 22, 138–143 [DOI] [PubMed] [Google Scholar]
- 4. Braakman I., Bulleid N. J. (2011) Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 80, 71–99 [DOI] [PubMed] [Google Scholar]
- 5. Priya S., Sharma S. K., Goloubinoff P. (2013) Molecular chaperones as enzymes that catalytically unfold misfolded polypeptides. FEBS Lett. 587, 1981–1987 [DOI] [PubMed] [Google Scholar]
- 6. Mizushima N., Levine B., Cuervo A. M., Klionsky D. J. (2008) Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Höhfeld J., Cyr D. M., Patterson C. (2001) From the cradle to the grave: molecular chaperones that may choose between folding and degradation. EMBO Rep. 2, 885–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kriegenburg F., Ellgaard L., Hartmann-Petersen R. (2012) Molecular chaperones in targeting misfolded proteins for ubiquitin-dependent degradation. FEBS J. 279, 532–542 [DOI] [PubMed] [Google Scholar]
- 9. Benyair R., Ron E., Lederkremer G. Z. (2011) Protein quality control, retention, and degradation at the endoplasmic reticulum. Int. Rev. Cell Mol. Biol. 292, 197–280 [DOI] [PubMed] [Google Scholar]
- 10. Vembar S. S., Brodsky J. L. (2008) One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thibault G., Ng D. T. (2012) The endoplasmic reticulum-associated degradation pathways of budding yeast. Cold Spring Harbor Perspect. Biol. 10.1101/cshperspect.a013193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Park S. H., Kukushkin Y., Gupta R., Chen T., Konagai A., Hipp M. S., Hayer-Hartl M., Hartl F. U. (2013) PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell 154, 134–145 [DOI] [PubMed] [Google Scholar]
- 13. Powers E. T., Balch W. E. (2013) Diversity in the origins of proteostasis networks–a driver for protein function in evolution. Nat. Rev. Mol. Cell Biol. 14, 237–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hartl F. U., Bracher A., Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 [DOI] [PubMed] [Google Scholar]
- 15. Walter P., Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 16. Stolz A., Wolf D. H. (2010) Endoplasmic reticulum associated protein degradation: a chaperone assisted journey to hell. Biochim. Biophys. Acta 1803, 694–705 [DOI] [PubMed] [Google Scholar]
- 17. Rose M. D., Misra L. M., Vogel J. P. (1989) KAR2, a karyogamy gene, is the yeast homolog of the mammalian BiP/GRP78 gene. Cell 57, 1211–1221 [DOI] [PubMed] [Google Scholar]
- 18. Normington K., Kohno K., Kozutsumi Y., Gething M. J., Sambrook J. (1989) S. cerevisiae encodes an essential protein homologous in sequence and function to mammalian BiP. Cell 57, 1223–1236 [DOI] [PubMed] [Google Scholar]
- 19. Bole D. G., Hendershot L. M., Kearney J. F. (1986) Posttranslational association of immunoglobulin heavy chain binding protein with nascent heavy chains in nonsecreting and secreting hybridomas. J. Cell Biol. 102, 1558–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dudek J., Benedix J., Cappel S., Greiner M., Jalal C., Müller L., Zimmermann R. (2009) Functions and pathologies of BiP and its interaction partners. Cell. Mol. Life Sci. 66, 1556–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Otero J. H., Lizák B., Hendershot L. M. (2010) Life and death of a BiP substrate. Semin. Cell Dev. Biol. 21, 472–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Steel G. J., Fullerton D. M., Tyson J. R., Stirling C. J. (2004) Coordinated activation of Hsp70 chaperones. Science 303, 98–101 [DOI] [PubMed] [Google Scholar]
- 23. Chung K. T., Shen Y., Hendershot L. M. (2002) BAP, a mammalian BiP-associated protein, is a nucleotide exchange factor that regulates the ATPase activity of BiP. J. Biol. Chem. 277, 47557–47563 [DOI] [PubMed] [Google Scholar]
- 24. Bies C., Blum R., Dudek J., Nastainczyk W., Oberhauser S., Jung M., Zimmermann R. (2004) Characterization of pancreatic ERj3p, a homolog of yeast DnaJ-like protein Scj1p. Biol. Chem. 385, 389–395 [DOI] [PubMed] [Google Scholar]
- 25. Weitzmann A., Volkmer J., Zimmermann R. (2006) The nucleotide exchange factor activity of Grp170 may explain the non-lethal phenotype of loss of Sil1 function in man and mouse. FEBS Lett. 580, 5237–5240 [DOI] [PubMed] [Google Scholar]
- 26. Shen Y., Meunier L., Hendershot L. M. (2002) Identification and characterization of a novel endoplasmic reticulum (ER) DnaJ homologue, which stimulates ATPase activity of BiP in vitro and is induced by ER stress. J. Biol. Chem. 277, 15947–15956 [DOI] [PubMed] [Google Scholar]
