

Background: Superoxide dismutases are inactivated by peroxynitrite.
Results: T. cruzi cytosolic Fe-SODB is highly resistant toward peroxynitrite-mediated tyrosine nitration and inactivation as compared with mitochondrial Fe-SODA.
Conclusion: Intramolecular electron transfer in Fe-SODB from Cys83 to critical Tyr35 prevents enzyme nitration and inactivation.
Significance: Disparate susceptibilities of Fe-SODs to peroxynitrite can influence parasite virulence during T. cruzi infection of mammalian cells.
Keywords: Free Radicals, Nitric Oxide, Oxidation-Reduction, Superoxide Dismutase (SOD), Trypanosome, Trypanosoma cruzi, Nitration, Peroxynitrite, Superoxide
Abstract
Trypanosoma cruzi, the causative agent of Chagas disease, contains exclusively iron-dependent superoxide dismutases (Fe-SODs) located in different subcellular compartments. Peroxynitrite, a key cytotoxic and oxidizing effector biomolecule, reacted with T. cruzi mitochondrial (Fe-SODA) and cytosolic (Fe-SODB) SODs with second order rate constants of 4.6 ± 0.2 × 104 m−1 s−1 and 4.3 ± 0.4 × 104 m−1 s−1 at pH 7.4 and 37 °C, respectively. Both isoforms are dose-dependently nitrated and inactivated by peroxynitrite. Susceptibility of T. cruzi Fe-SODA toward peroxynitrite was similar to that reported previously for Escherichia coli Mn- and Fe-SODs and mammalian Mn-SOD, whereas Fe-SODB was exceptionally resistant to oxidant-mediated inactivation. We report mass spectrometry analysis indicating that peroxynitrite-mediated inactivation of T. cruzi Fe-SODs is due to the site-specific nitration of the critical and universally conserved Tyr35. Searching for structural differences, the crystal structure of Fe-SODA was solved at 2.2 Å resolution. Structural analysis comparing both Fe-SOD isoforms reveals differences in key cysteines and tryptophan residues. Thiol alkylation of Fe-SODB cysteines made the enzyme more susceptible to peroxynitrite. In particular, Cys83 mutation (C83S, absent in Fe-SODA) increased the Fe-SODB sensitivity toward peroxynitrite. Molecular dynamics, electron paramagnetic resonance, and immunospin trapping analysis revealed that Cys83 present in Fe-SODB acts as an electron donor that repairs Tyr35 radical via intramolecular electron transfer, preventing peroxynitrite-dependent nitration and consequent inactivation of Fe-SODB. Parasites exposed to exogenous or endogenous sources of peroxynitrite resulted in nitration and inactivation of Fe-SODA but not Fe-SODB, suggesting that these enzymes play distinctive biological roles during parasite infection of mammalian cells.
Introduction
Superoxide dismutases (SODs5; EC 1.15.1.1) are metalloenzymes essential for all living aerobic organisms that catalyze the detoxification of superoxide radicals (O2⨪) to oxygen (O2) and hydrogen peroxide (H2O2). They are subdivided into three structurally distinct families, depending on the metal of the active site: (i) copper/zinc (Cu/Zn-SODs) existing in eukaryotes and bacteria; (ii) iron/manganese (Fe/Mn-SODs) found in bacteria and several higher plants; and (iii) Ni-SODs present in bacteria of the Streptomyces species (1). Fe- and Mn-SODs exhibit a high degree of sequence and structure similarity, strongly suggesting that these enzymes originate from a common ancestor (2).
Trypanosomatids of the genus Leishmania, Trypanosoma brucei, and Trypanosma cruzi are primitive eukaryotes containing exclusively Fe-SODs isoforms. For T. cruzi, the causative agent of Chagas disease, two SOD genes have been cloned and characterized: mitochondrial Fe-SODA and cytosolic Fe-SODB (3, 4). Chagas disease remains a major public health concern in Latin America, with an estimated total of 11 million people infected and 28 million at risk, according to the World Health Organization. Moreover, the disease is spreading worldwide as a result of migration (mammalian host and insect vectors), HIV co-infection, blood transfusion, and organ transplantation, as evidenced by the fact that 1 of every 4700 blood donors test positive for T. cruzi. The United States Centers for Disease Control estimates that there are 300,000 infected persons living in the United States as of 2010. The availability of drugs to treat the disease is currently minimal, and there is an urgent need for new drugs, which requires the proper identification of critical molecular targets and metabolic pathways that may differ from the mammalian host cells (5, 6). In this context, the antioxidant armamentarium of T. cruzi has been recently recognized to participate in parasite virulence (7); therefore, it is important to obtain a more detailed understanding of the interplay between oxidants formed during T. cruzi invasion to mammalian host cells and the parasite antioxidant enzyme network and how these processes influence the outcome of the infection.
During the acute infection, resident macrophages, at the site of parasite invasion, are among the first professional phagocytes to be invaded by T. cruzi (8, 9). To establish the infection, the invading T. cruzi needs to survive the highly oxidizing environment of the macrophage phagosome before escaping to the “safer” cytoplasmic milieu (10). The macrophage NADPH oxidase is activated during T. cruzi phagocytosis, resulting in a sustained (60–90-min) O2⨪ production toward the internalized parasite (10). Macrophage-derived O2⨪ is not particularly toxic per se to T. cruzi, in part because of its limited diffusion inside the parasite and also because of the ubiquitous presence of Fe-SODs, which promote its fast dismutation to hydrogen peroxide (H2O2). Alternatively, in immunostimulated macrophages, O2⨪ reacts with nitric oxide (•NO, derived from the inducible nitric-oxide synthase) at diffusion-controlled rates (k = 1 × 1010 m−1 s−1) to yield peroxynitrite, a strong oxidant and potent cytotoxic effector molecule against T. cruzi (10). In non-phagocytic cells, such as cardiomyocytes, mammalian cell-derived •NO may exert cytotoxic actions on T. cruzi associated with an increase in O2⨪ generation by the respiratory chain and peroxynitrite formation in mitochondria (11). Enhanced mitochondrial O2⨪ steady-state levels trigger T. cruzi programmed cell death (12). Importantly, Fe-SODA overexpressers are more resistant to apoptotic stimuli, revealing the participation of the enzyme in the fine tuning of the death signaling process (12).
The efficient removal of O2⨪ by Fe-SODs can attenuate peroxynitrite formation in different T. cruzi subcellular compartments. However, the •NO and O2⨪ reaction occurs in biological systems despite the presence of SODs because it can outcompete the enzyme-catalyzed O2⨪ dismutation (13). Moreover, in the case of mammalian Mn-SOD, it is well established that it can be efficiently inactivated by peroxynitrite (14), which increases O2⨪ levels, and subsequently peroxynitrite, creating a hazardous positive feedback loop that can impair mitochondrial energy metabolism (15) and the signaling of programmed cell death (16). In this scenario, it is reasonable to hypothesize that the contents of parasite Fe-SODs and the fluxes of O2⨪, •NO, and peroxynitrite may be important determinants in parasite survival or death during the infection process.
The mechanism of the peroxynitrite-dependent inactivation of mammalian Mn-SOD has been extensively studied and involves the preferential nitration of Tyr34, located 5 Å from the manganese ion of the active site (14, 17–20). This site-specific nitration has been ascribed to a kinetically favored reaction of peroxynitrite with the manganese ion, leading to the formation of oxidizing and nitrating species at the active site (18, 21). Due to the high degree of structural homology that Mn- and Fe-SODs share (2), a similar mechanism for peroxynitrite-dependent T. cruzi Fe-SODs enzyme inactivation may be expected. In this work, we have studied the reaction of peroxynitrite with the purified cytosolic and mitochondrial isoforms of T. cruzi Fe-SODs. The results obtained revealed outstanding differences between both T. cruzi SODs isoforms in terms of their susceptibility to peroxynitrite-mediated nitration and inactivation, with the cytosolic Fe-SODB being extremely resistant to oxidant treatment. The biochemical basis for the observed disparate oxidant susceptibility between both T. cruzi Fe-SODs was analyzed at the structural and molecular levels. Moreover, the occurrence of these biochemical events under cellular nitroxidative stress conditions was unambiguously established in T. cruzi Fe-SOD overexpressers, confirming the preferential nitration and inactivation of Fe-SODA in living parasites.
EXPERIMENTAL PROCEDURES
Chemicals
N-Ethylmaleimide (NEM), diethylenetriaminepentaacetic acid, 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB), manganese dioxide, Dulbecco's modified Eagle's medium (DMEM), lipopolysaccharide (LPS), Geneticin (G418), l-cysteine-methyl ester, 4-(2-pyridylazo)-resorcinol, and α-phenyl N-tertiary-butyl nitrone (PBN) were from Sigma. The nitric oxide donor 1-hydroxy-2-oxo-3-(N-ethyl-2-aminoethyl)-3-ethyl-1-triazene (NOC-12) was from Dojindo. Murine recombinant IFN-γ was from Calbiochem. Lab-Tek tissue culture chamber slides were from Nunc. All other chemicals were of reagent grade. Peroxynitrite was synthesized in a quenched flow reactor from sodium nitrite and hydrogen peroxide (H2O2) under acidic conditions and quantitated as described previously; excess H2O2 was removed by treatment with MnO2, and NO2− contamination was always <20% (22). Peroxynitrite concentration was determined spectrophotometrically at 302 nm (ϵ302 nm = 1670 m−1 cm−1) (23). The spin trap 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) and the anti-DMPO rabbit antiserum were kindly provided by Dr. Ronald Maison (NIEHS, National Institutes of Health, Bethesda, MD).
Expression, Purification, and Site-directed Mutagenesis of T. cruzi Cytosolic Fe-SODB and Mitochondrial Fe-SODA
Parasite SOD genes were amplified from T. cruzi genomic DNA (CL-Brener strain) using the following primers: Fe-SODA, 5′-GGATCCGCCCCGGCCGAGTTGCCCAA-3′ (forward) and 5′-GGAAGCTTTATTTTATTGCCTGCGCATG-3′ (reverse); Fe-SODB, 5′-GGGGATCCATGGTCTTCAGCATTCCTCC-3′ (forward) and 5′-GGAAGCTTCGTGGGTCAAAGTTGTCG-3′ (reverse) (restriction sites for BamHI and HindIII, respectively, are underlined). The purified PCR product (BIORON gel extraction kit) was ligated into the pGEM-T Easy vector (Promega) and transformed into electrocompetent Escherichia coli XL1 blue cells. The amplified T. cruzi Fe-SODA (without mitochondrial signal peptide) and Fe-SODB genes were cloned into the pQE-30 vector (Qiagen) between BamHI and HindIII. pQE30-Fe-SODs in E. coli M15 (pREP4) cells were grown at 37 °C in LB broth containing ampicillin (100 μg/ml) and kanamycin (50 μg/ml). Expression of recombinant Fe-SODs was induced with isopropyl-β-d-thiogalactopyranoside (0.8 mm) when the culture reached A600 = 0.6, and the temperature was lowered to 22 °C for overnight protein expression. The purification was performed in a 5-ml HiTrap affinity column (Amersham Biosciences) charged with Ni2+ and equilibrated with binding buffer (50 mm sodium phosphate, pH 7.6, containing imidazole (10 mm) and NaCl (500 mm) at a flow rate of 3 ml/min. Fe-SODs were eluted with a linear imidazole (10–500 mm) gradient in sodium phosphate (50 mm, pH 7.6) containing NaCl (500 mm). Imidazole was removed by buffer exchange with sodium phosphate buffer (50 mm, pH 7.4) using HiTrap desalting columns (Amersham Biosciences). Purity of T. cruzi Fe-SOD preparations was evaluated by SDS-PAGE, and protein concentration was measured by the Bradford method (24). Molecular weight of the purified enzymes was evaluated by gel filtration chromatography in a SuperdexTM-200 column (GE Healthcare) calibrated with the following protein standards: 13,700, 29,000, 43,000, and 75,000 Da (GE Healthcare). Iron content was measured by the 4-pyrydylazo-resorcinol method (25).
Site-directed mutagenesis of T. cruzi Fe-SODB (C159S) was performed by PCR from plasmidic DNA (pQE30-SODB) with Pfu Turbo DNA polymerase 1.25 units (Fermentas) using the following primers (0.4 μm): forward, 5′-ACTTGAAGCCTCTCCTTACAAGCGATGTATGGGAG-3′; reverse, 5′-CTCCCATACATCGCTTGTAAGGAGAGGCTTCAAGT-3′. The PCR was performed with an annealing temperature of 55 °C (16 cycles) and extension temperature of 68 °C (8 min). Following PCR, the mutated DNA was selected with DpnI (Biolabs), which degrades parental methylated DNA. Sequence fidelity was confirmed by DNA sequencing (Institut Pasteur, Montevideo, Uruguay). Fe-SODB C83S and N187D/K189E mutants were purchased from GenScript. Proteins were expressed and purified as described above.
Determination of T. cruzi Fe-SOD Rate Constants with O2⨪
The O2⨪ dismutase activity of the purified T. cruzi recombinant Fe-SODA and Fe-SODB was measured by the decrease in the rate of superoxide-dependent cytochrome c reduction at 550 nm using xanthine/xanthine oxidase as a superoxide source (26). The rate constants of T. cruzi Fe-SODA and Fe-SODB were determined by the kinetic competition method described previously. Xanthine-oxidase (6 milliunits) and xanthine (50 μm) were used for the generation of O2⨪ fluxes. The reduction of ferricytochrome c (20 μm) was monitored at 550 nm at 37 °C in phosphate buffer 50 mm, pH 7.8, using a 1-ml cuvette in the absence and presence of Fe-SODA or Fe-SODB (0–40 nm). The velocity of cytochrome c reduction in the absence (V0) and presence of Fe-SOD (VSOD) was employed to calculate an inhibition fraction (IF = 1 − (V0 − VSOD)/V0. Utilizing the reported bimolecular rate constant for the reaction of O2⨪ with ferricytochrome c at pH 7.8 (2.6 ± 0.1 × 105 m−1 s−1), the rate constants were calculated as kSOD = kcyt ([cyt]/[TcSOD]50). [TcSOD]50 (the concentration of SOD that caused an inhibition fraction of 0.5) was obtained from the fitting of experimental data to a rectangular hyperbolic curve (27).
Kinetics Studies of T. cruzi Fe-SOD Reactions with Peroxynitrite
The kinetics of peroxynitrite (10 and 17 μm for Fe-SODA and Fe-SODB, respectively) decomposition was monitored at 302 nm (ϵ302 = 1670 m−1 cm−1) in the presence or absence of control and NEM-treated Fe-SODA and Fe-SODB (0–15 μm) in a stopped-flow spectrophotometer (SX20, Applied Photophysics) with a mixing time of <2 ms. An initial rate approach was used to analyze the data (28); the first 0–0.15 s of peroxynitrite decomposition was fitted to a linear plot, and initial rates were calculated by dividing the slope of the absorbance time course by the peroxynitrite molar extinction coefficient at 302 nm and multiplying by a factor of 1.25 to account for the 20% fraction of peroxynitrite that is not deprotonated at pH 7.4 (because the absorption at 302 nm is due to the peroxynitrite anion). Second order rate constants (k2) were calculated according to the equation, v0 = (k1 + k2 × [Fe-SOD]0) × [peroxynitrite]0, were k1 is the rate constant of proton-catalyzed peroxynitrite decomposition (28). Reported values are the average of at least seven separate determinations. Temperature was maintained at 37 °C, and the pH was measured at the outlet.
Measurements of Protein Thiol Content and Thiol Alkylation
Protein thiols were quantitated using the DTNB assay (29). Protein thiols were reduced by incubation with DTT (1 mm) for 30 min at room temperature. Alkylation of T. cruzi Fe-SOD thiol groups by NEM was performed by incubation of Fe-SODA or Fe-SODB (200 μm) with NEM (10 mm) for 2 h in phosphate buffer (50 mm, pH 7.4) at 4 °C. Excess DTT and NEM were removed immediately after incubation using HiTrap desalting columns (Amersham Biosciences) as described previously (30).
