

Background: Fibroblasts from patients with idiopathic pulmonary fibrosis (IPF) overexpress the urokinase-type plasminogen activator receptor (uPAR) and are hypermotile.
Results: uPAR ligation increases fibroblast motility by localizing α5β1 integrin-Fyn signaling complexes to lipid rafts.
Conclusion: The hypermotile phenotype of IPF fibroblasts is due to lipid raft-localized uPAR-integrin-Fyn signaling complexes.
Significance: These unique lipid raft signals may be therapeutic targets for IPF.
Keywords: Fibroblast, Fibrosis, Integrin, Lipid Raft, Urokinase Receptor
Abstract
The urokinase-type plasminogen activator receptor (uPAR) is a glycosylphosphatidylinositol-linked membrane protein with no cytosolic domain that localizes to lipid raft microdomains. Our laboratory and others have documented that lung fibroblasts from patients with idiopathic pulmonary fibrosis (IPF) exhibit a hypermotile phenotype. This study was undertaken to elucidate the molecular mechanism whereby uPAR ligation with its cognate ligand, urokinase, induces a motile phenotype in human lung fibroblasts. We found that uPAR ligation with the urokinase receptor binding domain (amino-terminal fragment) leads to enhanced migration of fibroblasts on fibronectin in a protease-independent, lipid raft-dependent manner. Ligation of uPAR with the amino-terminal fragment recruited α5β1 integrin and the acylated form of the Src family kinase, Fyn, to lipid rafts. The biological consequences of this translocation were an increase in fibroblast motility and a switch of the integrin-initiated signal pathway for migration away from the lipid raft-independent focal adhesion kinase pathway and toward a lipid raft-dependent caveolin-Fyn-Shc pathway. Furthermore, an integrin homologous peptide as well as an antibody that competes with β1 for uPAR binding have the ability to block this effect. In addition, its relative insensitivity to cholesterol depletion suggests that the interactions of α5β1 integrin and uPAR drive the translocation of α5β1 integrin-acylated Fyn signaling complexes into lipid rafts upon uPAR ligation through protein-protein interactions. This signal switch is a novel pathway leading to the hypermotile phenotype of IPF patient-derived fibroblasts, seen with uPAR ligation. This uPAR dependent, fibrotic matrix-selective, and profibrotic fibroblast phenotype may be amenable to targeted therapeutics designed to ameliorate IPF.
Introduction
The urokinase-type plasminogen activator receptor (uPAR)2 is an external cell surface protein receptor that links to the plasma membrane by its glycosylphosphatidylinositol (GPI) side chain (1). GPI linkages, as well as protein acylation, will target proteins to lipid rafts, including specific Src family kinases (SFKs) (2–7). Lipid rafts are microdomains of the plasma membrane that are rich in cholesterol and sphingolipids (4, 6, 7). They exhibit unique biophysical properties and unique lipid and protein organization and are now thought to act as platforms for cell signaling (2, 4–8). However, the consequences of the raft localization of uPAR for cell physiology, cell signaling, and its interaction with integrins have yet to be fully elucidated.
uPAR binds its cognate ligand, urokinase-type plasminogen activator (uPA), with high affinity (Kd = 1 nm) and, upon doing so, activates several pathways (e.g. MAPK, JAK/STAT, and focal adhesion kinase (FAK)) with a host of biological responses, including adhesion, spreading, and migration, in a proteolytically independent manner (1, 9–11). Because uPAR lacks a cytoplasmic domain, the intracellular signal transduction of uPAR is effected through its association with other cell surface receptors, including epidermal growth factor receptor, G protein-coupled receptors, and integrins, to transduce signals intracellularly (1, 12). However, the regulatory triggers for uPAR signaling are not fully understood.