- 27. Silberstein S., Schlenstedt G., Silver P. A., Gilmore R. (1998) A role for the DnaJ homologue Scj1p in protein folding in the yeast endoplasmic reticulum. J. Cell Biol. 143, 921–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nishikawa S., Endo T. (1997) The yeast JEM1p is a DnaJ-like protein of the endoplasmic reticulum membrane required for nuclear fusion. J. Biol. Chem. 272, 12889–12892 [DOI] [PubMed] [Google Scholar]
- 29. Nishikawa S. I., Fewell S. W., Kato Y., Brodsky J. L., Endo T. (2001) Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J. Cell Biol. 153, 1061–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tyson J. R., Stirling C. J. (2000) LHS1 and SIL1 provide a lumenal function that is essential for protein translocation into the endoplasmic reticulum. EMBO J. 19, 6440–6452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gorbatyuk M. S., Gorbatyuk O. S. (2013) The molecular chaperone GRP78/BiP as a therapeutic target for neurodegenerative disorders: A mini review. J. Genet. Syndr. Gene Ther. 4, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tiffany-Castiglioni E., Qian Y. (2012) ER chaperone-metal interactions: links to protein folding disorders. Neurotoxicology 33, 545–557 [DOI] [PubMed] [Google Scholar]
- 33. Luo B., Lee A. S. (2013) The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 32, 805–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guerriero C. J., Brodsky J. L. (2012) The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 92, 537–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gestwicki J. E., Garza D. (2012) Protein quality control in neurodegenerative disease. Prog. Mol. Biol. Transl. Sci. 107, 327–353 [DOI] [PubMed] [Google Scholar]
- 36. Costanzo M., Zurzolo C. (2013) The cell biology of prion-like spread of protein aggregates: mechanisms and implication in neurodegeneration. Biochem. J. 452, 1–17 [DOI] [PubMed] [Google Scholar]
- 37. Cook C., Stetler C., Petrucelli L. (2012) Disruption of protein quality control in Parkinson's disease. Cold Spring Harbor Perspect. Med. 2, a009423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Morawe T., Hiebel C., Kern A., Behl C. (2012) Protein homeostasis, aging and Alzheimer's disease. Mol. Neurobiol. 46, 41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nassif M., Matus S., Castillo K., Hetz C. (2010) Amyotrophic lateral sclerosis pathogenesis: a journey through the secretory pathway. Antioxid. Redox Signal. 13, 1955–1989 [DOI] [PubMed] [Google Scholar]
- 40. Douglas P. M., Dillin A. (2010) Protein homeostasis and aging in neurodegeneration. J. Cell Biol. 190, 719–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bragg D. C., Armata I. A., Nery F. C., Breakefield X. O., Sharma N. (2011) Molecular pathways in dystonia. Neurobiol. Dis. 42, 136–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Blandy M. A. (1920) Short notes and clinical cases: A case of torsion-dystonia, or torsion-spasm. J. Neurol Psychopathol. 1, 148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ozelius L. J., Hewett J. W., Page C. E., Bressman S. B., Kramer P. L., Shalish C., de Leon D., Brin M. F., Raymond D., Corey D. P., Fahn S., Risch N. J., Buckler A. J., Gusella J. F., Breakefield X. O. (1997) The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat. Genet. 17, 40–48 [DOI] [PubMed] [Google Scholar]
- 44. Kramer P. L., Heiman G. A., Gasser T., Ozelius L. J., de Leon D., Brin M. F., Burke R. E., Hewett J., Hunt A. L., Moskowitz C. (1994) The DYT1 gene on 9q34 is responsible for most cases of early limb-onset idiopathic torsion dystonia in non-Jews. Am. J. Hum. Genet. 55, 468–475 [PMC free article] [PubMed] [Google Scholar]
- 45. Brüggemann N., Klein C. (2010) Genetics of primary torsion dystonia. Curr. Neurol. Neurosci. Rep. 10, 199–206 [DOI] [PubMed] [Google Scholar]
- 46. McNaught K. S., Kapustin A., Jackson T., Jengelley T. A., Jnobaptiste R., Shashidharan P., Perl D. P., Pasik P., Olanow C. W. (2004) Brainstem pathology in DYT1 primary torsion dystonia. Ann. Neurol. 56, 540–547 [DOI] [PubMed] [Google Scholar]
- 47. Rostasy K., Augood S. J., Hewett J. W., Leung J. C., Sasaki H., Ozelius L. J., Ramesh V., Standaert D. G., Breakefield X. O., Hedreen J. C. (2003) TorsinA protein and neuropathology in early onset generalized dystonia with GAG deletion. Neurobiol. Dis. 12, 11–24 [DOI] [PubMed] [Google Scholar]
- 48. Hanson P. I., Whiteheart S. W. (2005) AAA+ proteins: have engine, will work. Nat. Rev. Mol. Cell Biol. 6, 519–529 [DOI] [PubMed] [Google Scholar]