Peroxynitrite Treatment of T. cruzi Fe-SODs
Peroxynitrite (0–2000 μm) was added as a single dose under vortex to the purified enzymes (8 μm) in sodium phosphate buffer (200 mm, pH 7.4), and activity was measured as described above. Peroxynitrite addition was done to control or NEM-treated enzymes in the presence or absence of GSH (10 mm), uric acid (100 μm), l-cysteine methyl ester (8 μm), bicarbonate (25 mm), PBN (50 mm), and DMPO (100 mm). Peroxynitrite addition was also performed at different pH values (5.8–8.0), and T. cruzi Fe-SODs activity was measured as described above.
Western Blotting, Protein Nitrotyrosine, and Immunospin Trapping Analysis
After treatment, proteins were subjected to 15% SDS-PAGE, transferred to nitrocellulose membranes, and blocked in phosphate-buffered saline (PBS; 50 mm, pH 7.4) containing dry milk (5% w/v) for 1 h. Membranes were then probed with either rabbit polyclonal anti-nitrotyrosine antibody (1:2000 dilution raised in our laboratory (31)) or rabbit polyclonal anti-T. cruzi Fe-SODA or Fe-SODB (1:5000 dilution (32)) in PBS containing Tween 20 (0.1%, v/v) for 1 h. Membranes were washed and probed for 1 h with anti-rabbit-IgG (IR Dye-800- and IR Dye-680-conjugated (LI-COR Biosciences) or peroxidase-conjugated (Calbiochem), 1:15,000 dilution in PBS containing Tween 20 (0.1%, v/v)). After washing of the probed membranes, immunoreactive proteins were visualized with an infrared fluorescence detection system (Odyssey, LI-COR Biosciences) or using the Immun-StarTM chemiluminescence kit (Bio-Rad). For immune spin trapping, after exposure of T. cruzi Fe-SODB (50 μm) to peroxynitrite (5–20 μm) in the presence or absence of DMPO (100 mm), protein samples were subjected to Western blot analysis, and protein-DMPO nitrone adducts were detected using a rabbit polyclonal anti-DMPO-nitrone primary antibody (rabbit anti-DMPO serum, 1:2,000 dilution in PBS containing Tween 20 (0.1%, v/v) and bovine serum albumin (4%, v/v)) as described previously (33). Immunoreactive proteins were detected with the infrared system as described above.
EPR Studies
The EPR spectra were recorded at room temperature (25 °C) on a Bruker EMX EPR spectrometer. Wild type Fe-SODB or C83S single mutant (2 mm) were incubated with the spin trap PBN (50 mm) and exposed to peroxynitrite (500 μm). Immediately after oxidant addition, samples were transferred to a 200-μl flat cell, and the spectra were recorded within 1 min (15 spectrum acquisitions). Adducts between Fe-SODs and PBN were digested with Pronase (20 mg/ml) for 10 min, and spectra were recorded as above.
Peptide Mapping Analysis of Peroxynitrite-treated T. cruzi Fe-SODs
T. cruzi Fe-SODs (8 μm) were treated with peroxynitrite (0–300 μm) in potassium phosphate buffer (200 mm, pH 7.4) at 25 °C in the presence or absence of DMPO (100 mm). Protein samples were separated by one- or two-dimensional gel electrophoresis. Commercially available IPG strips (7 cm, linear 3–10, GE Healthcare) were used for the first dimensional separation. Enzymes were purified and concentrated with 2-D Clean-up (GE Healthcare) and dissolved in 125 μl of rehydration solution (7 m urea, 2 m thiourea, 2% (w/v) CHAPS, 0.5% (v/v) IPG buffer 3–10 (GE Healthcare), 0.002% (w/v) bromphenol blue, and 17 mm DTT). Samples in rehydration solution were loaded onto IPG strips by passive rehydration during 12 h at room temperature. The isoelectric focusing was done in an IPGphor unit (GE Healthcare) employing the following voltage profile: constant phase of 300 V for 30 min, linear increase to 1000 V in 30 min, linear increase to 5000 V in 80 min, and a final constant phase of 5000 V to reach a total of 2.0 kVh. Prior to running the second dimension, IPG strips were reduced for 15 min in equilibration buffer (6 m urea, 75 mm Tris-HCl, pH 8.8; 29.3% (v/v) glycerol, 2% (w/v) SDS, and 0.002% (v/v) bromphenol blue) supplemented with 10 mg/ml (DTT) and subsequently alkylated with iodoacetamide (25 mg/ml). The second dimensional separation was performed in 15% SDS-PAGE using an SE 260 minivertical gel electrophoresis unit (GE Healthcare). Gels were stained with Colloidal Coomassie Blue G-250. Images were digitized using a UMAX Power-Look 1120 scanner and LabScan version 5.0 software (GE Healthcare). Selected spots from two-dimensional gels or bands from one-dimensional gels were manually removed and in-gel digested with trypsin (sequencing grade; Promega) as described (34). Peptides were extracted from gels using aqueous acetonitrile (60%, v/v) containing trifluoroacetic acid (TFA; 0.1%, v/v) and concentrated by vacuum drying. Peptide mixtures were analyzed using a linear ion trap mass spectrometer (LTQ Velos, Thermo) coupled online with a nanoliquid chromatography system (Easy-nLC, Proxeon-Thermo) or using a MALDI-TOF/TOF mass spectrometer (4800 MALDI TOF/TOF, ABi Sciex). For the nano-LC/MS approach, peptides were separated on a reversed-phase column (Aquasil C18 100 mm, inner diameter 75 μm, 5 μm, PicoFRITTM column from New Objective) and eluted with a linear gradient of acetonitrile (0.1% formic acid (0–40% in 55 min) at a flow rate of 0.3 μl/min). Electrospray voltage was 1.40 kV, and capillary temperature was 200 °C. Peptides were detected in the positive ion mode using a mass range of 300–2000, and MS/MS of the top five peaks in each segment were acquired in a data-dependent manner. Peptides were identified by searching public databases (NCBInr, November 2012) and using the following Mascot MS/MS ion search mode parameters: peptide tolerance, 1.2 Da; MS/MS tolerance, 0.8 Da; methionine oxidation and tyrosine nitration as variable modifications. The significance limit for peptide identification was set at p < 0.05. MALDI TOF/TOF experiments were performed in the positive ion reflector mode, and mass spectra were externally calibrated using a mixture of peptide standards (Applied Biosystems). Peptide sequences were confirmed by MS/MS analysis of selected ions.
Nitrotyrosine Quantification
For total 3-nitrotyrosine quantification, peroxynitrite in a wide concentration range (0.05–3000 μm) was added to Fe-SODs (8–16 μm) in sodium phosphate buffer (200 mm, pH 8–5.8). NO2− removal from the samples after peroxynitrite treatment was carried out by two subsequent protein precipitation steps with acetonitrile (1 volume) for 40 min at 4 °C following centrifugation at 14,000 × g for 40 min at 4 °C. Proteins were resuspended in nanopure water (500 μl) containing the following internal standards: universal labeled tyrosine ([U-13C915N1]Tyr, 20 nmol); [NO2-13C6]Tyr (500 pmol); and [13C6]Tyr (35). Stable isotopically labeled precursors were used as internal standards for the quantification of total protein Tyr and NO2-Tyr ([U-13C915N1]Tyr and [NO2-3C6]Tyr, respectively) together with [13C6]Tyr to monitor for potential artifactual generation of NO2-Tyr during sample processing. Proteins were hydrolyzed overnight at 116 °C in HCl (6 n) using vacuum hydrolysis tubes (Pierce). After hydrolysis, samples were evaporated, and pellet was resuspended (35) in 200 μl of formic acid (0.1%, v/v) and analyzed by LC-MS/MS (Qtrap Applied Biosystems) as described previously (36).
Crystal Structure of T. cruzi Mitochondrial Fe-SODA
The crystal structure of the mitochondrial superoxide dismutase from T. cruzi (Fe-SODA) was determined at 2.23 Å resolution in the Protein Crystallography Facility of the Institut Pasteur de Montevideo (Protein Data Bank entry 4DVH). Fe-SODA crystals were grown in a hanging drop vapor diffusion setup, using the protein at 2.33 mg/ml in 50 mm sodium phosphate, pH 7.8. Protein (1 μl) was mixed in equal parts with mother liquor (1-ml reservoir), Tris-HCl (0.1 m, pH 8.5), and PEG 4000 (30%, w/v) and incubated at 291 K. Monoclinic crystals grew in a few days, were cryoprotected with mother liquor containing 25% (v/v) glycerol, and flash-frozen in liquid N2 until data collection. A complete data set was collected at 100 K with a copper rotating anode (Micromax-HF, Rigaku) and a Mar345DTB (Mar Research) image plate detector. Data were processed with MOSFLM (37) and Scala (38). The crystal structure was solved by molecular replacement with AMoRe (39) using the monomer of E. coli Fe-SOD (Protein Data Bank entry 2NYB) as a search probe. Restrained refinement was done with Buster-TNT (40) (BUSTER version 2.10.0, Global Phasing Ltd., Cambridge, UK), including a TLS model with one body per chain (one dimer in the asymmetric unit). Refinement was performed in iteration with manual model building using Coot (41). Structure validation was done with Molprobity (42).
Molecular Dynamics (MD) Simulations
Starting Structures
The crystal structure of the T. cruzi cytosolic wild type (WT) Fe-SODB was downloaded from the Protein Data Bank (Protein Data Bank entry 2GPC). Based on the WT structure, double mutant N187D/K189E was built in silico by replacing the corresponding residue side chains. Hydrogens were added using the Tleap module of the AMBER program package, considering standard protonation states for all titratable residues at physiological pH (Asp and Glu negatively charged, Lys and Arg positively charged, all of the rest neutral). Additionally, the tyrosyl radical (Tyr35-O•) and negatively charged cysteine (Cys-S−, thiolate)-containing system (tyrosyl radical-thiolate cytosolic Fe-SODB) was built by removing the corresponding phenolic and thiol hydrogens and changing the corresponding classical residue parameters (see below).
MD Simulation Parameters
Simulation of WT, mutant, and tyrosyl radical-thiolate cytosolic Fe-SODB homodimers was performed in an explicit solvent box (of ∼513-nm3 volume and containing ∼15,000 water molecules) using the TIP3P water model. Parameters for all standard residues were taken from the PARM99 force field (43). Classical parameters for Tyr-O• and Cys83-S− were taken from our previous work (44). Parameters for the Fe-SOD active site (the iron ion and its first sphere coordinating residues) were developed in the same way as those for Mn-SOD (20). All simulations were performed using the periodic boundary conditions approximation and the particle mesh Ewald summation method with a grid spacing of 1 Å for treating long range electrostatic interactions, whereas a direct cut-off distance of 8 Å was used for direct interactions. The Shake method was used to constrain the hydrogen atoms at their equilibrium distance, allowing the use of a 2-fs time step. The Berendsen thermostat was used to keep the temperature constant at 300 K (45). All MD simulations were performed with the AMBER program package (46). The equilibration protocol for all peptides consisted of slowly heating the optimized structures from 0 to 300 K during 0.1 ns while the volume of the system was kept constant (NVT). Next, a 0.2-ns-long density equilibrium simulation was performed using an NPT ensemble. Production simulations consisted of 10-ns-long NPT MD simulations for wild type and mutant cytosolic Fe-SODB and 20-ns-long NPT MD simulation for radical-thiolate cytosolic Fe-SODB. The stability of all simulations was assessed through root mean square deviation and root mean square fluctuation analysis (data not shown).
Analysis of the Intramolecular Electron Transfer (IET) Pathway(s)
IET pathways along the protein matrix were determined using the pathways algorithm developed previously (47, 48), with a specific set of parameters developed in our group to consider the Fe-SOD active site as well as recent observations of aromatic residues acting as stepping stones in long range electron transfer reactions (49–52). The method has been used successfully in previous work from our group (44, 51, 53). Pathways were computed between Cys83-S− and Tyr35-O• atoms located orbitals for 100 snapshots taken from the corresponding MD simulation. To estimate the pKa of the different Cys residues in WT and mutant cytosolic and mitochondrial Fe-SODs, the propKa pKa estimation software was used (54–57).
Parasites
T. cruzi epimastigotes (CL-Brener) were cultured at 28 °C in brain-heart infusion medium as described previously (12). T. cruzi CL-Brener (pTcINDEX-9E10) (Invitrogen) Fe-SODA overexpressers containing the bacteriophage T7 RNA polymerase, tetracycline (Tet) repressor genes, and the epitope (9E10) derived from the human c-Myc protein added to the C terminus of Fe-SODA were maintained as described previously in brain-heart infusion medium containing G418 and hygromycin (100 μg/ml) (12, 58). To induce Fe-SODA expression, epimastigotes were cultured in medium supplemented with tetracycline (2 μg/ml) for 3 days as described previously (12).
Nitroxidative Treatment of T. cruzi Epimastigotes
T. cruzi Fe-SODA overexpressers were used in order to identify, at the cellular level, peroxynitrite-dependent T. cruzi Fe-SOD nitroxidative modifications. For this, Fe-SOD parasites (5 × 108 cells/ml in Dulbecco's PBS, pH 7.4) were treated with the mitochondrial complex III inhibitor antimycin A (AA; 5 μm) in the presence of NOC-12 (5 mm, t½ = 100 min at pH 7.4), SIN-1 (5 mm), or peroxynitrite (300 μm in three subsequent additions of 100 μm) for 3 h at 28 °C under stirring (59). After treatment, parasites were washed three times in Dulbecco's PBS and resuspended in Tris-HCl (40 mm, pH 7.4) containing urea (7 m), thiourea (2 m), CHAPS (4%, w/v), PMSF (1 mm), and DTT (1%, w/v). Two-dimensional electrophoresis was performed with commercially available IPG strips (7 cm, non-linear 3–10, GE Healthcare) for the first dimensional separation. Protein samples (390 μg) were dissolved in rehydration solution (7 m urea, 2 m thiourea, 2% (w/v) CHAPS; 0.5% IPG buffer 3–10 (GE Healthcare), 0.002% (w/v) bromphenol blue, 17 mm DTT) and loaded onto IPG strips by passive rehydration during 12 h at room temperature. The isoelectric focusing was done in an IPGphor unit (GE Healthcare) employing the following voltage profile: linear increase to 150 V in 30 min; constant phase of 300 V for 2 h; constant phase of 500 V for 1 h; constant phase of 1000 V for 1 h; linear increase to 5000 V in 30 min; and a final constant phase of 5000 V for 6 h to reach a total of 33.6 kV-h. Prior to running the second dimension, IPG strips were incubated for 15 min, first in equilibration buffer (6 m urea, 75 mm Tris-HCl, pH 8.8, 29.3% (v/v) glycerol, 2% (w/v) SDS, 0.002% (w/v) bromphenol blue) supplemented with 10 mg/ml DTT for protein reduction and finally in equilibration buffer supplemented with 25 mg/ml iodoacetamide for protein thiol alkylation. The second dimensional separation was performed in 15% SDS-PAGE using an SE 260 minivertical gel electrophoresis unit (GE Healthcare). Proteins were transferred to nitrocellulose membranes (40 mA, overnight) and blocked with 5% milk in PBS for 1 h. Membranes were then probed with either rabbit polyclonal anti-T. cruzi Fe-SODA or Fe-SODB (1:2000 dilution), and immunoreactive proteins were visualized as described above.