Previous work from our laboratory and others has shown that uPAR interacts with multiple integrins to influence cell attachment, spreading, and migration, in part through MAPK (10, 13–19). Importantly, a complete and detailed understanding of the intracellular signaling pathway that mediates these physiologic effects, the role of uPAR ligation on inducing these effects, the location mapping of individual components of the intracellular pathway, and the role of uPAR ligation in cells that express native endogenous levels of uPAR and integrins have yet to be reported. Our current work addresses these questions by describing a novel uPAR ligation-dependent signaling switch.
Fibroblasts contribute to the pathological tissue scarring of the skin, heart, kidneys, and lung through multiple actions. These include their capacity to migrate into the damaged area, synthesize extracellular matrix, and remodel the tissue (9, 20, 21). Several studies have reported that lung fibroblasts derived from patients with idiopathic pulmonary fibrosis (IPF), a fatal scarring disease of the lung, have enhanced motility compared with their normal counterparts and that pathologic collections of fibroblasts can determine prognosis in IPF (22–28). However, the mechanisms that drive this hypermigratory fibroblast phenotype have not been fully elucidated.
Prior work implicates uPAR in several important wound healing functions, such as proliferation, adhesion, differentiation, and migration (1, 29, 30). We and others have shown that fibroblasts derived from patients with fibrotic lesions exhibit up-regulation of uPAR, and we have reported that uPAR-integrin interactions mediate selective fibroblast adherence to fibrotic lung tissue (10, 24). We therefore sought to determine the molecular mechanism whereby uPAR mediates the pathologically hypermigratory phenotype of fibrotic lung fibroblasts. Our novel signaling switch described herein drives the hypermigratory phenotype of fibrotic lung fibroblasts. These observations probably have implications for fibroproliferative diseases of the lung, skin, kidney, and heart as well as cancer cell invasion and metastasis (29–34).
EXPERIMENTAL PROCEDURES
Materials
Normal human lung fibroblasts (HLF, 19Lu) were purchased from ATCC (CCl-210). Primary isolates of HLF from IPF patients and normal controls were kindly provided by Dr. Patricia Sime, with the approval of the University of Rochester Institutional Review Board. Plasma from IPF (n = 25) and chronic obstructive pulmonary disease (n = 10) patients was provided by the Lung Tissue Research Consortium and supported by NHLBI, National Institutes of Health. Plasma from age- and gender-matched normal controls (n = 30) was generously provided by Dr. Stanley L. Hazen (Cleveland Clinic). Healthy control subjects gave written informed consent approved by the Cleveland Clinic Institutional Review Board. All heparinized plasma samples (both from the Lung Tissue Research Consortium and from Dr. Hazen) were prepared identically and frozen in aliquots at −80 °C.
Human fibronectin (FN; from plasma) was from Roche Applied Science. HRP-conjugated secondary antibodies were from Jackson Immunoresearch. Fluorochrome-conjugated secondary antibodies as well as the mouse mAb anti-human transferrin receptor were purchased from Invitrogen. The amino-terminal fragment (ATF) of human urokinase was from Molecular Innovations, whereas single chain human urokinase-type plasminogen activator (scuPA) was purchased from American Diagnostica. The SFK inhibitor, PP2, and its inactive analog, PP3, were from Calbiochem. All of the siRNAs were purchased from Dharmacon; the siLentFect lipid transfection reagent was from Bio-Rad; and the integrin homologous peptide, α-325, PRHRHMGAVFLLSQEAG, and the scrambled control peptide, S-325, HQLPGAHRGVEARFSML, were purchased from Anaspec (10, 35). Antibodies 1A8 (non-competitive control), 3C6 (which competes with α5β1 integrin for uPAR binding), and 2G10 (which competes with uPA for uPAR binding) were synthesized as described (36). Mouse mAb anti-human flotillin-1 and caveolin-1 were from BD Biosciences, rabbit polyclonal anti-human Fyn and Lyn were from Santa Cruz Biotechnology, Inc., rabbit polyclonal anti-human phospho-Fyn (Tyr-530, inactive) was from Novus Biologicals, rabbit mAb anti-human phospho-Fyn (Tyr-416, active) was from Cell Signaling Technology, and all other antibodies were purchased from Millipore, as reported previously (10). All other reagents were purchased from Sigma.