- 49. Weibezahn J., Bukau B., Mogk A. (2004) Unscrambling an egg: protein disaggregation by AAA+ proteins. Microb. Cell Fact 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wolf D. H., Stolz A. (2012) The Cdc48 machine in endoplasmic reticulum associated protein degradation. Biochim. Biophys. Acta 1823, 117–124 [DOI] [PubMed] [Google Scholar]
- 51. Janska H., Kwasniak M., Szczepanowska J. (2013) Protein quality control in organelles–AAA/FtsH story. Biochim. Biophys. Acta 1833, 381–387 [DOI] [PubMed] [Google Scholar]
- 52. Hewett J., Ziefer P., Bergeron D., Naismith T., Boston H., Slater D., Wilbur J., Schuback D., Kamm C., Smith N., Camp S., Ozelius L. J., Ramesh V., Hanson P. I., Breakefield X. O. (2003) TorsinA in PC12 cells: localization in the endoplasmic reticulum and response to stress. J. Neurosci. Res. 72, 158–168 [DOI] [PubMed] [Google Scholar]
- 53. Nery F. C., Armata I. A., Farley J. E., Cho J. A., Yaqub U., Chen P., da Hora C. C., Wang Q., Tagaya M., Klein C., Tannous B., Caldwell K. A., Caldwell G. A., Lencer W. I., Ye Y., Breakefield X. O. (2011) TorsinA participates in endoplasmic reticulum-associated degradation. Nat. Commun. 2, 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McLean P. J., Kawamata H., Shariff S., Hewett J., Sharma N., Ueda K., Breakefield X. O., Hyman B. T. (2002) TorsinA and heat shock proteins act as molecular chaperones: suppression of α-synuclein aggregation. J. Neurochem. 83, 846–854 [DOI] [PubMed] [Google Scholar]
- 55. Caldwell G. A., Cao S., Sexton E. G., Gelwix C. C., Bevel J. P., Caldwell K. A. (2003) Suppression of polyglutamine-induced protein aggregation in Caenorhabditis elegans by torsin proteins. Hum. Mol. Genet. 12, 307–319 [DOI] [PubMed] [Google Scholar]
- 56. Torres G. E., Sweeney A. L., Beaulieu J.-M., Shashidharan P., Caron M. G. (2004) Effect of torsinA on membrane proteins reveals a loss of function and a dominant-negative phenotype of the dystonia-associated ΔE-torsinA mutant. Proc. Natl. Acad. Sci. U.S.A. 101, 15650–15655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Goodchild R. E., Dauer W. T. (2004) Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc. Natl. Acad. Sci. U.S.A. 101, 847–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gordon K. L., Gonzalez-Alegre P. (2008) Consequences of the DYT1 mutation on torsinA oligomerization and degradation. Neuroscience 157, 588–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Granata A., Warner T. T. (2010) The role of torsinA in dystonia. Eur. J. Neurol. 17, Suppl. 1, 81–87 [DOI] [PubMed] [Google Scholar]
- 60. Naismith T. V., Dalal S., Hanson P. I. (2009) Interaction of torsinA with its major binding partners is impaired by the dystonia-associated ΔGAG deletion. J. Biol. Chem. 284, 27866–27874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vander Heyden A. B., Naismith T. V., Snapp E. L., Hodzic D., Hanson P. I. (2009) LULL1 retargets TorsinA to the nuclear envelope revealing an activity that is impaired by the DYT1 dystonia mutation. Mol. Biol. Cell 20, 2661–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Giles L. M., Chen J., Li L., Chin L.-S. (2008) Dystonia-associated mutations cause premature degradation of torsinA protein and cell-type-specific mislocalization to the nuclear envelope. Hum. Mol. Genet. 17, 2712–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhu L., Millen L., Mendoza J. L., Thomas P. J. (2010) A unique redox-sensing sensor II motif in TorsinA plays a critical role in nucleotide and partner binding. J. Biol. Chem. 285, 37271–37280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Granata A., Watson R., Collinson L. M., Schiavo G., Warner T. T. (2008) The dystonia-associated protein torsinA modulates synaptic vesicle recycling. J. Biol. Chem. 283, 7568–7579 [DOI] [PubMed] [Google Scholar]
- 65. Hewett J., Gonzalez-Agosti C., Slater D., Ziefer P., Li S., Bergeron D., Jacoby D. J., Ozelius L. J., Ramesh V., Breakefield X. O. (2000) Mutant torsinA, responsible for early-onset torsion dystonia, forms membrane inclusions in cultured neural cells. Hum. Mol. Genet. 9, 1403–1413 [DOI] [PubMed] [Google Scholar]
- 66. Hewett J. W., Zeng J., Niland B. P., Bragg D. C., Breakefield X. O. (2006) Dystonia-causing mutant torsinA inhibits cell adhesion and neurite extension through interference with cytoskeletal dynamics. Neurobiol. Dis. 22, 98–111 [DOI] [PubMed] [Google Scholar]
- 67. Konakova M., Pulst S. M. (2005) Dystonia-associated forms of torsinA are deficient in ATPase activity. J. Mol. Neurosci. 25, 105–117 [DOI] [PubMed] [Google Scholar]
- 68. Pham P., Frei K. P., Woo W., Truong D. D. (2006) Molecular defects of the dystonia-causing torsinA mutation. Neuroreport 17, 1725–1728 [DOI] [PubMed] [Google Scholar]