Immunoprecipitation of T. cruzi Fe-SODA
T. cruzi Fe-SODA overexpressers were incubated under different nitroxidative stress conditions described above. After treatment, parasites (1 × 109) were washed three times in Dulbecco's PBS and lysed in 250 μl of Tris-HCl (20 mm, pH 7.5) containing 150 mm NaCl, 1 mm EGTA, 1% Nonidet P-40, 1% sodium deoxycholate, and protease inhibitors (Sigma). Following incubation for 15 min in ice, samples were centrifuged at 4 °C and 13,000 × g, and supernatants were incubated under orbital shaking at 4 °C in the presence of monoclonal anti-c-Myc antibody (30 μg; Santa Cruz Biotechnology, Inc.) and Protein A/G Plus-agarose (20 μl; Santa Cruz Biotechnology). After incubation, samples were centrifuged at 800 × g, and agarose beads were washed four times in lysis buffer. Proteins bound to agarose were released by incubating the beads with 50 μl of Laemmli sample buffer (with 5% (v/v) β-mercaptoethanol) for 5 min at 100 °C. Proteins were separated on 15% SDS-polyacrylamide gels, transferred to nitrocellulose, and probed against anti-c-Myc (9E10) and anti-NO2-Tyr antibodies. Inmunoprecipitated proteins were developed as above using the anti-mouse IRE-680 (for Fe-SODA protein) and anti-rabbit IRE-800 (for 3-nitrotyrosine-containing proteins).
Data Analysis
All data are given as means ± S.D. unless otherwise noted, and p < 0.05 was considered significant. Means were compared using Student's t test.
RESULTS
Purification and Characterization of Fe-SODA and Fe-SODB
The recombinant T. cruzi Fe-SODA and Fe-SODB containing one atom of iron per monomer were purified to homogeneity as active enzymes homodimers as established by SDS-gel electrophoresis and gel filtration chromatography analysis. The apparent monomer molecular mass was 23 and 22.6 kDa for Fe-SODA and Fe-SODB, respectively (Fig. 1, A and B), consistent with the predicted values according to their primary structures. All of the enzyme preparations used in this study, including the mutants generated, were of high purity (>99%) with specific activities of 2647 and 3551 units/mg for Fe-SODA and Fe-SODB, respectively. These specific activities compare well with previously reported T. brucei Fe-SODs (60).
FIGURE 1.
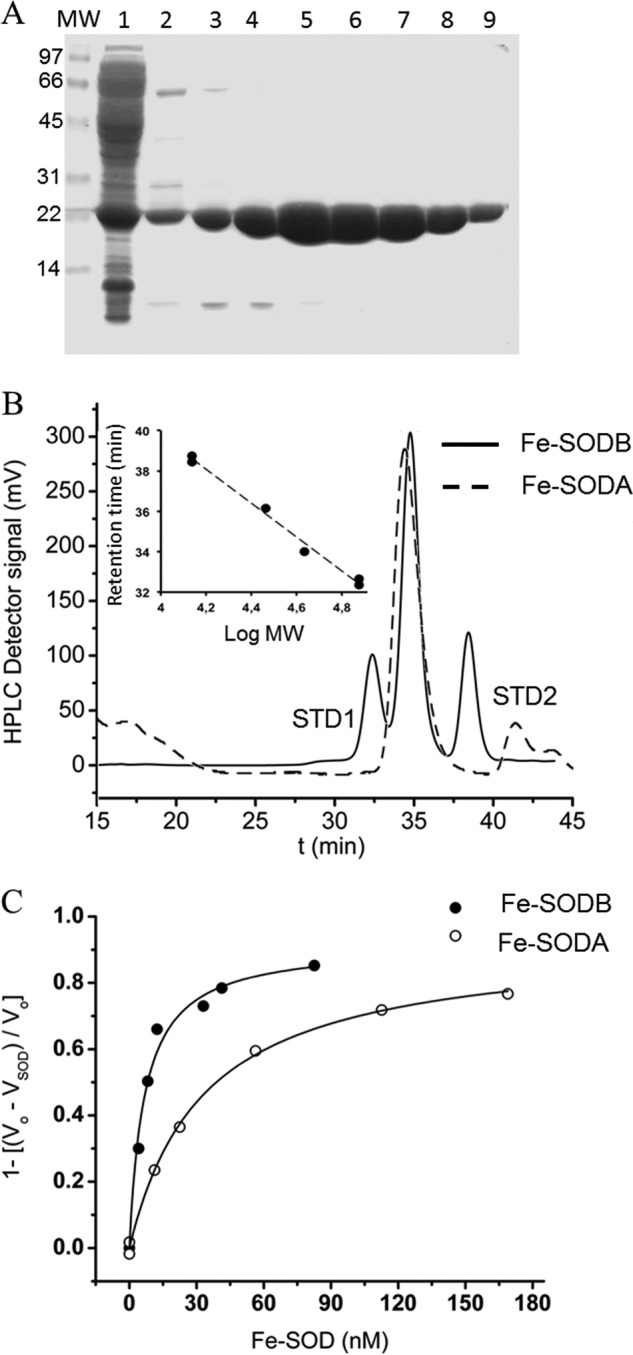
Purification of recombinant T. cruzi Fe-SODs and rate constants with O2⨪. A, SDS-gel electrophoresis (15% (w/v) under reducing conditions) of the eluted fractions from the Ni2+ affinity chromatography of Fe-SODA purification. Lane 1, soluble extract after isopropyl-β-d-thiogalactopyranoside (0.8 mm) induction; lanes 2–9, fractions eluted from the Ni2+ affinity chromatography using a linear imidazole gradient (10–500 mm). B, Fe-SODA pure fractions (lanes 5–9) and fractions from Fe-SODB purification were pooled and analyzed by gel filtration chromatography in Superdex 200 (solid line, Fe-SODB; dashed line, Fe-SODA). Inset, calibration of the Superdex S200 gel filtration column with molecular mass standards (13,000–75,000 Da). C, rate constants were determined by competition kinetics method. O2⨪-dependent reduction of ferricytochrome c (20 μm) was monitored at 550 nm at 37 °C in the absence and presence of T. cruzi Fe-SODA or Fe-SODB. Rate constants were calculated as described under “Experimental Procedures” at pH 7.8 and 37 °C. Inhibition fraction (1 − (v0 − vSOD/v0)) was plotted as a function of Fe-SOD concentrations and fitted to a rectangular hyperbolic function in order to calculate [Fe-SOD]50.
Determination of the Rate Constant with O2⨪ and Kinetics of the Reaction of T. cruzi Fe-SODs with Peroxynitrite
A competition kinetic approach was used to determine the second order rate constant of the reaction of T. cruzi Fe-SODs with O2⨪, and it was found to be 4.5 ± 1.8 × 108 m−1 s−1 for Fe-SODA and 7.6 ± 1.5 × 108 m−1 s−1 for Fe-SODB in 50 mm sodium phosphate, pH 7.8 (ionic strength (μ) = 0.14), in agreement with values obtained for other iron-containing SODs (Fig. 1C and Table 1) (61).
TABLE 1.
T. cruzi Fe-SOD rate constants with O2⨪ and peroxynitrite
Rate constants with O2⨪ were determined by the kinetic competition method as described in the legend to Fig. 1 at pH 7.8 and 37 °C. Rate constants with ONOO− were measured by an initial rate approach. ONOO− decomposition was measured at 302 nm, pH 7.4, and 37 °C as described in the legend to Fig. 2. ND, not determined.
| Enzyme preparation |
k |
|
|---|---|---|
| O2⨪ | ONOO− | |
| m−1 s−1 | ||
| Fe-SODA | 4.5 ± 1.8 × 108 | 4.6 ± 0.2 × 104 |
| Fe-SODB | 7.6 ± 1.5 × 108 | 4.3 ± 0.4 × 104 |
| NEM-treated Fe-SODB | ND | 4.3 ± 0.2 × 104 |
The rate constant for peroxynitrite reaction with T. cruzi Fe-SODs was measured following the decay of peroxynitrite at increasing concentrations of enzyme (0–15 μm), as described previously (18). A linear correlation was obtained after plotting the initial velocity of peroxynitrite decomposition (v0) (Fig. 2, inset) as a function of Fe-SOD concentrations (Fig. 2), with the y axis intercept reflecting the proton-catalyzed decomposition of peroxynitrite (as described under “Experimental Procedures”). By dividing the slope of the plot over peroxynitrite concentration, the second order rate constant was determined as 4.6 ± 0.2 × 104 m−1 s−1 for Fe-SODA and 4.3 ± 0.4 × 104 m−1 s−1 for Fe-SODB per monomer at pH 7.4 and 37 °C, in agreement with that obtained for other SODs (Table 1) (18, 62, 63). When the rate constant was determined in the presence of Fe-SOD in which the solvent-accessible cysteines were blocked by NEM alkylation, the same value was obtained (Table 1), indicating a negligible contribution of cysteine residues (kONOO− = 6 × 103 m−1 s−1 for free cysteine at pH 7.4 and 37 °C) to the global rate constant observed. Because the reactivity of cysteine toward peroxynitrite is typically the largest among all amino acids (28), the data support the existence of a fast direct reaction of peroxynitrite with the iron atom of the Fe-SODs.
FIGURE 2.
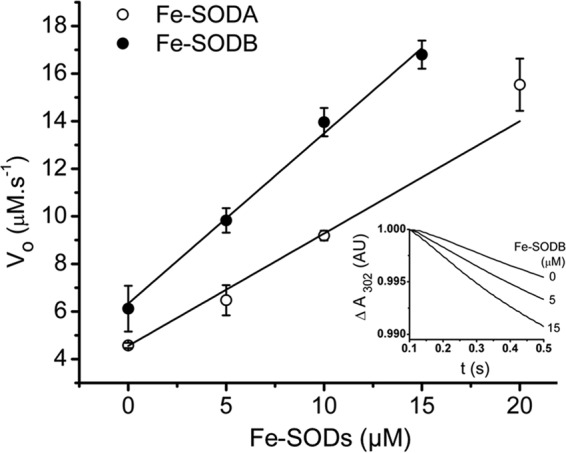
T. cruzi Fe-SODs rate constants with peroxynitrite. The kinetics of peroxynitrite decomposition (10 and 17 μm peroxynitrite for Fe-SODA and Fe-SODB, respectively) was monitored at 302 nm at pH 7.4 and 37 °C in the presence of Fe-SOD (0–15 μm) in a stopped-flow spectrophotometer. An initial rate approach (first 0–0.15 s) was used to analyze the data as described under “Experimental Procedures.” Reported values are the average of at least seven separate determinations. Inset, peroxynitrite (ONOO−) decomposition in the presence of Fe-SODB (0, 5, and 15 μm) measured at pH 7.4 and 37 °C using a stopped-flow spectrophotometer. AU, absorbance units. Error bars, S.D.
Peroxynitrite-dependent Inactivation and Nitration of T. cruzi Fe-SODs
Peroxynitrite addition (0–1200 μm) to purified Fe-SODA and Fe-SODB (8 μm) at pH 7.4 led to a dose-dependent inhibition of the superoxide dismutase activity although with quite disparate susceptibilities (Fig. 3A). Indeed, Fe-SODB was largely resistant to peroxynitrite-mediated inactivation, in contrast with the highly sensitive Fe-SODA, an unexpected result taking into account the structural similarities between both isoforms (3, 4). Enzyme inactivation was associated to protein tyrosine nitration, with the higher nitration yields in Fe-SODA correlating well with the level of inactivation observed (Fig. 3B).
FIGURE 3.

Peroxynitrite-mediated inactivation and nitration of T. cruzi Fe-SODA and Fe-SODB. A, inactivation of Fe-SODs. Peroxynitrite (ONOO−, 0–1200 μm) was added to the purified enzymes (8 μm) at pH 7.4, and residual Fe-SOD activity was measured. Activity is expressed as a percentage relative to the control enzyme incubated in the absence of peroxynitrite (100% activity). B, nitration of Fe-SODs. Immunochemical detection of protein 3-nitrotyrosine (black) was performed after peroxynitrite (0–2000 μm) exposure to either Fe-SODA or Fe-SODB (8 μm) using the Immun-StarTM chemiluminescence kit. Fe-SOD loading was evaluated by Ponceau-S protein staining (pink). Error bars, S.E.
Fe-SODA and Fe-SODB B contain seven and nine tyrosine residues respectively, some of which are solvent-exposed and others of which are buried in the protein structure. Notably, Tyr35 is part of the active site and the closest to the iron metal centers. Thus, in principle, tyrosine nitration could affect non-critical tyrosines as well as Tyr35, with oxidative modification of the latter being responsible for enzyme inactivation. Under the experimental conditions of Fig. 3, in which biologically relevant micromolar concentrations of enzyme were used, there are two possible mechanisms by which peroxynitrite can generate nitrating intermediates to mediate Fe-SOD tyrosine nitration (64, 65): (i) the proton-catalyzed homolysis of peroxynitrous acid (ONOOH, pKa = 6.8) to yield nitrogen dioxide (•NO2) and hydroxyl radical (•OH) or (ii) the reaction of peroxynitrite anion with the iron center to yield the oxo-Fe complex and •NO2. To define which of the nitration mechanisms was better coupled to enzyme inactivation, we performed peroxynitrite exposures at different pH values and in the presence of CO2 (Fig. 4), as in previous reports (66). Treatment of T. cruzi Fe-SODA with peroxynitrite (150 μm) at acidic pH values (where most peroxynitrite decays via homolysis) led to mild enzyme inactivation, whereas protein nitration was high; on the contrary, at basic pH values, enzyme inactivation was more pronounced, but the yields of protein nitration were less (Fig. 4, A and B). Thus, overall, these data indicate that the reactive species leading to enzyme inactivation is peroxynitrite anion via its reaction with the metal center and the site-specific nitration of active site tyrosine (see below). In support of this contention, the presence of bicarbonate (25 mm, 1.3 mm CO2) protected Fe-SODA from peroxynitrite-mediated inactivation (Fig. 4C), as was previously seen for E. coli Mn-SOD (18). The reaction of peroxynitrite anion (ONOO−) with CO2 (k = 3 × 104 m−1 s−1 at pH 7.4 and 37 °C) yields the nitrosoperoxocarboxylate adduct (ONOOCO2−) that rapidly (<1 μs) decomposes by homolysis, resulting in the generation of •NO2 and CO3⨪ radicals (67, 68). Thus, CO2 competes with the metal of Fe-SOD for ONOO−, yielding the secondary derived radicals that may react with solvent-accessible non-critical protein tyrosine residues without causing enzyme inactivation (19, 69, 70).
FIGURE 4.