Preparation of Lipid Rafts
HLF were plated on FN, serum-starved, and treated with or without the indicated agonists or inhibitors in 1% BSA, serum-free medium. Lipid rafts were prepared by sucrose density centrifugation of cold detergent cell lysates as described (37). The buoyant fractions (lipid raft) and non-buoyant fractions (non-raft) were pooled and analyzed by Western blot or ELISA as described below. The separation method was validated as in Fig. 5A.
FIGURE 5.

uPAR ligation rescues the integrin activation and migration defect upon lipid raft disruption by CD. Lipid rafts were disrupted by cholesterol depletion using treatment with or without CD (10 mm, 30 min) followed by treatment with or without CD and with or without ATF (10 nm, 60 min). A, validation of the lipid raft isolation procedure. Cells were treated with or without CD followed by Western blot for caveolin-1 (raft marker) or transferrin receptor (TsfR) (non-raft marker). B and C, filipin staining of cholesterol. B, pseudocolored photomicrographs of filipin staining (red/yellow, high; blue/green, low). C, quantification of plasma membrane/perinuclear area ratio of filipin staining. *, p < 0.05 comparing conditions with or without CD. D, effect of CD on the integrin activation/FN-coated bead binding assay as above. +, p < 0.05 comparing values with or without CD under basal (−uPA) conditions. E, monolayer migration assay as above. +, p < 0.05 comparing values with or without CD under basal (−uPA) conditions. Error bars, S.E.
ELISA and Western Blot Analysis
Same volume equivalents of lipid raft or non-raft fractions were used in ELISA (R&D Systems) and Western blot analysis, and immunoprecipitates from caveolin-1 or a control antibody (granulocyte colony-stimulating factor) were Western blotted for the indicated proteins. Solubilized proteins were separated by SDS-PAGE and then immunoblotted on Immobilon-P membranes as described (10, 38). Specific protein bands were detected using ECL Western blot reagents and directly quantified using UVP with VisionWorksLS software (Upland, CA).
Co-localization of uPAR and Lipid Rafts by Immunofluorescence
Lipid rafts were localized by staining for lipid GM1 with cholera toxin as suggested by the manufacturer (Invitrogen). Staining for uPAR was performed as described previously (10). Co-localization of uPAR and lipid rafts at the cell perimeter was determined on a pixel by pixel basis using Compix Software analysis of digital ×40 photomicrographs (>50 cells/condition).
Filipin Staining
HLF were plated on FN, serum-starved, and incubated in 1% BSA, serum-free medium with or without methyl-β-cyclodextrin (CD; 10 mm) for 30 min followed by treatment with or without CD and with or without ATF (10 nm) for 60 min. Cells were stained for filipin (50 μg/ml, 30 min, room temperature) as described (39). Fluorescence intensity pseudocolors were added with ImageJ. The intensity ratio between the cell edge and the perinuclear area was determined for >40 cells/condition.
Cell Morphology Assay
HLF were plated on FN and treated as described under “Filipin Staining.” The percentage of cells with protrusive structures was determined by direct cell counting (>200 cells/condition) of fluorescently labeled cells (phalloidin or PKH-26) at ×20 original magnification.
Fibroblast Bead Attachment Assay
FN-coated beads (5.0 μm, Thermo Scientific) were allowed to bind to serum-starved (0.4% SCM, 24 h) adherent HLF monolayers for 20 min at 37 °C in attachment assay buffer containing calcium, magnesium, and manganese in triplicate as described (40). Bead number per surface area of cells was determined by counting of beads and manual tracing of cell boundaries in low power photomicrographs using ImageJ software.
In Vitro Migration Assay
In vitro migration assays on tissue culture plates were performed as described (10). Wounded cell monolayers were incubated in 1% BSA, serum-free medium with or without the indicated agonists or inhibitors. Digital pictures of the wounds were taken at 0.5 h (time 0) and 24 h later, and areas devoid of cells (“wounds”) were measured using the ImageJ software.