- 69. Bharadwaj P., Martins R., Macreadie I. (2010) Yeast as a model for studying Alzheimer's disease. FEMS Yeast Res. 10, 961–969 [DOI] [PubMed] [Google Scholar]
- 70. Gelling C. L., Brodsky J. L. (2010) Mechanisms underlying the cellular clearance of antitrypsin Z: lessons from yeast expression systems. Proc. Am. Thorac. Soc. 7, 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tenreiro S., Outeiro T. F. (2010) Simple is good: yeast models of neurodegeneration. FEMS Yeast Res. 10, 970–979 [DOI] [PubMed] [Google Scholar]
- 72. Miller-Fleming L., Giorgini F., Outeiro T. F. (2008) Yeast as a model for studying human neurodegenerative disorders. Biotechnol. J. 3, 325–338 [DOI] [PubMed] [Google Scholar]
- 73. Mumberg D., Müller R., Funk M. (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122 [DOI] [PubMed] [Google Scholar]
- 74. Gerami-Nejad M., Zacchi L. F., McClellan M., Matter K., Berman J. (2013) Shuttle vectors for facile gap repair cloning and integration into a neutral locus in Candida albicans. Microbiology 159, 565–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Liu Z., Zolkiewska A., Zolkiewski M. (2003) Characterization of human torsinA and its dystonia-associated mutant form. Biochem. J. 374, 117–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Seguí-Real B., Martinez M., Sandoval I. V. (1995) Yeast aminopeptidase I is post-translationally sorted from the cytosol to the vacuole by a mechanism mediated by its bipartite N-terminal extension. EMBO J. 14, 5476–5484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Taxis C., Hitt R., Park S. H., Deak P. M., Kostova Z., Wolf D. H. (2003) Use of modular substrates demonstrates mechanistic diversity and reveals differences in chaperone requirement of ERAD. J. Biol. Chem. 278, 35903–35913 [DOI] [PubMed] [Google Scholar]
- 78. Finger A., Knop M., Wolf D. H. (1993) Analysis of two mutated vacuolar proteins reveals a degradation pathway in the endoplasmic reticulum or a related compartment of yeast. Eur. J. Biochem. 218, 565–574 [DOI] [PubMed] [Google Scholar]
- 79. Becker D. M., Lundblad V. (2008) Yeast-manipulation of yeast genes. Curr. Protoc. Mol. Biol. Chapter 13, Unit 13.7 [Google Scholar]
- 80. Hoffman C. S. (2008) Yeast-preparation of yeast DNA, RNA, and proteins. Curr. Protoc. Mol. Biol. Chapter 13 Unit 13.11 [DOI] [PubMed] [Google Scholar]
- 81. Adams A., Gotschling D., Kaiser C., Stearns T. (1997) Methods in Yeast Genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 82. Gelling C. L., Dawes I. W., Perlmutter D. H., Fisher E. A., Brodsky J. L. (2012) The endosomal protein-sorting receptor sortilin has a role in trafficking α-1 antitrypsin. Genetics 192, 889–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhang Y., Michaelis S., Brodsky J. L. (2002) CFTR expression and ER-associated degradation in yeast. Methods Mol. Med. 70, 257–265 [DOI] [PubMed] [Google Scholar]
- 84. Brodsky J. L., Schekman R. (1993) A Sec63p-BiP complex from yeast is required for protein translocation in a reconstituted proteoliposome. J. Cell Biol. 123, 1355–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Stirling C. J., Rothblatt J., Hosobuchi M., Deshaies R., Schekman R. (1992) Protein translocation mutants defective in the insertion of integral membrane proteins into the endoplasmic reticulum. Mol. Biol. Cell 3, 129–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Amberg D. C., Burke D., Strathern J. N., and Cold Spring Harbor Laboratory (2005) Yeast immunofluorescence. in Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual, pp. 149–152, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 87. Paton A. W., Beddoe T., Thorpe C. M., Whisstock J. C., Wilce M. C., Rossjohn J., Talbot U. M., Paton J. C. (2006) AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature 443, 548–552 [DOI] [PubMed] [Google Scholar]
- 88. Vander Heyden A. B., Naismith T. V., Snapp E. L., Hanson P. I. (2011) Static retention of the lumenal monotopic membrane protein torsinA in the endoplasmic reticulum. EMBO J. 30, 3217–3231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Callan A. C., Bunning S., Jones O. T., High S., Swanton E. (2007) Biosynthesis of the dystonia-associated AAA+ ATPase torsinA at the endoplasmic reticulum. Biochem. J. 401, 607–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Needham P. G., Mikoluk K., Dhakarwal P., Khadem S., Snyder A. C., Subramanya A. R., Brodsky J. L. (2011) The thiazide-sensitive NaCl cotransporter is targeted for chaperone-dependent ER-associated degradation. J. Biol. Chem. 286, 43611–43621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kustedjo K., Deechongkit S., Kelly J. W., Cravatt B. F. (2003) Recombinant expression, purification, and comparative characterization of torsinA and its torsion dystonia-associated variant ΔE-torsinA. Biochemistry 42, 15333–15341 [DOI] [PubMed] [Google Scholar]