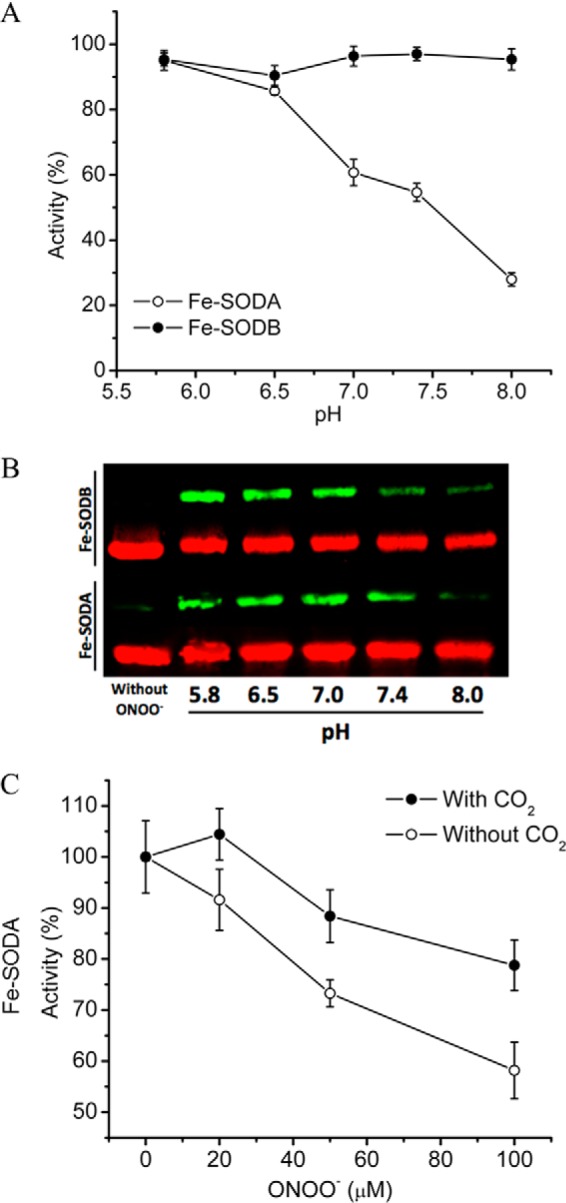
Effects of pH and CO2 on the peroxynitrite-dependent inactivation of T. cruzi Fe-SODs. A, pH-dependent peroxynitrite-mediated inactivation. Peroxynitrite (150 μm) was added to Fe-SODA (empty circles) and Fe-SODB (filled circles) (8 μm) in sodium phosphate buffer (100 mm) at different pH values (5.5–8.0), and residual Fe-SOD activity was measured. Activity is expressed relative to the native enzyme incubated in the absence of peroxynitrite (100% activity) at the indicated pH values. B, pH-dependent peroxynitrite-mediated nitration. Immunochemical detection of 3-nitrotyrosine was performed after peroxynitrite (150 μm) exposure to either Fe-SODA or Fe-SODB (8 μm) in sodium phosphate buffer (100 mm) at different pH values using specific anti-NO2-Tyr antibodies (green). Equal loading of T. cruzi Fe-SOD samples were evaluated by specific Fe-SODA and Fe-SODB antibodies (red). C, effect of CO2 in the peroxynitrite reaction with Fe-SODA. Peroxynitrite (0–100 μm) was added to Fe-SODA (8 μm) in sodium phosphate buffer 100 mm, pH 7.4, in the presence (filled circles) or absence (empty circles) of bicarbonate (24 mm; CO2 = 1.3 mm)), and residual SOD activity was measured. Activity is expressed relative to the non-treated enzyme in the presence or absence of bicarbonate. Error bars, S.E.
Glutathione (10 mm) known to react with peroxynitrite (k = 1.35 × 103 m−1 s−1 (71)) and •NO2 (72) and uric acid (100 μm), a known •OH and •NO2 scavenger (k = 7.2 × 109 and 1.9 × 107 m−1 s−1, respectively) (73, 74)) fully protected Fe-SODA from enzyme inactivation and nitration (not shown).
Peroxynitrite-dependent Inactivation of T. cruzi Fe-SOD by the Selective Nitration of Tyr35
In order to confirm the preferential nitration of Tyr35 after peroxynitrite treatment (0–300 μm) of T. cruzi Fe-SODs (8 μm), control and treated enzymes were separated by two-dimensional gel electrophoresis, and protein spots were analyzed by nano-LC-nano-SI-MS. One major spot for both Fe-SODA and Fe-SODB and other minor ones were identified in the native enzymes, indicating post-translational modifications that affect the protein isoelectric point. In the case of the peroxynitrite-treated Fe-SODs, the two-dimensional gels revealed the generation of several more acidic spots in both Fe-SODs with a more drastic shift in the case Fe-SODA, in agreement with its higher susceptibility to peroxynitrite (Fig. 5A). Peroxynitrite-treated proteins are expected to have more acidic pI values due to the change of the pKa of 3-nitrotyrosine (7.3) with respect to Tyr (10.3) (with the consequent gain of a negative charge) (21, 75), among other possible oxidative modifications. Mass spectrometry analysis of the major spot (pI = 7.5) of Fe-SODB generated after peroxynitrite treatment (150 μm) revealed the presence of only one nitrated peptide (m/z = 790) corresponding to the triple charged molecular ion, assigned to the sequence 31HHQG35YVTKLNAAAQTNSALATK52, which contained Tyr35 (Fig. 5B), revealing the preferential nitration of this residue under these experimental conditions. At higher concentrations of peroxynitrite (600–1000 μm), other peptides containing Tyr177 (solvent-exposed) were also found nitrated (Table 2). In the case of Fe-SODA, the critical Tyr (Tyr36 in the crystal structure; see below) was also preferentially nitrated after exposure to 100 μm peroxynitrite, being detected on a dinitrated peptide containing Tyr36 and Tyr29 (Table 2).
FIGURE 5.
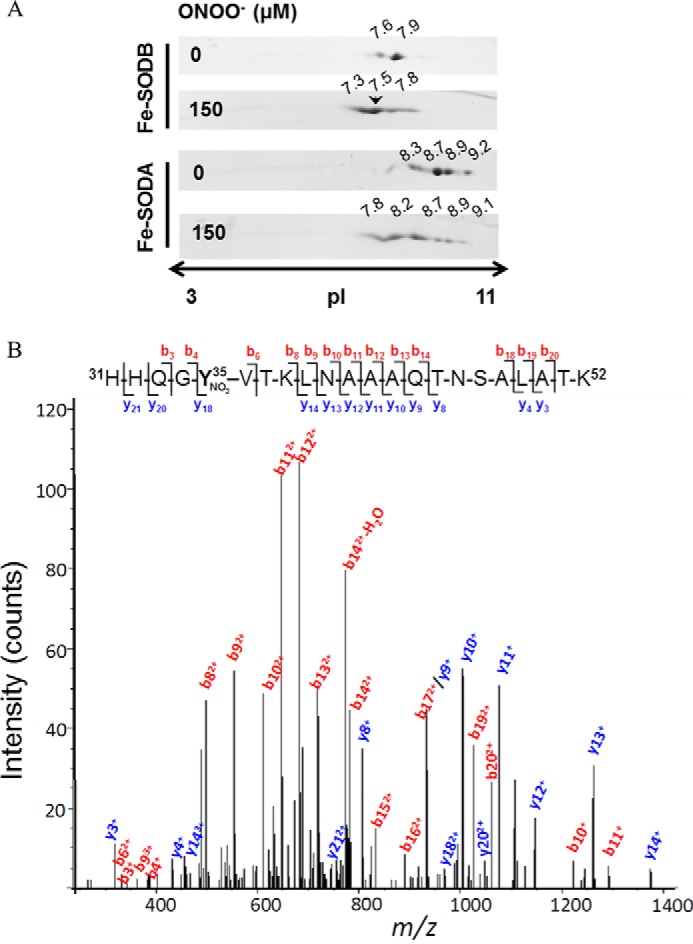
Peptide mapping of T. cruzi Fe-SODs after peroxynitrite treatment. A, T. cruzi Fe-SODs (8 μm) were treated with peroxynitrite (150 μm) in sodium phosphate buffer (0.2 m) at pH 7.4 and 25 °C. Two-dimensional gel electrophoresis was performed as described under “Experimental Procedures.” The arrowhead shows the selected Fe-SODB spot of pI 7.5 analyzed by mass spectrometry. B, MS/MS spectrum of triply charged ion at m/z 790.6 (MH+ 2369.7, retention time = 30.3 min) from a tryptic digestion of peroxynitrite-treated Fe-SODB spot (pI = 7.5, indicated with an arrow in A). The major N-terminal (b, red-labeled) and C-terminal (y, blue-labeled) fragment ions that allowed the sequence 31–52 assignment that includes a nitrated tyrosine residue (mascot ion score = 61; p < 0.05) are shown. Inset, amino acid sequence of peptide 31–52, indicating major b and y ions detected by full-scan MS/MS.
TABLE 2.
Peroxynitrite-modified tyrosine-containing peptides in T. cruzi Fe-SODs detected by MALDI-TOF mass spectrometry
MALDI-TOF/TOF mass spectrometry analysis was carried out in control and peroxynitrite-treated purified recombinant T. cruzi Fe-SODs. Fe-SODB and Fe-SODA were treated with increasing peroxynitrite concentrations (0–1000 μm), and enzyme activity was assayed. After treatment, control and treated enzymes were subjected to SDS-gel electrophoresis, and protein bands were in-gel digested with trypsin (sequence grade). Digested proteins were analyzed by MALDI-TOF mass spectrometry, and peptides were searched in the MASCOT database. Ox, oxidation; Mo, monoisotopic. Numbering corresponds to the position observed in the crystal structure of both T. cruzi Fe-SODs.
| Peroxynitrite | Mo Observed mass ((M + H)+) | Mo Theoretical mass ((M + H)+) | Assigned sequence |
|---|---|---|---|
| μm | Da | Da | |
| Fe-SODB | |||
| 150 | 2127.09 | 2127.04 | 21K.QQVTLH28YDKHHQG35YVTK38 + Nitro(Y) |
| 600 | 2127.00 | 2127.04 | 21K.QQVTLH28YDKHHQG35YVTK38 + Nitro(Y) |
| 1014.46 | 1014.47 | 30K.HHQG35YVTK38 + Nitro(Y) | |
| 2156.00 | 2156.03 | 172K.NDRAA177YVQTFWNVVNWK188 + Nitro(Y) | |
| 1770.82 | 1770.86 | 175R.AA177YVQTFWNVVNWK188 + Nitro(Y) | |
| 2269.10 | 2269.11 | 175R.AA177YVQTFWNVVNWKNVER192 + Nitro(Y) | |
| 1000 | 2127.08 | 2127.04 | 21K.QQVTLH28YDKHHQG35YVTK38 + Nitro(Y) |
| 1770.88 | 1770.86 | 175R.AA177YVQTFWNVVNWK188 + Nitro(Y) | |
| 2269.17 | 2269.11 | 175R.AA177YVQTFWNVVNWKNVER192 + Nitro(Y) | |
| Fe-SODA | |||
| 100 | 3095.54 | 3095.50 | 14DGCAPVLSPRQLELH29YTKHHKA36YVDK40 + 2Nitro(Y) |
| 2096.02 | 2096.06 | 155GLRPVFTVDVWEHA169Y170YK171 + Ox(Y) | |
| 3337.64 | 3337.66 | 177RVD180YLKEIWTIVDWEFVSRT127YEQAMK132 + Ox(Y) + Ox(M) | |
| 150 | 2868.39 | 2868.39 | 40LNALAGAT48YDGKT53MEDIIVALANDSEK66 + Nitro(Y) |
| 2884.37 | 2884.39 | 40LNALAGAT48YDGKT53MEDIIVALANDSEK66 + Nitro(Y); Ox(M) | |
| 2096.03 | 2096.06 | 155GLRPVFTVDVWEHA169Y170YK171 + Ox(Y) | |
| 2125.03 | 2125.04 | 155GLRPVFTVDVWEHA169Y170YK171 + Nitro(Y) | |
| 2112.03 | 2112.06 | 155GLRPVFTVDVWEHA169Y170YK171 + 2Ox(Y) | |
| 2314.16 | 2314.17 | 178VD180YLKEIWTIVDWEFVSR195 + Ox(Y) | |
Total 3-nitrotyrosine quantification was performed for both T. cruzi Fe-SODs isoforms after peroxynitrite treatment (0–3000 and 0–100 μm peroxynitrite for Fe-SODB and Fe-SODA, respectively) in order to accurately correlate enzyme inactivation with Tyr nitration. Control and peroxynitrite-treated Fe-SODB were analyzed, and the results are shown in Table 3. Results are expressed as the ratio of 3-nitrotyrosine/SOD monomer, and theoretical 3-nitrotyrosine values were calculated assuming enzyme inactivation to be due to the sole nitration of one Tyr per Fe-SOD monomer (i.e. Tyr35 in Fe-SODB), and thus 100% inactivation must yield a 3-nitrotyrosine/SOD ratio of 1. For Fe-SODB treated with peroxynitrite at pH 8, a linear correlation between the percentage of enzyme inactivation and total 3-nitrotyrosine quantification was observed, clearly indicating that peroxynitrite-mediated enzyme inactivation is due to the selective nitration of Tyr35 (although the solvent-accessible Tyr177 was also found nitrated but had a small contribution to total 3-nitrotyrosine). In the case of Fe-SODA, a larger yield of tyrosine nitration and the extent of enzyme inactivation was observed at a smaller peroxynitrite concentration at pH 8 in comparison with Fe-SODB (Table 3). Under this condition, the quantitated experimental value of 3-nitrotyrosine by LC-MS/MS was somewhat less than that predicted from the loss of activity, which may be due to the fact that other Tyr oxidative modifications were observed by MALDI-TOF/MS (Table 2) and may contribute to inactivation, a hypothesis that needs further confirmation. Interestingly, peroxynitrite treatment of Fe-SODA at pH 5.8 revealed experimental 3-nitrotyrosine quantitation higher than the predicted one from the observed 16% of enzyme inactivation (Table 3), in good agreement with the nitration of non-critical solvent-exposed tyrosines via •NO2 and •OH formed in the bulk from ONOOH homolysis.
TABLE 3.
3-Nitrotyrosine quantification of peroxynitrite-treated T. cruzi Fe-SODs by LC-MS/MS
After peroxynitrite treatment (0–1500 μm), T. cruzi Fe-SODs were hydrolyzed overnight at 116 °C in HCl (6 n), and pellet was resuspended in formic acid (0.1%, v/v) and analyzed by LC-MS/MS as described under “Experimental Procedures.” Theoretical NO2-Tyr was calculated assuming that enzyme inactivation is due to the selective nitration of only one Tyr per enzyme monomer (i.e. Tyr35 in Fe-SODB or Tyr36 in Fe-SODA). Results are expressed as NO2-Tyr/SOD monomer.