In Vitro Migration Assay on Mouse Lung Tissue
All animal protocols were performed as approved by the Cleveland Clinic institutional animal care and use committee and using methods in the guidelines for the humane care of animals by the American Physiological Society. Lungs from female C57Bl/6 mice (18–20 g) that had received intratracheal instillation of 4 units/kg bleomycin, which is used to induce lung fibrosis in mice, 2 weeks prior were inflated with OCT (Sakura Finetek, Torrance, CA) as described (10, 41, 42). PKH-labeled normal HLF were allowed to attach to preblocked, 10 μm sections containing areas of normal and fibrotic lung, as published (10). After washing off unattached cells, the sections/cells were observed in 1% BSA, serum-free medium (5% CO2 at 37 °C) with or without the indicated additives on an inverted (Leica DM IRBE) microscope. Time lapse video microscopy was performed, and migration velocity was analyzed using HC Image software (Hamamatsu, Bridgewater, NJ).
Statistical Analysis
All data are means ± S.E. unless otherwise indicated. Continuous variables from more than two groups were compared by means of an analysis of variance with a post hoc analysis (Dunnett's test or Student-Newman-Keuls). Significance was accepted at the p ≤ 0.05 level.
RESULTS
uPAR Plasma Levels Are Elevated in IPF Patients and Correlate with Disease Severity
In order to evaluate whether uPAR can be physiologically linked to IPF, we measured the plasma uPAR levels in a small cohort of highly characterized patients with IPF from a national database. All plasma samples (controls and diseased) were heparinized and processed in an identical manner. IPF patients had higher plasma uPAR levels (2-fold; p < 0.01) compared with age- and gender-matched healthy controls or those with chronic obstructive pulmonary disease (COPD; Fig. 1A). Furthermore, among those with IPF, the extent of the plasma uPAR elevation positively correlated with disease severity, as measured by a low diffusion capacity (diffusion capacity for carbon monoxide (DLCO), a measure of diffusion of gas across the capillary-alveolar tissue) (Fig. 1B, Pearson correlation coefficient (r) = −0.534). Taken together, these data, if validated prospectively in a larger cohort, suggest that plasma uPAR values may be a useful indicator of disease state.
FIGURE 1.

Elevated plasma uPAR levels correlate with IPF disease and its severity. A, uPAR ELISA on plasma from healthy controls and from patients with IPF or chronic obstructive pulmonary disease (COPD). *, p < 0.05 as compared with healthy controls. +, p < 0.05 IPF patients versus chronic obstructive pulmonary disease patients. B, plasma uPAR levels correlate with DLCO in IPF patients. The top and bottom curved lines represent the 95% confidence intervals, whereas the middle line is the regression line. C, IPF cells are hyperresponsive to ATF-induced migration. *, p < 0.05 denotes difference in response of normal and IPF fibroblasts to uPAR ligation with uPAR siRNA and control siRNA (GAPDH siRNA). D, human lung fibroblasts were seeded on 10-μm fibrotic or normal mouse lung sections and treated with 10 nm ATF. Data are plotted as percentage increase in migration velocity upon uPAR ligation. *, p < 0.05 fibrotic versus normal tissue. Error bars, S.E.
The Hypermigratory Phenotype in IPF Patient-derived Fibroblasts Is Mediated by uPAR Ligation
We and others have noted that primary isolates of lung fibroblasts from IPF patients overexpress uPAR (10, 24). The physiological consequences of this observation for migration have not been elucidated. Herein, we demonstrate that primary isolates of IPF fibroblasts are hyperresponsive to the promigratory effects of uPAR ligation, relative to that of fibroblasts isolated from non-fibrotic control lungs (Fig. 1C). Throughout this work, uPAR ligation occurs either with its cognate ligand, proteolytically inactive uPA, or the uPA ligand binding domain (ATF; 10 nm). We extend these observations to show that the hyperresponsive phenotype in IPF fibroblasts is concordant with the significantly higher uPAR expression levels in the fibrotic lung-derived cells (over 31% higher by ELISA; data not shown). Importantly, knockdown of uPAR using siRNA (more than 80% knockdown by ELISA; data not shown), demonstrates that both the hyperresponsive phenotype in IPF fibroblasts and the uPA-induced migration are overwhelmingly dependent on uPAR (Fig. 1C).