- 92. Kruse K. B., Brodsky J. L., McCracken A. A. (2006) Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human α-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol. Biol. Cell 17, 203–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chen P., Burdette A. J., Porter J. C., Ricketts J. C., Fox S. A., Nery F. C., Hewett J. W., Berkowitz L. A., Breakefield X. O., Caldwell K. A., Caldwell G. A. (2010) The early-onset torsion dystonia-associated protein, torsinA, is a homeostatic regulator of endoplasmic reticulum stress response. Hum. Mol. Genet. 19, 3502–3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kock N., Naismith T. V., Boston H. E., Ozelius L. J., Corey D. P., Breakefield X. O., Hanson P. I. (2006) Effects of genetic variations in the dystonia protein torsinA: identification of polymorphism at residue 216 as protein modifier. Hum. Mol. Genet. 15, 1355–1364 [DOI] [PubMed] [Google Scholar]
- 95. Hewett J. W., Tannous B., Niland B. P., Nery F. C., Zeng J., Li Y., Breakefield X. O. (2007) Mutant torsinA interferes with protein processing through the secretory pathway in DYT1 dystonia cells. Proc. Natl. Acad. Sci. U.S.A. 104, 7271–7276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Valastyan J. S., Lindquist S. (2011) TorsinA and the torsinA-interacting protein printor have no impact on endoplasmic reticulum stress or protein trafficking in yeast. PLoS One 6, e22744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Naismith T. V., Heuser J. E., Breakefield X. O., Hanson P. I. (2004) TorsinA in the nuclear envelope. Proc. Natl. Acad. Sci. U.S.A. 101, 7612–7617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Goodchild R. E., Kim C. E., Dauer W. T. (2005) Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron 48, 923–932 [DOI] [PubMed] [Google Scholar]
- 99. Zolkiewski M., Wu H.-C. (2011) Emerging Area: TorsinA, a Novel ATP-dependent Factor Linked to Dystonia, in Protein Chaperones and Protection from Neurodegenerative Diseases (Witt S. N., ed) John Wiley and Sons, Inc., Hoboken, NJ [Google Scholar]
- 100. Plemper R. K., Böhmler S., Bordallo J., Sommer T., Wolf D. H. (1997) Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature 388, 891–895 [DOI] [PubMed] [Google Scholar]
- 101. Kabani M., Kelley S. S., Morrow M. W., Montgomery D. L., Sivendran R., Rose M. D., Gierasch L. M., Brodsky J. L. (2003) Dependence of endoplasmic reticulum-associated degradation on the peptide binding domain and concentration of BiP. Mol. Biol. Cell 14, 3437–3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Simons J. F., Ferro-Novick S., Rose M. D., Helenius A. (1995) BiP/Kar2p serves as a molecular chaperone during carboxypeptidase Y folding in yeast. J. Cell Biol. 130, 41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Polaina J., Conde J. (1982) Genes involved in the control of nuclear fusion during the sexual cycle of Saccharomyces cerevisiae. Mol. Gen. Genet. 186, 253–258 [DOI] [PubMed] [Google Scholar]
- 104. Kimata Y., Kimata Y. I., Shimizu Y., Abe H., Farcasanu I. C., Takeuchi M., Rose M. D., Kohno K. (2003) Genetic evidence for a role of BiP/Kar2 that regulates Ire1 in response to accumulation of unfolded proteins. Mol. Biol. Cell 14, 2559–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Brodsky J. L., Werner E. D., Dubas M. E., Goeckeler J. L., Kruse K. B., McCracken A. A. (1999) The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J. Biol. Chem. 274, 3453–3460 [DOI] [PubMed] [Google Scholar]
- 106. Bragg D. C., Kaufman C. A., Kock N., Breakefield X. O. (2004) Inhibition of N-linked glycosylation prevents inclusion formation by the dystonia-related mutant form of torsinA. Mol. Cell. Neurosci. 27, 417–426 [DOI] [PubMed] [Google Scholar]
- 107. Takeuchi M., Kimata Y., Hirata A., Oka M., Kohno K. (2006) Saccharomyces cerevisiae Rot1p is an ER-localized membrane protein that may function with BiP/Kar2p in protein folding. J. Biochem. 139, 597–605 [DOI] [PubMed] [Google Scholar]