| ONOOH | Fe-SOD activity | Experimental nitrotyrosine/SOD monomer | Theoretical nitrotyrosine/SOD monomer | |
|---|---|---|---|---|
| μm | % | |||
| Fe-SODB (pH 8) | 0 | 100 | 0 | 0 |
| 500 | 82.8 | 0.18 | 0.17 | |
| 1500 | 74.5 | 0.29 | 0.26 | |
| 3000 | 35 | 0.66 | 0.65 | |
| Fe-SODA | ||||
| pH 8 | 0 | 100 | 0.03 | 0 |
| 100 | 56 | 0.26 | 0.43 | |
| pH 5.8 | 100 | 84 | 0.33 | 0.16 |
Crystal Structure of T. cruzi Mitochondrial Fe-SODA and Differences from Cytosolic Fe-SODB
In order to understand the contrasting behaviors of both Fe-SOD isoforms toward peroxynitrite-mediated inactivation, we solved the crystal structure of the mitochondrial T. cruzi Fe-SODA (Table 4) and compared it with the previously reported structure of cytosolic Fe-SODB (Protein Data Bank entry 2GPC) (76). The Fe-SODA structure revealed a tightly bound dimer in the asymmetric unit (Fig. 6A), the two protomers related by a strong non-crystallographic 2-fold axis. The total buried area due to dimerization is high (∼1900 Å2), consistent with the dimeric behavior of Fe-SODA in solution. The overall structure of Fe-SODA is similar to those of previously solved iron SODs (76, 77). The nomenclature of secondary structure elements is depicted on one of the Fe-SODA monomers in Fig. 6B. Each monomer binds one iron cation within the metal-binding pocket between the two domains. The metal is pentacoordinated to His28-Nϵ2 (in helix α1), His79-Nϵ2 (α3), Asp163-Oδ2 (β3), His167-Nϵ2 (in the helical linker loop connecting β3 with α6), and a water molecule (HOH7 in chain A and HOH6 in chain B). As in other iron SODs, the metal coordination geometry is trigonal bipyramidal, with His79, His167, and Asp163 as equatorial ligands and the His28 and water oxygen axial. T. cruzi mitochondrial Fe-SODA is structurally very similar to cytosolic Fe-SODB with a root mean square deviation of 0.75 Å, superimposing one monomer (843 atoms) and 1.14 Å aligning the whole dimer (1843 atoms). In particular, the identities and positions of the iron-binding residues in the active sites are absolutely conserved, including the axial water molecules. In the folded state of Fe-SOD, as represented by the crystal structure, the metal cation is seen completely buried within its binding pocket, with no connection to the solvent-accessible surface of the protein. An outer, second shell of residues with respect to the ones acting as direct iron ligands can be identified, interacting with the coordinating shell, and/or delimiting bulk solvent channels toward the metal pocket (Fig. 6C). These second shell residues are also positioned in the same way in both Fe-SOD isoforms, and they include critical Tyr35 (78) at 5.6 Å from the metal center. Further extending our structural analysis, there are differences between Fe-SODA and Fe-SODB, which concern Cys and Trp amino acids. Among the four cysteine residues present in Fe-SODA (Cys16, Cys85, Cys131, and Cys150) and the three in Fe-SODB (Cys83 (which is observed in its oxidized sulfonic state in the structure), Cys146, and Cys159), only one is conserved; Cys150 in Fe-SODA is structurally equivalent to Cys146 in Fe-SODB (Fig. 7). The four Cys side chains in mitochondrial Fe-SODA are solvent-accessible in the crystal structure, in agreement with the data obtained by sulfhydryl titration (four DTNB-reactive thiols per monomer). In contrast, Fe-SODB displays only two of its Cys thiols accessible to bulk solvent (Cys83 and Cys146). Fe-SODB Cys159 is instead buried within the folded protein core, at 9.9 Å from the iron atom, again consistent with the thiol titration (2 DTNB-reactive thiols/monomer). Analysis of the Trp residues reveals that, apart from a conserved core of five tryptophans in both isoforms, key differences are uncovered in a critical region: the funnel entrance toward the metal-binding pocket, in the interface between helices α1 and α3, including also the rather long initial N-terminal loop that precedes α1. In the cytosolic Fe-SODB structure, Trp9 and Trp79 (Fig. 7B) are observed located at 11.6 and 6.6 Å, respectively, from the active site iron. These two residues are substituted by phenylalanines in the mitochondrial Fe-SODA. Instead, Fe-SODA locates Trp12 (substituted by Tyr11 in Fe-SODB) within the N-terminal loop but pointing its side chain away with respect to the entrance channel. Overall, the structural differences in Cys and Trp residues comparing the two T. cruzi Fe-SOD isoforms could explain their different susceptibilities to peroxynitrite-mediated inactivation and nitration. The structural data are thus consistent with the existence of an IET mechanism in Fe-SODB involving Cys, Trp and the active site tyrosine residue, as observed previously for other proteins (33, 79, 80).
TABLE 4.
Crystal structure of mitochondrial Fe-SODA
Shown are data collection and refinement statistics.
| Space group | P21 |
| Protein molecules per asymmetric unit | 2 |
| Solvent content (%) | 43.4 |
| Wavelength (Å) | 1.5418 |
| Data resolution (Å)a | 24.17-2.23 (2.35-2.23) |
| Measured reflections | 133337 |
| Multiplicitya | 7.1 (6.8) |
| Completeness (%)a | 98.8 (95.7) |
| Rmeas (%)a,b | 9.9 (43.2) |
| 〈I/σ(I)〉a | 18.5 (5.4) |
| a, b, c (Å) | 47.17, 74.55, 56.85 |
| β (degrees) | 96.8 |
| Refinement resolution (Å) | 24.17-2.23 |
| Rcrystc (no. of reflections) | 0.168 (17,522) |
| Rfreec (no. of reflections) | 0.219 (1368) |
| Root mean square, bonds (Å) | 0.01 |
| Root mean square, angles (degrees) | 1.1 |
| Protein non-hydrogen atoms | 3227 |
| Water atoms | 102 |
| Iron atoms | 2 |
| Mean B factor, overall: chain A/chain B (Å2) | 28/29 |
| Mean B factor, main chain: chain A/chain B (Å2) | 26/27 |
| Mean B factor, side chains: chain A/chain B (Å2) | 30/31 |
| Mean B factor, waters (Å2) | 30 |
| Mean B factor, liganded iron (Å2) | 17.8 |
| Map versus model correlation coefficient (overall/local)d | 0.889/0.919 |
| No. of residues in Ramachandran plot regionse (allowed/favored/outliers) | 402/390/0 |
| Protein Data Bank code | 4DVH |
a Values in parentheses apply to the high resolution shell.
b Rmeas = Σh √Nh/(Nh − 1) Σi |Ii − 〈I±〉|/ΣhΣiI±, where Nh represents multiplicity for each reflection; Ii is the ith observation of reflection h; and 〈I〉 is the mean of the intensity of all observations of reflection h, with I± = 1/NhΣi(I(−) or I(−)). Σh is taken over all reflections. Σi is taken over all observations of each reflection.
c R = Σh|F(h)obs − F(h)calc|/Σh|F(h)obs|; Rcryst and Rfree were calculated using the working and test hkl reflection sets, respectively.
d Calculated with Phenix get_cc_mtz_pdb (92).
e Calculated with Molprobity (42).
FIGURE 6.

Crystal structure of T. cruzi mitochondrial Fe-SODA. A, solvent-accessible surface representation of the T. cruzi Fe-SODA dimer. The surface is rendered semitransparent to observe the secondary structure organization underneath, drawn as a schematic. Each protomer is highlighted with different colors. The active sites are depicted with spheres (iron atoms in purple and coordinating waters in red) and sticks for the coordinating amino acids. B, schematic representation of one Fe-SODA monomer (chain B) with a color ramp to indicate the N terminus (blue) to C terminus (red) direction. Helices are labeled from α1 to α7, and β-sheets are labeled from β1 to β3. Note the conserved bulge in α1. The metal-binding active site is shown as in A, with the coordinating residues labeled (water HOH6, red sphere, is unlabeled for clarity). C, stereo view of the active site, highlighting the first sphere of direct iron coordination residues in labeled sticks (colored by atom, with carbon in gray, nitrogen in blue, and oxygen in red). The coordinating water (red sphere) is not labeled for clarity. A second sphere of residues, in contact with the first sphere and/or defining entrance channels to the iron-binding pocket, are shown as lines colored by atom (with carbon in yellow, for the same monomer, or orange for the other monomer in the dimer). The critical Tyr36 is observed on the left. A more distant shell is also shown as lines (His33, Tyr169, and Tyr180), with carbon atoms in blue.
FIGURE 7.
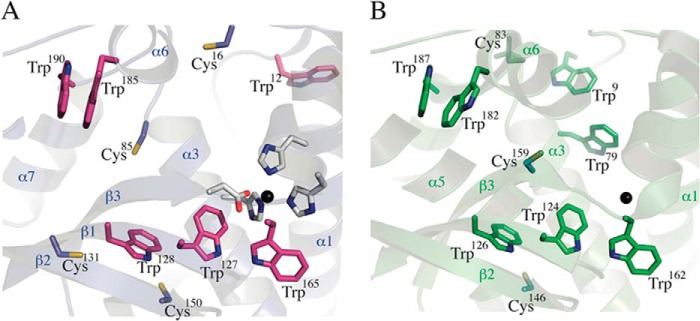
Comparative positions of cysteines and tryptophans in Fe-SODA and Fe-SODB. A, T. cruzi Fe-SODA structure. All of the Cys and Trp residues are labeled and highlighted in stick representation, colored by atom (nitrogen in blue, sulfur in yellow, oxygen in red, and carbon differentially slate blue for Cys and magenta for Trp). As a reference, the active site is shown with the iron atom in dark gray and the first shell of coordinating residues in sticks (colored by atom with carbon in white). The coordinating water is not shown. An underlying schematic is drawn semitransparent, with labels for some of the secondary structure elements. B, T. cruzi Fe-SODB structure (Protein Data Bank entry 2GPC). In the same orientation as Fe-SODA (A), Cys and Trp are labeled and highlighted in stick representation, colored by atom (nitrogen in blue, sulfur in yellow, carbon in green). Only the iron atom is shown as a reference in spheres (dark gray), and underlying semitransparent schematic to localize secondary structure elements.
Insights for the Resistance of Cytosolic Fe-SODB to Peroxynitrite-mediated Inactivation
Cysteine residues can repair tyrosyl radicals in proteins via IET (44). The overall reaction with a peptide sequence can be exemplified as follows.
By this mechanism, the radical character initially located in a tyrosine residue is transferred to a cysteine residue, and therefore further tyrosine oxidative modifications, including tyrosine nitration (by the combination reaction of tyrosyl radical with •NO2), are inhibited. In the case of Fe-SODB, the possible contribution of Cys residues to the resistance to peroxynitrite was explored at different levels. There are two solvent-accessible thiols (Cys83 and Cys146) in Fe-SODB from a total of three (as measured with DTNB and by crystal structure analysis; Fig. 7). Blockage of the accessible thiols in Fe-SODB with NEM rendered the enzyme significantly more susceptible to peroxynitrite-mediated inactivation (Fig. 8A), whereas no alterations in sensitivity were observed for NEM-treated Fe-SODA (all four Cys residues in Fe-SODA are solvent-accessible as measured with DTNB and by crystal structure analysis; Fig. 7). These results support the participation of the Fe-SODB solvent-accessible Cys (Cys83 and/or Cys146) in the resistance toward peroxynitrite. Previous mechanistic studies using Tyr-Cys-containing peptides (44, 80, 81) showed that IET (shown in Reaction 1) is mediated by a proton-coupled electron transfer mechanism that involves as a first key step the deprotonation of the Cys residue, which then in the thiolate form acts as the electron donor, followed by IET, yielding the final radical Cys-S• (44). To analyze the probability that any of the Cys residues present in T. cruzi Fe-SODs could participate in Tyr-O• repair involving an IET process, we first estimated each Cys pKa value using the propKa software (54–57). The results obtained show that the predicted pKa values of the Cys present in mitochondrial Fe-SODA (Cys16, Cys85, Cys131, and Cys150) and Fe-SODB (Cys146 and Cys159) are similar to that observed for free Cys (∼8.2–8.3), making less likely the existence of deprotonated forms at pH 7.4. Interestingly, the pKa value predicted for Fe-SODB Cys83 is approximately 2 pH units lower than the one obtained for the other Cys residues. This lower pKa can be explained by the presence in its immediate environment of two positively charged residues, Lys188 and Arg192. The Lys188-Nξ is located at 5.5 Å from the Cys83-Sγ and Arg192-Cξ at 8.2 Å, increasing the chance for the thiol group to be partially charged at pH 7.4. The Fe-SODB critical Tyr35 is located >20 Å away from Cys83; thus, to repair Tyr35-O•, a long range IET needs to occur. In this case, relay amino acids (e.g. Trp9 and Trp79) in the electron transfer pathway are required to improve the efficiency of the process (82, 83). To analyze possible IET pathways in cytosolic Fe-SODB, we performed 20-ns-long MD simulations of the corresponding homodimer, with Tyr35 described as radical, and Cys83 as thiolate (as predicted for its lower pKa) (i.e. tyrosyl radical-thiolate Fe-SODB system). We then computed possible IET paths between both residues using the pathways algorithm as described under “Experimental Procedures” (44). The resulting most probable IET path comprises the participation of key residues that contributes to the process: Trp79 and His32 (Fig. 8B). The presence of aromatic residues along the IET pathway (Trp79 and Trp9, absent in the Fe-SODA crystal structure) could increase the overall IET rate, as was recently shown (49, 52). In summary, the in silico analysis of the tyrosyl radical-thiolate Fe-SODB system strongly suggests that the partially charged Cys83-S− acts as the electron source to rescue critical Tyr35-O• formed after peroxynitrite treatment, preventing nitration and inactivation of cytosolic Fe-SODB.
FIGURE 8.
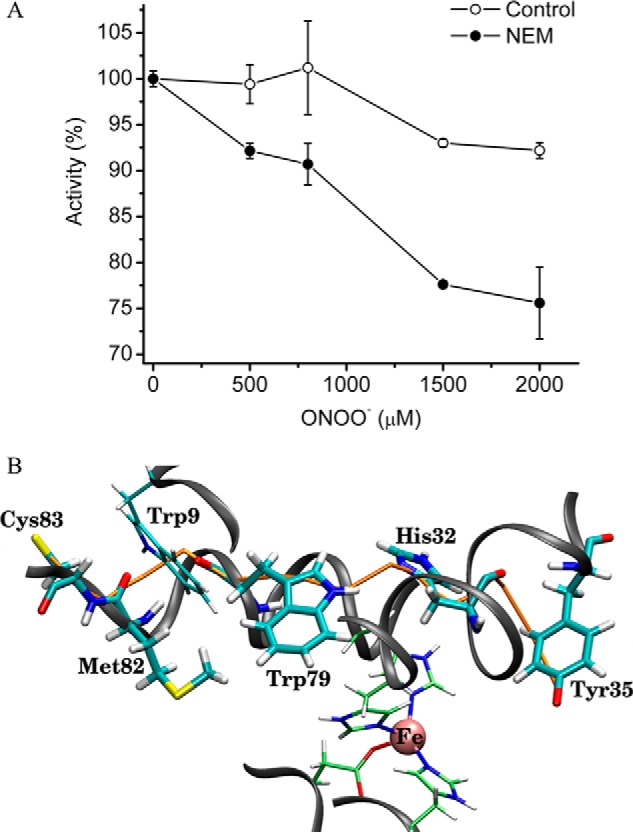
Participation of solvent-accessible Fe-SODB thiols in the resistance to peroxynitrite and role of intramolecular electron transfer. A, effects of sulfhydryl-blocked Fe-SODB in the reaction with peroxynitrite. Peroxynitrite (0–2000 μm) was added to control (empty circles) or NEM-blocked sulfhydryl Fe-SODB (filled circles) (8 μm) in sodium phosphate buffer (100 mm) at pH 7.4. Activity is expressed as percentage activity relative to the native enzyme or NEM-treated enzyme in the absence of peroxynitrite (100% activity). B, selected snapshot for Fe-SODB intramolecular electron transfer pathway from Cys83-S− to Tyr35-O•. The figure shows the path for IET as predicted with the pathways algorithm and taken from the explicit water MD simulation (see “Experimental Procedures” for details). Fe-SODB is shown as a black ribbon representation, and the residues Cys83, Met82, Trp9, Trp79, His32, and Tyr35 are shown as boldface sticks. The iron atom is shown as a pink sphere with the coordination residues as cylindrical representations. The predicted IET path is shown in orange and involves electron transfer starting on the Cys83-S atom sequentially to Met82, Trp79, and His32 and ending in the aromatic ring of Tyr35-O•. Parts of the pathway occur through the backbone, and others occur through space. Surrounding waters were omitted for clarity. Error bars, S.E.
Detection of Cys83-S• after the Reaction of Fe-SODB with Peroxynitrite
If an IET mechanism is operative after peroxynitrite reaction with Fe-SODB, a protein radical corresponding to Fe-SODB-Cys83-S• generated in this process should be experimentally detected (Reaction 1). Immunospin trapping and EPR spin trapping analysis were performed in order to detect the protein thiyl radical. Control or NEM-treated Fe-SODB (5 μm) was exposed to peroxynitrite (0–20 μm) in the presence of the spin trap DMPO (100 mm). Importantly, low concentrations of peroxynitrite were used to favor its preferential reaction with the metal center in Fe-SOD and to minimize homolysis because the latter can render thiyl radical by reactions of •OH and •NO2 with cysteine independent of the IET mechanism. DMPO-nitrone adducts were detected by Western blot using the anti-DMPO-nitrone antibody as described previously (33, 80). Results show that the covalently DMPO-nitrone adducts detected in Fe-SODB control conditions were significantly inhibited in NEM-treated Fe-SODB, strongly suggesting the generation of Cys83-S• after peroxynitrite reaction with the enzyme (Fig. 9). Moreover, the shift to acidic pI values of native Fe-SODB (major spot pI = 7.5) after peroxynitrite treatment (<7.2) was prevented in the presence of DMPO with a new more basic spot (pI = 7.8) probably corresponding to the Fe-SODB-DMPO adduct (Fig. 9B).