uPAR Ligation Induces a Hypermigratory Response in Fibroblasts Plated on Fibrotic Lung Sections
Given the emerging understanding of the significance of cell migration in relation to the biophysical and biochemical composition of the substrate, we have developed a model system to test fibroblast migration on a physiologically relevant substrate. The effect of uPAR ligation on fibroblast migration on unfixed, normal, and fibrotic lung tissue sections from Bleomycin-injured mice was assessed. The migration response to uPAR ligation was greater in fibroblasts interacting with fibrotic areas of lung tissue, as compared with that on normal lung tissue (a 40% increase, p < 0.01; Fig. 1D). These data demonstrate that uPAR ligation plays a key role in fibroblast migration, selectively on fibrotic lesional matrix, adding additional physiological relevance to uPAR ligation in lung fibrosis.
Ligation of uPAR with uPA Induces a Motile Phenotype in Human Lung Fibroblasts
We have previously shown that uPAR interacts with multiple integrins on the surface of non-transformed human lung fibroblasts and, in doing so, up-regulates the integrin functions of adhesion and migration (10). We now show that ligation of uPAR induces a motile fibroblast phenotype. This was quantified by a greater than 3-fold increase in the percentage of cells with protrusions (3.6 ± 0.12-fold; p < 0.05), a decrease in actin stress fiber density (by 58 ± 8%; p < 0.05), an increase in integrin α5β1-dependent (10) monolayer motility on FN (75 ± 15%; p < 0.05), and a redistribution of uPAR to the leading edge of motile cells (80 ± 22%; p < 0.05) (Fig. 2, A and B). In order to confirm the strict uPAR dependence of the motile phenotype, knockdown of uPAR expression (by 90% as quantified via ELISA) with uPAR-directed siRNA (but not control siRNA) led to an almost complete abrogation of the uPAR ligation-dependent enhancing effect on monolayer motility and also reduced basal monolayer motility in the absence of uPAR ligation (by 40%, p < 0.05; Fig. 2C). In summary, uPAR ligation induces a motile phenotype in lung fibroblasts.
FIGURE 2.
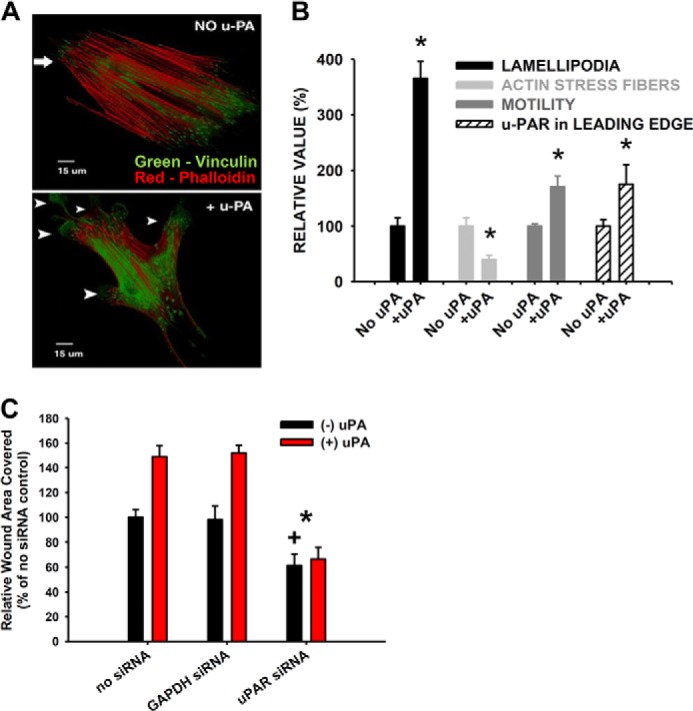
uPAR ligation induces a motile phenotype in lung fibroblasts. Normal human lung fibroblasts were plated on FN and incubated with 10 nm scuPA (A and B, +uPA), 10 nm ATF (C, +uPA), or vehicle (No uPA) for 90 min at 37 °C. A, immunofluorescence micrograph of vinculin (green) and actin stress fiber-stained (phalloidin; red) cells. The arrowheads point to protrusive structures. B, quantification of motile phenotype as indicated. *, p < 0.05, compared with no uPA. C, fibroblast monolayer migration ± uPAR or GAPDH siRNA ± 10 nm ATF. *, p < 0.05 denotes loss of uPA-induced hypermotility versus GAPDH siRNA control; +, p < 0.05 denotes decrease in basal (−uPA) migration compared with no siRNA and GAPDH siRNA controls. Error bars, S.E.