- 108. Newman A. P., Groesch M. E., Ferro-Novick S. (1992) Bos1p, a membrane protein required for ER to Golgi transport in yeast, co-purifies with the carrier vesicles and with Bet1p and the ER membrane. EMBO J. 11, 3609–3617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Brodsky J. L. (2007) The protective and destructive roles played by molecular chaperones during ERAD (endoplasmic-reticulum-associated degradation). Biochem. J. 404, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Han S., Liu Y., Chang A. (2007) Cytoplasmic Hsp70 promotes ubiquitination for endoplasmic reticulum-associated degradation of a misfolded mutant of the yeast plasma membrane ATPase, PMA1. J. Biol. Chem. 282, 26140–26149 [DOI] [PubMed] [Google Scholar]
- 111. Nakatsukasa K., Huyer G., Michaelis S., Brodsky J. L. (2008) Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell 132, 101–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Molinari M., Helenius A. (2000) Chaperone selection during glycoprotein translocation into the endoplasmic reticulum. Science 288, 331–333 [DOI] [PubMed] [Google Scholar]
- 113. Ng D. T., Randall R. E., Lamb R. A. (1989) Intracellular maturation and transport of the SV5 type II glycoprotein hemagglutinin-neuraminidase: specific and transient association with GRP78-BiP in the endoplasmic reticulum and extensive internalization from the cell surface. J. Cell Biol. 109, 3273–3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kampinga H. H., Craig E. A. (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11, 579–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Buck T. M., Plavchak L., Roy A., Donnelly B. F., Kashlan O. B., Kleyman T. R., Subramanya A. R., Brodsky J. L. (2013) The Lhs1/GRP170 chaperones facilitate the endoplasmic reticulum-associated degradation of the epithelial sodium channel. J. Biol. Chem. 288, 18366–18380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Jakob C. A., Bodmer D., Spirig U., Battig P., Marcil A., Dignard D., Bergeron J. J., Thomas D. Y., Aebi M. (2001) Htm1p, a mannosidase-like protein, is involved in glycoprotein degradation in yeast. EMBO Rep. 2, 423–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Gordon K. L., Glenn K. A., Gonzalez-Alegre P. (2011) Exploring the influence of torsinA expression on protein quality control. Neurochem. Res. 36, 452–459 [DOI] [PubMed] [Google Scholar]
- 118. Urano F., Bertolotti A., Ron D. (2000) IRE1 and efferent signaling from the endoplasmic reticulum. J. Cell Sci. 113, 3697–3702 [DOI] [PubMed] [Google Scholar]
- 119. Leppert G., McDevitt R., Falco S. C., Van Dyk T. K., Ficke M. B., Golin J. (1990) Cloning by gene amplification of two loci conferring multiple drug resistance in Saccharomyces. Genetics 125, 13–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Fleming J. A., Lightcap E. S., Sadis S., Thoroddsen V., Bulawa C. E., Blackman R. K. (2002) Complementary whole-genome technologies reveal the cellular response to proteasome inhibition by PS-341. Proc. Natl. Acad. Sci. U.S.A. 99, 1461–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hemmings B. A., Zubenko G. S., Hasilik A., Jones E. W. (1981) Mutant defective in processing of an enzyme located in the lysosome-like vacuole of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 78, 435–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Simon K., Lingappa V. R., Ganem D. (1988) Secreted hepatitis B surface antigen polypeptides are derived from a transmembrane precursor. J. Cell Biol. 107, 2163–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kustedjo K., Bracey M. H., Cravatt B. F. (2000) Torsin A and its torsion dystonia-associated mutant forms are lumenal glycoproteins that exhibit distinct subcellular localizations. J. Biol. Chem. 275, 27933–27939 [DOI] [PubMed] [Google Scholar]
- 124. Babst M., Wendland B., Estepa E. J., Emr S. D. (1998) The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J. 17, 2982–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Whiteheart S. W., Rossnagel K., Buhrow S. A., Brunner M., Jaenicke R., Rothman J. E. (1994) N-Ethylmaleimide-sensitive fusion protein: a trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J. Cell Biol. 126, 945–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Weibezahn J., Schlieker C., Bukau B., Mogk A. (2003) Characterization of a trap mutant of the AAA+ chaperone ClpB. J. Biol. Chem. 278, 32608–32617 [DOI] [PubMed] [Google Scholar]