FIGURE 9.
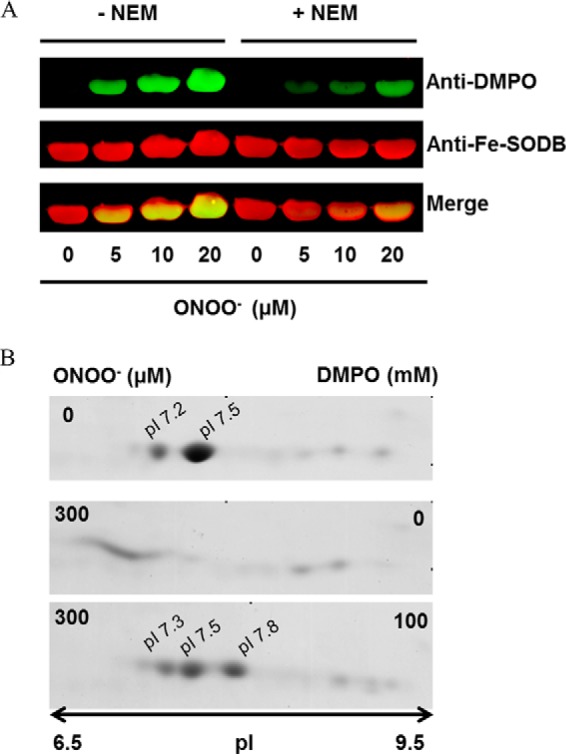
Immunospin trapping of protein thiyl radical in the peroxynitrite reaction with Fe-SODB. A, control or thiol-blocked NEM-treated Fe-SODB (50 μm) was exposed to peroxynitrite (0–20 μm) in sodium phosphate buffer (100 mm, pH 7.4) in the presence of the spin trap DMPO (100 mm). Immunoreactive proteins were detected with anti-DMPO nitrone antibody (green) and specific anti-Fe-SODB antibody (red). Merged bands are shown in yellow. B, two-dimensional gel electrophoresis of T. cruzi Fe-SODB (8 μm) treated with peroxynitrite (300 μm) in sodium phosphate buffer (0.2 m) at pH 7.4 and 25 °C in the presence and absence of DMPO (100 mm). Two-dimensional gel electrophoresis was performed as described under “Experimental Procedures.”
Furthermore, EPR spin trapping analysis of the reaction of Fe-SODB (2 mm) with peroxynitrite (500 μm) was performed in the presence of the spin trap PBN (50 mm). Again, reaction conditions were optimized to favor the direct reaction of peroxynitrite with Fe-SODB and to obtain detectable EPR signals. The addition of peroxynitrite to Fe-SODB led to the detection of an EPR spectrum characteristic of a strongly immobilized nitroxide adduct (Fig. 10A). The spin adduct was subjected to nonspecific proteolysis with Pronase, resulting in the conversion of the spectrum into an isotropic six-line spectrum characteristic of PBN-protein thiyl adducts (Fig. 10B) (84–86). In order to confirm that the EPR signal observed was due to the formation of Cys83-S• after peroxynitrite reaction, the Fe-SODB mutant C83S was generated. In this case, the PBN-protein radical adduct was almost completely inhibited (Fig. 10, C and D), demonstrating the generation of Cys83-S•.
FIGURE 10.
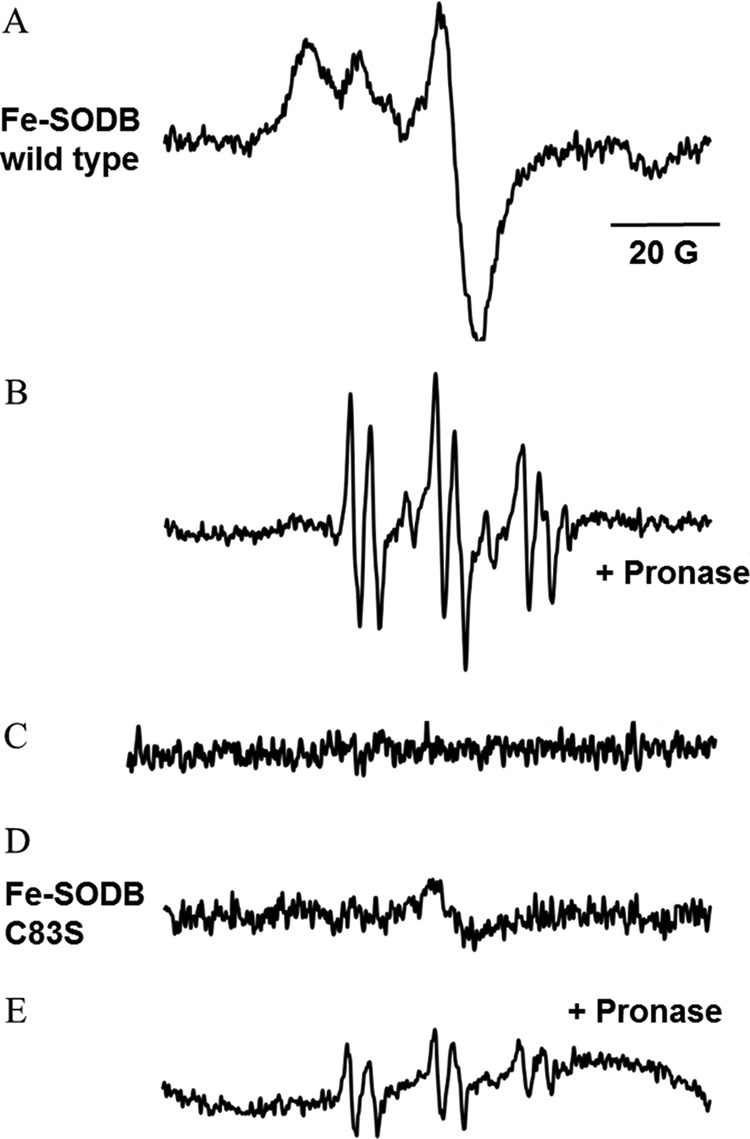
EPR spectra of PBN-protein radical adducts obtained in the peroxynitrite reaction with Fe-SODB. The spectra were obtained after a 1-min incubation at room temperature of wild type Fe-SODB or C83S mutant (2 mm) in the presence of PBN (50 mm) and peroxynitrite (500 μm) in phosphate buffer (0.1 m). A, wild type Fe-SODB; B, wild type Fe-SODB treated with Pronase (20 mg/ml for 10 min). C, wild type Fe-SODB without peroxynitrite. D, C83S Fe-SODB; E, C83S treated with Pronase (20 mg/ml for 10 min). Instrumental conditions were as follows: microwave power, 20 milliwatts; modulation amplitude, 1.0 G; time constant, 164 ms; gain, 5 × l05.
The Role of Cys83 in the Inhibition of Tyr35 Nitration via IET; Studies with Mutant Fe-SODB and Free Thiols
In order to confirm the participation of Cys83 in the IET mechanism proposed, site-directed mutagenesis experiments were performed. First, our primary candidate for the IET process was the Cys159 located at 9.9 Å from the iron atom of the active site of Fe-SODB and not present in the crystal structure of Fe-SODA. Mutation of Cys159 (C159S) did not affect the resistance of Fe-SODB to peroxynitrite-dependent inactivation, indicating that this residue was not participating in the IET process (Fig. 11B). Importantly, mutation of the single Cys83 (C83S) rendered the enzyme more susceptible to peroxynitrite-mediated inactivation as compared with wild type Fe-SODB (Fig. 11C). Moreover, we also generated the double mutant N187D/K189E, which completely modifies the protein environment adjacent to Cys83 from a net positively charged to a negatively charged one. This change of microenvironment is expected to result in a significant increase in Cys83 pKa, with a consequent decrease of the amount of Cys-S− necessary to repair the critical Tyr35-O•. Indeed, N187D/K189E Fe-SODB significantly lost the resistance against peroxynitrite treatment when compared with the wild type protein, reinforcing the role of Cys83 in the IET proposed mechanism (Fig. 11D).
FIGURE 11.

Peroxynitrite reaction with Fe-SODB mutants; role of Cys83. Fe-SODB wild type (A) or mutants (C159S (B), C83S (C), and N187D/K189E (D)) (8 μm) were exposed to peroxynitrite (0–1200 μm) in sodium phosphate buffer (100 mm, pH 7.4), and residual SOD activity was measured as previously. In C, C83S Fe-SODB was also exposed to peroxynitrite in the presence (empty circles) of l-cysteine-methyl ester (8 μm, pKa = 6.5). Error bars, S.E.
It has been shown that thiols are the dominant “sink” for peroxynitrite and •NO2 in cells (28, 87). The calculated rate constant of •NO2 with GSH and Cys is 2 and 5 × 107 m−1 s−1, respectively (28). This scavenging reaction may prevent the •NO2-dependent tyrosine nitration and thus enzyme inactivation (28, 72). In order to examine whether Cys83 could contribute to the inhibition of enzyme inactivation via direct •NO2 scavenging, we performed experiments using the Fe-SODB C83S mutant in the presence of equimolecular amounts of the more reactive l-cysteine methyl ester (8 μm; pKa ∼6.7 (88)). The presence of this •NO2 radical scavenger failed to protect Fe-SODB-C83S mutant from peroxynitrite inactivation, strongly suggesting that the protection observed in wild type Fe-SODB by Cys83 was mainly mediated by an IET and not by •NO2 free radical scavenging (Fig. 11C). It is important to note that although mutation of Cys83 in Fe-SODB renders the enzyme more susceptible to oxidant-dependent inactivation, it was still more resistant than Fe-SODA. This result indicates that other residues, probably Trp79 and Trp9, may be additionally participating in Tyr35-O• repair after peroxynitrite treatment.
Fe-SODA Modifications during Cellular Nitroxidative Stress Conditions to T. cruzi
Fe-SODA T. cruzi overexpressers (12, 58) were used to search for modifications of Fe-SOD occurring during nitroxidative stress conditions in living parasites (epimastigote stage). Following the induction of Fe-SODA expression by tetracycline (4–6-fold increase respect to wild type), parasites were incubated in the presence of antimycin A (complex III electron chain inhibitor) plus a •NO donor, in order to specifically generate peroxynitrite at the mitochondrial cell compartment (59). Following treatment, parasites protein extracts were separated by two-dimensional electrophoresis and probed with anti-Fe-SODs antibodies. After AA/•NO treatment, an important shift toward more acidic pH values in the pI of Fe-SODA was evident (Fig. 12). This pI shift was also observed during exogenous peroxynitrite challenge to parasites (Fig. 12A). The pI changes observed in Fe-SODA obtained from living parasites during exposure to either endogenous (AA/•NO) or exogenous peroxynitrite were similar to those observed for the recombinant Fe-SODA after peroxynitrite treatment (Fig. 5A). Notably, Fe-SODB was not significantly altered under these cellular nitroxidative conditions, in agreement with its high resistance to peroxynitrite.
FIGURE 12.
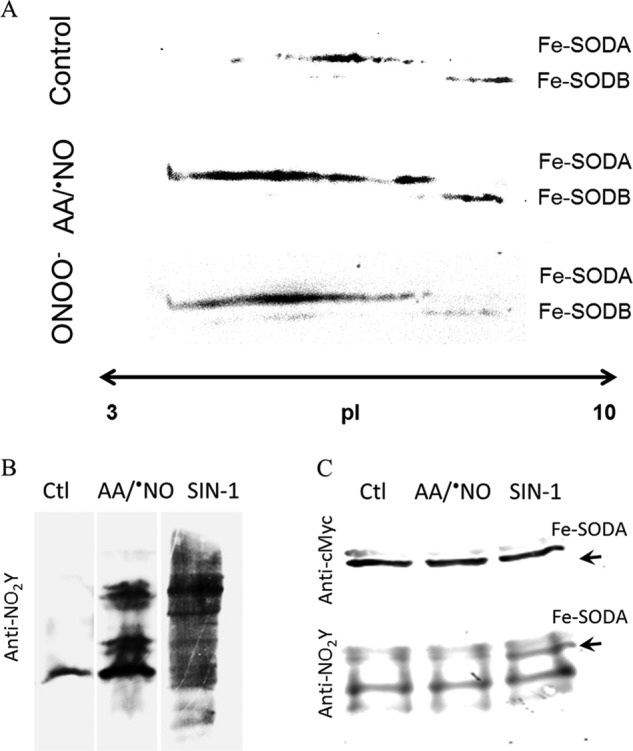
Cellular detection of Fe-SODA nitroxidative modifications. A, two-dimensional gel electrophoresis. T. cruzi Fe-SODA overexpressers were treated with AA (5 μm) plus NOC-12 (5 mm; t½ = 100 min at pH 7.4), SIN-1 (5 mm), or ONOO− (300 μm) for 3 h at room temperature. After treatment, samples were processed as described under “Experimental Procedures” and subjected to two-dimensional electrophoresis. Membranes were probed with the specific Fe-SODA and Fe-SODB antibodies. B, 3-nitrotyrosine detection in parasites. Following treatment in the conditions described in A, parasite extracts were separated in 15% SDS gels and transferred to nitrocellulose membranes. Membranes were probed with the specific anti-3-nitrotyrosine antibody. C, immunoprecipitation of nitrated Fe-SODA. Parasite extracts as above were incubated overnight at 4 °C in the presence of the monoclonal c-Myc antibody that recognized the 9E10 epitope of Fe-SODA in the presence of protein A/G-agarose as described under “Experimental Procedures.” Immunoprecipitated proteins were run in 15% SDS-gel electrophoresis, electrotransferred to nitrocellulose, and revealed using anti-c-Myc antibody and anti-3-nitrotyrosine antibody and as described under “Experimental Procedures.”
The pI changes in Fe-SODA during nitroxidative challenge to living parasites correlated reasonably well with the extents of protein tyrosine nitration (Fig. 12B). Indeed, endogenous and exogenous fluxes of peroxynitrite (AA/•NO or the peroxynitrite donor SIN-1, respectively) caused nitration of parasite proteins. Immunoprecipitation analysis of Fe-SODA revealed the presence of nitrated enzyme in the SIN-1-treated parasites, unambiguously revealing the reaction of nitrating species and subsequent oxidative postranslational modifications of Fe-SODA in living parasites (Fig. 12C). Overall, the data in Fig. 12 confirm the feasibility of these biochemical events in Fe-SODA as biologically relevant processes.
DISCUSSION
T. cruzi mitochondrial and cytosolic Fe-SODs were purified to homogeneity as active enzymes with specific activities and O2⨪ dismutation rates comparable with those of other Mn- and Fe-SODs. Similarly, both SODs readily reacted with peroxynitrite at comparable second order rate constants (∼4.5 × 104 m−1 s−1) (Table 1) (18, 33, 62, 63) and were dose-dependently inactivated and nitrated by peroxynitrite (Fig. 3).
Peptide mapping by mass spectrometry analysis of the peroxynitrite-treated enzymes together with 3-nitrotyrosine quantification revealed that peroxynitrite-dependent inactivation of T. cruzi Fe-SODs is due to the selective nitration of the universally conserved Tyr35 located near the iron atom of the active site, as was previously observed for Mn-SOD and E. coli SODs (Fig. 5 and Table 2) (17, 18). The proximal reactive species was the anionic form of peroxynitrite (ONOO−), and the primary target at the enzyme was the active site iron atom, as was revealed by pH studies, cysteine alkylation, and CO2 competition experiments (Figs. 4 and 8 and Table 1).