Ligation of uPAR Enhances uPAR Interactions with α5β1 Integrin-initiated Activation and Migration Signaling through the Src Family Kinase Fyn
We have previously shown basal fibroblast motility to be largely dependent on α5β1 (10). Binding of FN-coated beads to immobilized fibroblasts was used as a more direct and proximate measure of α5β1 activation under conditions with and without uPAR ligation. Ligation of uPAR with ATF enhances α5β1 activation (2-fold; p < 0.05) in a manner that is completely inhibitable by α5β1 function-blocking antibodies or α5 siRNA (Fig. 3, A and B). (Integrin α5 only pairs with β1, so α5 siRNA is also blocking α5β1 function). Concordantly, siRNA to α5 completely abrogates the uPAR ligation-induced enhancement of migration on FN as well as that seen under unligated uPAR conditions (Fig. 3, C and D). We and others have previously found that a peptide homologous to the extracellular domain of the α integrin chain (α325 peptide) disrupts uPAR-α integrin interactions either by altering the conformation of the integrin or by competing with a uPAR binding site in both solid phase assays and on the cell surface (10, 35). Disrupting uPAR-α5β1 interactions with this promiscuously acting peptide (10) completely abrogates the uPAR ligation-enhancing effect on motility in a dose-dependent manner while having minimal effect on basal motility (Fig. 3E). In contrast, a control, scrambled peptide had no effect.
FIGURE 3.

Integrin α5β1-uPAR interactions are required for uPAR ligation-induced integrin function and motility. A and B, FN-coated beads were applied to fibroblasts that were pretreated with or without ATF (10 nm, 30 min) and with or without α5 function-blocking antibody (α5 Ab, 10 μg/ml), and the number of beads/cell area was counted. A–D, *, p < 0.05 denotes loss of uPA-induced attachment or hypermotility versus increase under control conditions (IgG, scrambled siRNA). +, p < 0.05 denotes comparison among basal (−uPA) conditions. A, representative photomicrographs. B, quantification of bead attachment. C, fibroblast monolayer migration with or without α5 or scrambled siRNA with or without ATF (10 nm). D, validation of α5 siRNA knockdown by Western blot. E, fibroblast monolayer migration with or without α325 or scrambled peptide with or without 10 nm ATF. Conditions were as indicated. *, p < 0.05 denotes loss of uPA-induced hypermotility with α325 peptide compared with scrambled peptide under +uPA (10 nm ATF) conditions. F, fibroblast monolayer migration with or without antibodies (10 μg/ml each) 1A8 (control antibody), 3C6 (which blocks uPAR-β1 interactions), or 2G10 (which blocks uPAR-uPA interactions) with or without 10 nm ATF. *, p < 0.05 denotes loss of uPA-induced hypermotility with 3C6 or 2G10 antibody versus 1A8 under +uPA (10 nm ATF) conditions. Error bars, S.E.