- 127. Zhao C., Brown R. S., Chase A. R., Eisele M. R., Schlieker C. (2013) Regulation of Torsin ATPases by LAP1 and LULL1. Proc. Natl. Acad. Sci. U.S.A. 110, E1545–E1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Giles L. M., Li L., Chin L.-S. (2009) Printor, a novel torsinA-interacting protein implicated in dystonia pathogenesis. J. Biol. Chem. 284, 21765–21775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Goodchild R. E., Dauer W. T. (2005) The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J. Cell Biol. 168, 855–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Vale R. D. (2000) AAA proteins. Lords of the ring. J. Cell Biol. 150, F13–F19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Kamm C., Fischer H., Garavaglia B., Kullmann S., Sharma M., Schrader C., Grundmann K., Klein C., Borggraefe I., Lobsien E., Kupsch A., Nardocci N., Gasser T. (2008) Susceptibility to DYT1 dystonia in European patients is modified by the D216H polymorphism. Neurology 70, 2261–2262 [DOI] [PubMed] [Google Scholar]
- 132. Risch N. J., Bressman S. B., Senthil G., Ozelius L. J. (2007) Intragenic cis and trans modification of genetic susceptibility in DYT1 torsion dystonia. Am. J. Hum. Genet. 80, 1188–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Benitez E. M., Stolz A., Wolf D. H. (2011) Yos9, a control protein for misfolded glycosylated and non-glycosylated proteins in ERAD. FEBS Lett. 585, 3015–3019 [DOI] [PubMed] [Google Scholar]
- 134. Spear E. D., Ng D. T. (2005) Single, context-specific glycans can target misfolded glycoproteins for ER-associated degradation. J. Cell Biol. 169, 73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Eletto D., Maganty A., Eletto D., Dersh D., Makarewich C., Biswas C., Paton J. C., Paton A. W., Doroudgar S., Glembotski C. C., Argon Y. (2012) Limitation of individual folding resources in the ER leads to outcomes distinct from the unfolded protein response. J. Cell Sci. 125, 4865–4875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Luo S., Mao C., Lee B., Lee A. S. (2006) GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol. Cell. Biol. 26, 5688–5697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Gordon K. L., Glenn K. A., Bode N., Wen H. M., Paulson H. L., Gonzalez-Alegre P. (2012) The ubiquitin ligase F-box/G-domain protein 1 promotes the degradation of the disease-linked protein torsinA through the ubiquitin-proteasome pathway and macroautophagy. Neuroscience 224, 160–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Helenius A., Aebi M. (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 [DOI] [PubMed] [Google Scholar]
- 139. Dwek R. A., Butters T. D., Platt F. M., Zitzmann N. (2002) Targeting glycosylation as a therapeutic approach. Nat. Rev. Drug Discov. 1, 65–75 [DOI] [PubMed] [Google Scholar]
- 140. Freeze H. H. (2013) Understanding human glycosylation disorders: biochemistry leads the charge. J. Biol. Chem. 288, 6936–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Kornfeld S. (1998) Diseases of abnormal protein glycosylation: an emerging area. J. Clin. Invest. 101, 1293–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Jones J., Krag S. S., Betenbaugh M. J. (2005) Controlling N-linked glycan site occupancy. Biochim. Biophys. Acta 1726, 121–137 [DOI] [PubMed] [Google Scholar]
- 143. Zhu L., Wrabl J. O., Hayashi A. P., Rose L. S., Thomas P. J. (2008) The torsin-family AAA+ protein OOC-5 contains a critical disulfide adjacent to Sensor-II that couples redox state to nucleotide binding. Mol. Biol. Cell 19, 3599–3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Schulz B. L., Stirnimann C. U., Grimshaw J. P., Brozzo M. S., Fritsch F., Mohorko E., Capitani G., Glockshuber R., Grütter M. G., Aebi M. (2009) Oxidoreductase activity of oligosaccharyltransferase subunits Ost3p and Ost6p defines site-specific glycosylation efficiency. Proc. Natl. Acad. Sci. U.S.A. 106, 11061–11066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Parlati F., Dominguez M., Bergeron J. J., Thomas D. Y. (1995) Saccharomyces cerevisiae CNE1 encodes an endoplasmic reticulum (ER) membrane protein with sequence similarity to calnexin and calreticulin and functions as a constituent of the ER quality control apparatus. J. Biol. Chem. 270, 244–253 [DOI] [PubMed] [Google Scholar]
- 146. Shenkman M., Groisman B., Ron E., Avezov E., Hendershot L. M., Lederkremer G. Z. (2013) A shared endoplasmic reticulum-associated degradation pathway involving the EDEM1 protein for glycosylated and nonglycosylated proteins. J. Biol. Chem. 288, 2167–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Bucciantini M., Giannoni E., Chiti F., Baroni F., Formigli L., Zurdo J., Taddei N., Ramponi G., Dobson C. M., Stefani M. (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511 [DOI] [PubMed] [Google Scholar]