In the most likely reaction mechanism, the metal-based Lewis adduct formed in the reaction (i.e. SOD-FeIII-OONO (89)) undergoes homolysis to yield •NO2 and the corresponding oxo-metal complex (SOD-FeIV=O) (89). This SOD-FeIV=O complex is strongly oxidizing and promotes the oxidation of Tyr35 to Tyr35-O• that rapidly combines with •NO2 generated in situ to yield a tyrosine-nitrated enzyme, as proposed in Scheme I. Thus, site specificity is provided by a combination of kinetic factors (fast reaction of peroxynitrite with the iron center in contrast to the much slower proton-catalyzed homolysis) and close structural relationships between the iron atom and the active site tyrosine. Once Tyr35 is nitrated, O2⨪ dismutation is impeded by both a steric effect (as the nitro group located in the access channel impedes O2⨪ diffusion) and the electrical charge repulsion provided by the ionization of the phenolic group in Tyr, by analogy with the recently reported data for mammalian Mn-SOD (20, 90).
SCHEME 1.
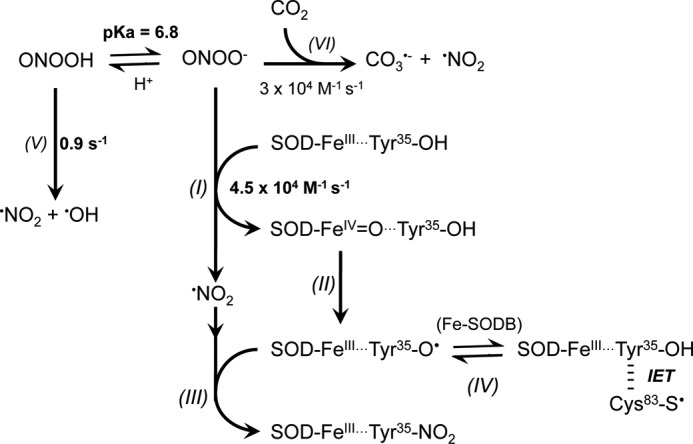
Proposed reaction mechanisms for the reaction of T. cruzi Fe-SODs with peroxynitrite. Peroxynitrite anion (ONOO−, 80% at pH 7.4) reacts with the FeIII atom of the Fe-SODs, yielding the corresponding oxo-metal complex (SOD-FeIV=O) with •NO2 generation (reaction I). SOD-FeIV=O oxidizes the active site Tyr (Tyr35 in Fe-SODB and Tyr36 in Fe-SODA) to its corresponding tyrosyl radical (SOD-FeIII···Tyr35-O•) (reaction II). SOD-FeIII···Tyr35-O• rapidly combines with •NO2 (I), yielding the nitrated and inactivated enzyme SOD-FeIII···Tyr35-NO2 (reaction III). In T. cruzi Fe-SODB (reaction IV), Cys83-S− repairs FeIII···Tyr35-O•, regenerating FeIII···Tyr35-OH through an IET process. The homolytic cleavage of ONOOH (reaction V) and the nucleophilic addition of ONOO− to CO2 (reaction VI) (reviewed in Ref. 89) compete with reaction I and decrease the extents of Tyr35 nitration and enzyme inactivation.
Although both isoforms were inactivated by peroxynitrite, Fe-SODB was extremely resistant to nitration and inactivation as compared with its mitochondrial counterpart (Fig. 3). For example, only 20% of Fe-SODB became inactivated at pH 7.4 with high peroxynitrite concentrations (1500 μm), whereas most of the Fe-SODA activity was already lost at 200 μm peroxynitrite, a similar susceptibility to what was reported for E. coli Mn- and Fe-SODs and mammalian Mn-SOD (18, 91).
Due to the apparent sequence and structural homology among Fe-SODs of different species, this disparate susceptibility on peroxynitrite-mediated inactivation of T. cruzi Fe-SODs was intriguing. Thus, in order to search for structural differences that may explain the high intrinsic resistance to oxidant inactivation of the Fe-SODB isoform, we solved the crystal structure of T. cruzi mitochondrial Fe-SODA at 2.2 Å resolution. Comparison of the crystal structures of both isoforms revealed a number of potentially relevant differences to explain the results obtained in our study. Two tryptophans (Trp9 and Trp79) and two cysteines (Cys83 and Cys159) are located near the active site of Fe-SODB and are absent in the crystal structure of the mitochondrial counterpart (Fig. 7). It was previously shown that the presence of a cysteine residue in the proximity of a tyrosine might enable radical transfer during one-electron oxidation processes, with the cysteine acting as electron donor (44). In this regard, following peroxynitrite reaction with the iron center of Fe-SODB, the Tyr35-O• generated could be quickly reduced by a cysteine residue back to Tyr35 prior to the nitration step, therefore preventing enzyme inactivation. Of the three cysteines present in Fe-SODB, only Cys83, located 22 Å away from Tyr35, is predicted to have a lower pKa value favoring the electron transfer process to Tyr35-O• (44). Moreover, MD simulations of the tyrosyl radical-thiolate system (Tyr35-O•···Cys83-S−) in Fe-SODB revealed Trp79 and His32 as key residues acting as “relay” amino acids or “stepping stones” for the electron transfer (Fig. 8B). Ultimately, this type of IET process generates a thiyl radical, as observed previously for the hydrogen peroxide-mediated oxidation of myoglobin (80, 81) and peroxynitrite-mediated oxyhemoglobin oxidation (33, 80).
In this work, using different experimental approaches, the Cys83-S• generated in the Fe-SODB after the reaction with peroxynitrite was identified (Figs. 9 and 10), demonstrating the participation of an IET pathway in the observed resistance to peroxynitrite. A further proof of the key role of Cys83 in modulating the redox chemistry at the active site is that the Fe-SODB C83S mutant significantly increased the sensitivity to peroxynitrite-mediated inactivation (Fig. 11C). Future experiments, including additional enzyme mutants, are needed in order to determine the precise amino acids involved in the IET pathway(s) and whether other amino acids can also participate in tyrosyl radical repair. From a biological perspective, it is conceivable that the solvent-accessible Cys83-S• can be repaired in living cells by low molecular weight reductants. For instance, molecules, such as glutathione, trypanothione, or ascorbate, present in the T. cruzi cytosol may provide a sustained protection to Fe-SODB at the expense of these “sacrificial” antioxidant molecules. In this regard, we were able to detect nitrated Fe-SODA in living parasites exposed to exogenous (SIN-1) or endogenous (AA/•NO) fluxes of peroxynitrite. Whereas in the case of SIN-1, peroxynitrite will be formed and react in various cellular compartments, including cytosol and mitochondria, the AA/•NO treatment leads to the mitochondrial formation of peroxynitrite. In both cases, only substantial post-translational oxidative modifications were observed in Fe-SODA, underscoring the capacity of Fe-SODB to resist peroxynitrite-mediated inactivation. Both an important shift in the pI of Fe-SODA and the detection of nitrated protein (after immunoprecipitation) are indicative of oxidative modifications due to peroxynitrite reactions in mitochondria (Fig. 12). Future experiments using Fe-SODA and Fe-SODB T. cruzi overexpressers in infections to macrophages and cardiomyocytes will allow us to evaluate in more detail the biological significance of our results. In particular, the peroxynitrite-mediated inactivation of Fe-SODA may be secondarily responsible for parasite programmed cell death during the infection process. Indeed, the inactivation of Fe-SODA would raise the intramitochondrial O2⨪ state concentration, which is an apoptotic signal in T. cruzi (12). On the other hand, the robust Fe-SODB would resist the oxidative challenge promoted by mammalian host cells and contribute to the neutralization of the oxidative challenge. Thus, as was observed previously for other T. cruzi antioxidant enzymes (7), T. cruzi Fe-SODs may function as virulence factors, a hypothesis that is currently under investigation in our laboratories both in the cellular and animal models of Chagas disease. From a chemistry-oriented point of view the data we are now reporting underscore that the extent of peroxynitrite-mediated oxidative modifications in genetically engineered proteins can be modulated taking advantage of long range intramolecular electron transfer processes.
This work was supported, in whole or in part, by National Institutes of Health Grant 1R01AI095173. This work was also supported by a grant from the Universidad de la República (CSIC, Uruguay) (to R. R.) and by PEDECIBA (Progama de Desarrollo de Ciencias Básicas, Uruguay) and CeBEM (Centro de Biología Estructural del Mercosur).
- SOD
- superoxide dismutase
- NEM
- N-ethylmaleimide
- DTNB
- 5,5′-dithiobis-(2-nitrobenzoic acid)
- PBN
- α-phenyl N-tertiary-butyl nitrone
- NOC-12
- 1-hydroxy-2-oxo-3-(N-ethyl-2-aminoethyl)-3-ethyl-1-triazene
- DMPO
- 5,5-dimethyl-1-pyrroline-N-oxide
- IPG
- immobilized pH gradient
- AA
- antimycin A
- IET
- intramolecular electron transfer.
REFERENCES
- 1. Fridovich I. (1995) Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 64, 97–112 [DOI] [PubMed] [Google Scholar]
- 2. Stallings W. C., Pattridge K. A., Strong R. K., Ludwig M. L. (1984) Manganese and iron superoxide dismutases are structural homologs. J. Biol. Chem. 259, 10695–10699 [PubMed] [Google Scholar]
- 3. Ismail S. O., Paramchuk W., Skeiky Y. A., Reed S. G., Bhatia A., Gedamu L. (1997) Molecular cloning and characterization of two iron superoxide dismutase cDNAs from Trypanosoma cruzi. Mol. Biochem. Parasitol. 86, 187–197 [DOI] [PubMed] [Google Scholar]
- 4. Temperton N. J., Wilkinson S. R., Kelly J. M. (1996) Cloning of an Fe-superoxide dismutase gene homologue from Trypanosoma cruzi. Mol. Biochem. Parasitol. 76, 339–343 [DOI] [PubMed] [Google Scholar]
- 5. Urbina J. A. (2010) Specific chemotherapy of Chagas disease: relevance, current limitations and new approaches. Acta Trop. 115, 55–68 [DOI] [PubMed] [Google Scholar]
- 6. Lepesheva G. I., Villalta F., Waterman M. R. (2011) Targeting Trypanosoma cruzi sterol 14α-demethylase (CYP51). Adv. Parasitol. 75, 65–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Piacenza L., Zago M. P., Peluffo G., Alvarez M. N., Basombrio M. A., Radi R. (2009) Enzymes of the antioxidant network as novel determiners of Trypanosoma cruzi virulence. Int. J. Parasitol. 39, 1455–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kierszenbaum F., Knecht E., Budzko D. B., Pizzimenti M. C. (1974) Phagocytosis: a defense mechanism against infection with Trypanosoma cruzi. J. Immunol. 112, 1839–1844 [PubMed] [Google Scholar]
- 9. Muñoz-Fernández M. A., Fernández M. A., Fresno M. (1992) Synergism between tumor necrosis factor-α and interferon-γ on macrophage activation for the killing of intracellular Trypanosoma cruzi through a nitric oxide-dependent mechanism. Eur. J. Immunol. 22, 301–307 [DOI] [PubMed] [Google Scholar]
- 10. Alvarez M. N., Peluffo G., Piacenza L., Radi R. (2011) Intraphagosomal peroxynitrite as a macrophage-derived cytotoxin against internalized Trypanosoma cruzi: consequences for oxidative killing and role of microbial peroxiredoxins in infectivity. J. Biol. Chem. 286, 6627–6640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Valez V., Cassina A., Batinic-Haberle I., Kalyanaraman B., Ferrer-Sueta G., Radi R. (2013) Peroxynitrite formation in nitric oxide-exposed submitochondrial particles: detection, oxidative damage and catalytic removal by Mn-porphyrins. Arch. Biochem. Biophys. 529, 45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Piacenza L., Irigoín F., Alvarez M. N., Peluffo G., Taylor M. C., Kelly J. M., Wilkinson S. R., Radi R. (2007) Mitochondrial superoxide radicals mediate programmed cell death in Trypanosoma cruzi: cytoprotective action of mitochondrial iron superoxide dismutase overexpression. Biochem. J. 403, 323–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Demicheli V., Quijano C., Alvarez B., Radi R. (2007) Inactivation and nitration of human superoxide dismutase (SOD) by fluxes of nitric oxide and superoxide. Free Radic. Biol. Med. 42, 1359–1368 [DOI] [PubMed] [Google Scholar]
- 14. MacMillan-Crow L. A., Crow J. P., Kerby J. D., Beckman J. S., Thompson J. A. (1996) Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc. Natl. Acad. Sci. U.S.A. 93, 11853–11858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Radi R., Rodriguez M., Castro L., Telleri R. (1994) Inhibition of mitochondrial electron transport by peroxynitrite. Arch. Biochem. Biophys. 308, 89–95 [DOI] [PubMed] [Google Scholar]
- 16. Ghafourifar P., Schenk U., Klein S. D., Richter C. (1999) Mitochondrial nitric-oxide synthase stimulation causes cytochrome c release from isolated mitochondria. Evidence for intramitochondrial peroxynitrite formation. J. Biol. Chem. 274, 31185–31188 [DOI] [PubMed] [Google Scholar]
- 17. Yamakura F., Taka H., Fujimura T., Murayama K. (1998) Inactivation of human manganese-superoxide dismutase by peroxynitrite is caused by exclusive nitration of tyrosine 34 to 3-nitrotyrosine. J. Biol. Chem. 273, 14085–14089 [DOI] [PubMed] [Google Scholar]
- 18. Quijano C., Hernandez-Saavedra D., Castro L., McCord J. M., Freeman B. A., Radi R. (2001) Reaction of peroxynitrite with Mn-superoxide dismutase. Role of the metal center in decomposition kinetics and nitration. J. Biol. Chem. 276, 11631–11638 [DOI] [PubMed] [Google Scholar]
- 19. Surmeli N. B., Litterman N. K., Miller A. F., Groves J. T. (2010) Peroxynitrite mediates active site tyrosine nitration in manganese superoxide dismutase: evidence of a role for the carbonate radical anion. J. Am. Chem. Soc. 132, 17174–17185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moreno D. M., Martí M. A., De Biase P. M., Estrin D. A., Demicheli V., Radi R., Boechi L. (2011) Exploring the molecular basis of human manganese superoxide dismutase inactivation mediated by tyrosine 34 nitration. Arch. Biochem. Biophys. 507, 304–309 [DOI] [PubMed] [Google Scholar]
- 21. Radi R. (2013) Protein tyrosine nitration: biochemical mechanisms and structural basis of functional effects. Acc. Chem. Res. 46, 550–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Radi R., Beckman J. S., Bush K. M., Freeman B. A. (1991) Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch. Biochem. Biophys. 288, 481–487 [DOI] [PubMed] [Google Scholar]
- 23. Hughes M. N., Nicklin H. G. (1970) A possible role for the species peroxonitrite in nitrification. Biochim. Biophys. Acta 222, 660–661 [DOI] [PubMed] [Google Scholar]
- 24. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 25. Crow J. P., Sampson J. B., Zhuang Y., Thompson J. A., Beckman J. S. (1997) Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J. Neurochem. 69, 1936–1944 [DOI] [PubMed] [Google Scholar]
- 26. McCord J. M., Fridovich I. (1969) Superoxide dismutase: an enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 244, 6049–6055 [PubMed] [Google Scholar]
- 27. Peskin A. V., Winterbourn C. C. (2006) Taurine chloramine is more selective than hypochlorous acid at targeting critical cysteines and inactivating creatine kinase and glyceraldehyde-3-phosphate dehydrogenase. Free Radic. Biol. Med. 40, 45–53 [DOI] [PubMed] [Google Scholar]