To further substantiate that the interaction between α5β1 integrin and uPAR is crucial to the uPAR ligation enhancement of migration, we utilized a second complementary technique. Namely, we compared the effect of a previously characterized antibody that specifically competes with β1 integrin for uPAR binding with that of control antibodies that only block uPA binding to uPAR or a non-blocking control antibody that has no effect (36). We found that the increase in migration upon uPAR ligation was selectively inhibited by the uPAR-β1-blocking antibody (3C6), whereas the non-blocking control (1A8) had no effect (Fig. 3F). The uPAR-β1-blocking antibody had no effect on migration in the absence of uPAR ligation (basal migration). As expected, the uPAR-uPA interaction-blocking antibody (2G10) abrogated the uPAR ligation-induced migration. These data clearly demonstrate the significant contribution of uPAR interactions with α5β1 to uPAR-induced migration.
Because we and others have shown the importance of SFKs to migration signaling downstream of integrins in lung fibroblasts, we hypothesized that they may also be involved in the uPAR ligation enhancement of migration (25, 41). The SFK inhibitor PP2 (100 nm) selectively blocked (by 85%, p < 0.05) the uPAR ligation-induced enhancement of migration, whereas the inactive analog (PP3; 100 nm) had no effect (Fig. 4A). Down-regulation of the three SFKs expressed in our cells (44) individually identified Fyn as the key SFK mediating the increase in migration upon uPAR ligation (Fig. 4, B and C). In contrast, c-Src was the key SFK mediating migration under basal conditions while still supporting uPAR ligation-induced migration (Fig. 4, B and C). In addition, the uPAR ligation-induced increase in migration was also abrogated in IPF fibroblasts that were treated with Fyn siRNA (data not shown). Together, these data identify Fyn to be the key SFK that mediates uPAR ligation-induced migration.
FIGURE 4.

The Src family kinase family (SFK) member Fyn is critical for uPAR ligation-dependent motility. A, fibroblast monolayer migration with or without SFK inhibitor PP2 (100 nm) or its inactive analog, PP3 (100 nm) with or without ATF (10 nm). *, p < 0.05 denotes loss of uPA-induced hypermotility versus PP3 control. B, monolayer migration with or without Lyn, Fyn, c-Src, or scrambled siRNA with or without ATF (10 nm). *, p < 0.05 denotes loss of uPA-induced hypermotility versus scrambled siRNA. +, p < 0.05 denotes decrease in basal (−uPA) migration versus scrambled siRNA. #, p < 0.05 denotes difference in basal (−uPA) c-Src migration versus c-Src migration under conditions of ligated uPAR (+uPA). C, validation of SFK siRNA knockdown by Western blot. The individual proteins with their corresponding GAPDH loading controls are shown. Error bars, S.E.
uPAR Ligation Rescues the Migration Defect Due to Lipid Raft Disruption
As documented above, the uPAR ligation response in migration was largely dependent on Fyn, α5β1, and uPAR. Because uPAR is a GPI-linked protein, we determined the effect of lipid raft microdomain disruption by depletion of cholesterol on the uPAR ligation-dependent signal. Lipid raft disruption by CD was validated by the loss of caveolin-1 in buoyant lipid raft fractions (fractions 2–6) (Fig. 5A), whereas validation of cholesterol depletion by CD is shown by the loss of filipin staining in the cell plasma membrane (Fig. 5, B and C). In the absence of uPAR ligation, disruption of lipid rafts inhibited α5β1 integrin activation (FN-coated bead binding by >90%), and monolayer migration (by 50%) (Fig. 5, D and E). A general cell toxic effect of CD is unlikely to explain these findings because lactate dehydrogenase release into the medium was not detected. Surprisingly, uPAR ligation was able to completely rescue the inhibitory effect of lipid raft disruption on α5β1 activation and partially rescue its effects on migration (Fig. 5, D and E). Cholesterol staining with filipin (Fig. 5, B and C) validates that the rescue effects of uPAR ligation are independent of cholesterol in the plasma membrane. These data demonstrate that uPAR ligation can restore α5β1 function, even under conditions of reduced cholesterol-induced lipid raft disruption.