- 148. Maisonneuve E., Fraysse L., Moinier D., Dukan S. (2008) Existence of abnormal protein aggregates in healthy Escherichia coli cells. J. Bacteriol. 190, 887–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Shashidharan P., Good P. F., Hsu A., Perl D. P., Brin M. F., Olanow C. W. (2000) TorsinA accumulation in Lewy bodies in sporadic Parkinson's disease. Brain Res. 877, 379–381 [DOI] [PubMed] [Google Scholar]
- 150. Jungwirth M., Dear M. L., Brown P., Holbrook K., Goodchild R. (2010) Relative tissue expression of homologous torsinB correlates with the neuronal specific importance of DYT1 dystonia-associated torsinA. Hum. Mol. Genet. 19, 888–900 [DOI] [PubMed] [Google Scholar]
- 151. Bonifacino J. S., Suzuki C. K., Lippincott-Schwartz J., Weissman A. M., Klausner R. D. (1989) Pre-Golgi degradation of newly synthesized T-cell antigen receptor chains: intrinsic sensitivity and the role of subunit assembly. J. Cell Biol. 109, 73–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Mogk A., Schlieker C., Strub C., Rist W., Weibezahn J., Bukau B. (2003) Roles of individual domains and conserved motifs of the AAA+ chaperone ClpB in oligomerization, ATP hydrolysis, and chaperone activity. J. Biol. Chem. 278, 17615–17624 [DOI] [PubMed] [Google Scholar]
- 153. Matsumura Y., David L. L., Skach W. R. (2011) Role of Hsc70 binding cycle in CFTR folding and endoplasmic reticulum-associated degradation. Mol. Biol. Cell 22, 2797–2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Beggah A., Mathews P., Beguin P., Geering K. (1996) Degradation and endoplasmic reticulum retention of unassembled α- and β-subunits of Na,K-ATPase correlate with interaction of BiP. J. Biol. Chem. 271, 20895–20902 [DOI] [PubMed] [Google Scholar]
- 155. Forsayeth J. R., Gu Y., Hall Z. W. (1992) BiP forms stable complexes with unassembled subunits of the acetylcholine receptor in transfected COS cells and in C2 muscle cells. J. Cell Biol. 117, 841–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Knittler M. R., Haas I. G. (1992) Interaction of BiP with newly synthesized immunoglobulin light chain molecules: cycles of sequential binding and release. EMBO J. 11, 1573–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Schmitz A., Maintz M., Kehle T., Herzog V. (1995) In vivo iodination of a misfolded proinsulin reveals co-localized signals for Bip binding and for degradation in the ER. EMBO J. 14, 1091–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Ni M., Zhang Y., Lee A. S. (2011) Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 434, 181–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Panayi G. S., Corrigall V. M. (2008) BiP, an anti-inflammatory ER protein, is a potential new therapy for the treatment of rheumatoid arthritis. Novartis Found. Symp. 291, 212–216 [DOI] [PubMed] [Google Scholar]
- 160. Ermakova S. P., Kang B. S., Choi B. Y., Choi H. S., Schuster T. F., Ma W. Y., Bode A. M., Dong Z. (2006) (−)-Epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res. 66, 9260–9269 [DOI] [PubMed] [Google Scholar]
- 161. Brodsky J. L., Chiosis G. (2006) Hsp70 molecular chaperones: emerging roles in human disease and identification of small molecule modulators. Curr. Top. Med. Chem. 6, 1215–1225 [DOI] [PubMed] [Google Scholar]
- 162. Qiu W., Kohen-Avramoglu R., Mhapsekar S., Tsai J., Austin R. C., Adeli K. (2005) Glucosamine-induced endoplasmic reticulum stress promotes ApoB100 degradation: evidence for Grp78-mediated targeting to proteasomal degradation. Arterioscler. Thromb. Vasc. Biol. 25, 571–577 [DOI] [PubMed] [Google Scholar]
- 163. Duan W., Mattson M. P. (1999) Dietary restriction and 2-deoxyglucose administration improve behavioral outcome and reduce degeneration of dopaminergic neurons in models of Parkinson's disease. J. Neurosci. Res. 57, 195–206 [DOI] [PubMed] [Google Scholar]
- 164. Kammoun H. L., Chabanon H., Hainault I., Luquet S., Magnan C., Koike T., Ferré P., Foufelle F. (2009) GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Invest. 119, 1201–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Inokuchi Y., Nakajima Y., Shimazawa M., Kurita T., Kubo M., Saito A., Sajiki H., Kudo T., Aihara M., Imaizumi K., Araie M., Hara H. (2009) Effect of an inducer of BiP, a molecular chaperone, on endoplasmic reticulum (ER) stress-induced retinal cell death. Invest. Ophthalmol. Vis. Sci. 50, 334–344 [DOI] [PubMed] [Google Scholar]
- 166. Lim H. A., Kim J. H., Kim J. H., Sung M. K., Kim M. K., Park J. H., Kim J. S. (2006) Genistein induces glucose-regulated protein 78 in mammary tumor cells. J. Med. Food 9, 28–32 [DOI] [PubMed] [Google Scholar]