- 28. Alvarez B., Ferrer-Sueta G., Freeman B. A., Radi R. (1999) Kinetics of peroxynitrite reaction with amino acids and human serum albumin. J. Biol. Chem. 274, 842–848 [DOI] [PubMed] [Google Scholar]
- 29. Ellman G. L. (1959) Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82, 70–77 [DOI] [PubMed] [Google Scholar]
- 30. Trujillo M., Budde H., Piñeyro M. D., Stehr M., Robello C., Flohé L., Radi R. (2004) Trypanosoma brucei and Trypanosoma cruzi tryparedoxin peroxidases catalytically detoxify peroxynitrite via oxidation of fast reacting thiols. J. Biol. Chem. 279, 34175–34182 [DOI] [PubMed] [Google Scholar]
- 31. Brito C., Naviliat M., Tiscornia A. C., Vuillier F., Gualco G., Dighiero G., Radi R., Cayota A. M. (1999) Peroxynitrite inhibits T lymphocyte activation and proliferation by promoting impairment of tyrosine phosphorylation and peroxynitrite-driven apoptotic death. J. Immunol. 162, 3356–3366 [PubMed] [Google Scholar]
- 32. Peloso E. F., Gonçalves C. C., Silva T. M., Ribeiro L. H., Piñeyro M. D., Robello C., Gadelha F. R. (2012) Tryparedoxin peroxidases and superoxide dismutases expression as well as ROS release are related to Trypanosoma cruzi epimastigotes growth phases. Arch. Biochem. Biophys. 520, 117–122 [DOI] [PubMed] [Google Scholar]
- 33. Romero N., Radi R., Linares E., Augusto O., Detweiler C. D., Mason R. P., Denicola A. (2003) Reaction of human hemoglobin with peroxynitrite: isomerization to nitrate and secondary formation of protein radicals. J. Biol. Chem. 278, 44049–44057 [DOI] [PubMed] [Google Scholar]
- 34. Hellman U. (2000) Sample preparation by SDS/PAGE and in-gel digestion. EXS 88, 43–54 [DOI] [PubMed] [Google Scholar]
- 35. Nicholls S. J., Shen Z., Fu X., Levison B. S., Hazen S. L. (2005) Quantification of 3-nitrotyrosine levels using a benchtop ion trap mass spectrometry method. Methods Enzymol. 396, 245–266 [DOI] [PubMed] [Google Scholar]
- 36. Turko I. V., Murad F. (2005) Mapping sites of tyrosine nitration by matrix-assisted laser desorption/ionization mass spectrometry. Methods Enzymol. 396, 266–275 [DOI] [PubMed] [Google Scholar]
- 37. Leslie A. G. W. (2007) Processing difraction data with mosflm. in Evolving Methods for Macromolecular Crystallography (Read R. J., Sussman J. L., eds) pp. 41–51, Springer, New York [Google Scholar]
- 38. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 39. Trapani S., Navaza J. (2008) AMoRe: classical and modern. Acta Crystallogr. D Biol. Crystallogr. 64, 11–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., Bricogne G. (2011) Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 67, 293–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cheatham T. E., 3rd, Cieplak P., Kollman P. A. (1999) A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn. 16, 845–862 [DOI] [PubMed] [Google Scholar]
- 44. Petruk A. A., Bartesaghi S., Trujillo M., Estrin D. A., Murgida D., Kalyanaraman B., Marti M. A., Radi R. (2012) Molecular basis of intramolecular electron transfer in proteins during radical-mediated oxidations: computer simulation studies in model tyrosine-cysteine peptides in solution. Arch. Biochem. Biophys. 525, 82–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Leach A. (2001) Molecular Modelling: Principles and Applications, Prentice Hall, New York [Google Scholar]
- 46. Case D., Darden T., Cheatham T., Simmerling C., Wang J., Duke R., Luo R., Merz K., Pearlman D., Crowley M., Walker R., Zhang W., Wang W., Hayik S., Roitberg A., Seabra G., Wong K., Paesani F., Wu X., Brozell S., Tsui V., Gohlke H., Yang L., Tan C., Mongan J., Hornak V., Cui G., Beroza P., Mathews D., Schafmeister C., Ross W., Kollman P. A. (2006) AMBER 9, University of California, San Francisco, California [Google Scholar]
- 47. Beratan D. N., Betts J. N., Onuchic J. N. (1991) Protein electron transfer rates set by the bridging secondary and tertiary structure. Science 252, 1285–1288 [DOI] [PubMed] [Google Scholar]
- 48. Beratan D. N., Onuchic J. N., Winkler J. R., Gray H. B. (1992) Electron-tunneling pathways in proteins. Science 258, 1740–1741 [DOI] [PubMed] [Google Scholar]
- 49. Shih C., Museth A. K., Abrahamsson M., Blanco-Rodriguez A. M., Di Bilio A. J., Sudhamsu J., Crane B. R., Ronayne K. L., Towrie M., Vlcek A., Jr., Richards J. H., Winkler J. R., Gray H. B. (2008) Tryptophan-accelerated electron flow through proteins. Science 320, 1760–1762 [DOI] [PubMed] [Google Scholar]
- 50. Stubbe J., Nocera D. G., Yee C. S., Chang M. C. (2003) Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer? Chem. Rev. 103, 2167–2201 [DOI] [PubMed] [Google Scholar]
- 51. Alvarez-Paggi D., Martín D. F., DeBiase P. M., Hildebrandt P., Martí M. A., Murgida D. H. (2010) Molecular basis of coupled protein and electron transfer dynamics of cytochrome c in biomimetic complexes. J. Am. Chem. Soc. 132, 5769–5778 [DOI] [PubMed] [Google Scholar]
- 52. Aubert C., Mathis P., Eker A. P., Brettel K. (1999) Intraprotein electron transfer between tyrosine and tryptophan in DNA photolyase from Anacystis nidulans. Proc. Natl. Acad. Sci. U.S.A. 96, 5423–5427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ly H. K., Marti M. A., Martin D. F., Alvarez-Paggi D., Meister W., Kranich A., Weidinger I. M., Hildebrandt P., Murgida D. H. (2010) Thermal fluctuations determine the electron-transfer rates of cytochrome c in electrostatic and covalent complexes. Chemphyschem 11, 1225–1235 [DOI] [PubMed] [Google Scholar]
- 54. Li H., Robertson A. D., Jensen J. H. (2005) Very fast empirical prediction and rationalization of protein pKa values. Proteins 61, 704–721 [DOI] [PubMed] [Google Scholar]
- 55. Bas D. C., Rogers D. M., Jensen J. H. (2008) Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins 73, 765–783 [DOI] [PubMed] [Google Scholar]
- 56. Olsson M. H. M., Søndergaard C. R., Rostkowski M., Jensen J. H. (2011) PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537 [DOI] [PubMed] [Google Scholar]
- 57. Søndergaard C. R., Olsson M. H. M., Rostkowski M., Jensen J. H. (2011) Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 7, 2284–2295 [DOI] [PubMed] [Google Scholar]
- 58. Taylor M. C., Kelly J. M. (2006) pTcINDEX: a stable tetracycline-regulated expression vector for Trypanosoma cruzi. BMC Biotechnol. 6, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Piacenza L., Peluffo G., Alvarez M. N., Kelly J. M., Wilkinson S. R., Radi R. (2008) Peroxiredoxins play a major role in protecting Trypanosoma cruzi against macrophage- and endogenously-derived peroxynitrite. Biochem. J. 410, 359–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wilkinson S. R., Prathalingam S. R., Taylor M. C., Ahmed A., Horn D., Kelly J. M. (2006) Functional characterisation of the iron superoxide dismutase gene repertoire in Trypanosoma brucei. Free Radic. Biol. Med. 40, 198–209 [DOI] [PubMed] [Google Scholar]
- 61. Forman H. J., Fridovich I. (1973) Superoxide dismutase: a comparison of rate constants. Arch. Biochem. Biophys. 158, 396–400 [DOI] [PubMed] [Google Scholar]
- 62. Crow J. P., Beckman J. S., McCord J. M. (1995) Sensitivity of the essential zinc-thiolate moiety of yeast alcohol dehydrogenase to hypochlorite and peroxynitrite. Biochemistry 34, 3544–3552 [DOI] [PubMed] [Google Scholar]
- 63. Zou M. H., Daiber A., Peterson J. A., Shoun H., Ullrich V. (2000) Rapid reactions of peroxynitrite with heme-thiolate proteins as the basis for protection of prostacyclin synthase from inactivation by nitration. Arch. Biochem. Biophys. 376, 149–155 [DOI] [PubMed] [Google Scholar]
- 64. Ferrer-Sueta G., Radi R. (2009) Chemical biology of peroxynitrite: kinetics, diffusion, and radicals. ACS Chem. Biol. 4, 161–177 [DOI] [PubMed] [Google Scholar]
- 65. Radi R. (2004) Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. U.S.A. 101, 4003–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tien M., Berlett B. S., Levine R. L., Chock P. B., Stadtman E. R. (1999) Peroxynitrite-mediated modification of proteins at physiological carbon dioxide concentration: pH dependence of carbonyl formation, tyrosine nitration, and methionine oxidation. Proc. Natl. Acad. Sci. U.S.A. 96, 7809–7814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Denicola A., Freeman B. A., Trujillo M., Radi R. (1996) Peroxynitrite reaction with carbon dioxide/bicarbonate: kinetics and influence on peroxynitrite-mediated oxidations. Arch. Biochem. Biophys. 333, 49–58 [DOI] [PubMed] [Google Scholar]
- 68. Lymar S. V., Hurst J. K. (1996) Carbon dioxide: physiological catalyst for peroxynitrite-mediated cellular damage or cellular protectant? Chem. Res. Toxicol. 9, 845–850 [DOI] [PubMed] [Google Scholar]
- 69. Pryor W. A., Lemercier J. N., Zhang H., Uppu R. M., Squadrito G. L. (1997) The catalytic role of carbon dioxide in the decomposition of peroxynitrite. Free Radic. Biol. Med. 23, 331–338 [DOI] [PubMed] [Google Scholar]
- 70. Koppenol W. H., Kissner R. (1998) Can O=NOOH undergo homolysis? Chem. Res. Toxicol. 11, 87–90 [DOI] [PubMed] [Google Scholar]
- 71. Koppenol W. H., Moreno J. J., Pryor W. A., Ischiropoulos H., Beckman J. S. (1992) Peroxynitrite, a cloaked oxidant formed by nitric oxide and superoxide. Chem. Res. Toxicol. 5, 834–842 [DOI] [PubMed] [Google Scholar]
- 72. Ford E., Hughes M. N., Wardman P. (2002) Kinetics of the reactions of nitrogen dioxide with glutathione, cysteine, and uric acid at physiological pH. Free Radic. Biol. Med. 32, 1314–1323 [DOI] [PubMed] [Google Scholar]
- 73. Reiter C. D., Teng R. J., Beckman J. S. (2000) Superoxide reacts with nitric oxide to nitrate tyrosine at physiological pH via peroxynitrite. J. Biol. Chem. 275, 32460–32466 [DOI] [PubMed] [Google Scholar]
- 74. Sawa T., Akaike T., Maeda H. (2000) Tyrosine nitration by peroxynitrite formed from nitric oxide and superoxide generated by xanthine oxidase. J. Biol. Chem. 275, 32467–32474 [DOI] [PubMed] [Google Scholar]
- 75. Abriata L. A., Cassina A., Tórtora V., Marín M., Souza J. M., Castro L., Vila A. J., Radi R. (2009) Nitration of solvent-exposed tyrosine 74 on cytochrome c triggers heme iron-methionine 80 bond disruption: nuclear magnetic resonance and optical spectroscopy studies. J. Biol. Chem. 284, 17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bachega J. F., Navarro M. V., Bleicher L., Bortoleto-Bugs R. K., Dive D., Hoffmann P., Viscogliosi E., Garratt R. C. (2009) Systematic structural studies of iron superoxide dismutases from human parasites and a statistical coupling analysis of metal binding specificity. Proteins 77, 26–37 [DOI] [PubMed] [Google Scholar]
- 77. Boucher I. W., Brzozowski A. M., Brannigan J. A., Schnick C., Smith D. J., Kyes S. A., Wilkinson A. J. (2006) The crystal structure of superoxide dismutase from Plasmodium falciparum. BMC Struct. Biol. 6, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Guan Y., Hickey M. J., Borgstahl G. E., Hallewell R. A., Lepock J. R., O'Connor D., Hsieh Y., Nick H. S., Silverman D. N., Tainer J. A. (1998) Crystal structure of Y34F mutant human mitochondrial manganese superoxide dismutase and the functional role of tyrosine 34. Biochemistry 37, 4722–4730 [DOI] [PubMed] [Google Scholar]
- 79. Reece S. Y., Hodgkiss J. M., Stubbe J., Nocera D. G. (2006) Proton-coupled electron transfer: the mechanistic underpinning for radical transport and catalysis in biology. Philos. Trans. R. Soc. Lond B Biol. Sci. 361, 1351–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bhattacharjee S., Deterding L. J., Jiang J., Bonini M. G., Tomer K. B., Ramirez D. C., Mason R. P. (2007) Electron transfer between a tyrosyl radical and a cysteine residue in hemoproteins: spin trapping analysis. J. Am. Chem. Soc. 129, 13493–13501 [DOI] [PubMed] [Google Scholar]
- 81. Witting P. K., Mauk A. G. (2001) Reaction of human myoglobin and H2O2. Electron transfer between tyrosine 103 phenoxyl radical and cysteine 110 yields a protein-thiyl radical. J. Biol. Chem. 276, 16540–16547 [DOI] [PubMed] [Google Scholar]
- 82. Giese B., Graber M., Cordes M. (2008) Electron transfer in peptides and proteins. Curr. Opin. Chem. Biol. 12, 755–759 [DOI] [PubMed] [Google Scholar]
- 83. Cordes M., Köttgen A., Jasper C., Jacques O., Boudebous H., Giese B. (2008) Influence of amino acid side chains on long-distance electron transfer in peptides: electron hopping via “stepping stones”. Angew Chem. Int. Ed. Engl. 47, 3461–3463 [DOI] [PubMed] [Google Scholar]
- 84. Graceffa P. (1983) Spin labeling of protein sulfhydryl groups by spin trapping a sulfur radical: application to bovine serum albumin and myosin. Arch. Biochem. Biophys. 225, 802–808 [DOI] [PubMed] [Google Scholar]
- 85. Maples K. R., Jordan S. J., Mason R. P. (1988) In vivo rat hemoglobin thiyl free radical formation following administration of phenylhydrazine and hydrazine-based drugs. Drug Metab. Dispos. 16, 799–803 [PubMed] [Google Scholar]
- 86. Gatti R. M., Radi R., Augusto O. (1994) Peroxynitrite-mediated oxidation of albumin to the protein-thiyl free radical. FEBS Lett. 348, 287–290 [DOI] [PubMed] [Google Scholar]
- 87. Carballal S., Bartesaghi S., Radi R. (2014) Kinetic and mechanistic considerations to assess the biological fate of peroxynitrite. Biochim. Biophys. Acta 1840, 768–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Fasman G. D. (1976). in Handbook of Biochemistry and Molecular Biology: Physical Data and Chemical Data, pp. 305–351, CRC Press, Inc., Cleveland, OH [Google Scholar]
- 89. Radi R. (2013) Peroxynitrite, a stealthy biological oxidant. J. Biol. Chem. 288, 26464–26472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Quint P., Reutzel R., Mikulski R., McKenna R., Silverman D. N. (2006) Crystal structure of nitrated human manganese superoxide dismutase: mechanism of inactivation. Free Radic. Biol. Med. 40, 453–458 [DOI] [PubMed] [Google Scholar]
- 91. Ischiropoulos H., Zhu L., Chen J., Tsai M., Martin J. C., Smith C. D., Beckman J. S. (1992) Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch. Biochem. Biophys. 298, 431–437 [DOI] [PubMed] [Google Scholar]
- 92. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]