uPAR Ligation Induces the Translocation of a “Migration Signaling Complex” Consisting of uPAR, α5β1 Integrin, and Fyn into Lipid Rafts
In order to understand the mechanism whereby uPAR ligation restores α5β1 function and signaling in a cholesterol-independent manner, we first determined the biochemical and spatial relationships of the key signaling components before and after uPAR ligation and/or cholesterol depletion. Approximately 30–50% of the total cellular uPAR was found to be in lipid rafts under basal conditions by biochemical and spatial localization techniques (data not shown). The addition of CD disrupted the co-localization of uPAR with lipid rafts, as measured by the raft-localized lipid, GM1. However, the addition of CD followed by uPAR ligation restored the co-localization of uPAR with lipid rafts (Fig. 6, A and B). Furthermore, uPAR ligation increased the absolute amount of uPAR in lipid rafts, even under conditions of reduced cholesterol (+CD, +uPA conditions; Fig. 6C), suggesting that protein-protein or protein-lipid interactions can supersede cholesterol depletion in recruiting GPI-linked proteins to lipid rafts.
FIGURE 6.

uPAR ligation restores uPAR localization to lipid rafts after disruption. Lipid rafts were disrupted with CD as above followed by treatment with or without scuPA (A and B) or ATF (C; 10 nm, 60 min). +, p < 0.05 comparing values with or without CD under basal (−uPA) conditions. A, immunofluorescence micrographs. Lipid rafts were stained with cholera toxin (GM1; green) and uPAR (red). The arrows point to areas of lipid raft/uPAR co-localization. B, quantification of the co-localization of lipid rafts and uPAR in the plasma membrane. C, uPAR ELISA from lipid raft fractions. Error bars, S.E.
In contrast to reports in other cells, integrin α5β1 was all but excluded from rafts under basal conditions. Surprisingly, ligation of uPAR induces a specific translocation of α5β1 integrin (but not αV or α3 integrins; data not shown) into the raft microdomains, and as above, α5β1 recruitment can also be partially restored in rafts by uPAR ligation, even after cholesterol depletion (Fig. 7, A and B). A similar recruitment effect of uPAR ligation on Fyn, caveolin-1, and flotillin was noted but not Lyn (Fig. 7, A and B). In addition, only the active phosphorylated Fyn (p-Fyn, Tyr(P)-416) was recruited to lipid rafts upon uPAR ligation, whereas the inactive phosphorylated Fyn (Tyr(P)-530) was not (Fig. 7, C and D). Taken together, these data demonstrate that uPAR ligation recruits uPAR itself, Fyn, caveolin-1, and α5β1 integrin into lipid rafts, thereby co-localizing the key migration signaling intermediates, even under cholesterol-depleted conditions.
FIGURE 7.

uPAR ligation recruits a migration signaling complex (α5β1 integrin, Fyn, and caveolin) to lipid rafts. Lipid rafts were disrupted with CD followed by uPAR ligation with ATF (+uPA) as above. A and C, representative Western blots from lipid raft fractions. Experiments were repeated in triplicate. UT, untreated cells. B and D, average band densities from at least three independent experiments. Band densities were taken with respect to the value under untreated conditions for each blot. *, p < 0.05 comparing untreated versus +uPA conditions; +, p < 0.05 comparing UT versus +CD conditions; #, p < 0.05 comparing +CD versus +CD, +uPA conditions. E, Western blots from caveolin-1 and control antibody (granulocyte colony-stimulating factor) immunoprecipitates (IP) from lipid raft fractions. Experiments were performed in triplicate. F, average band densities from at least three independent experiments. *, p < 0.05 comparing UT versus +uPA conditions. Band densities were taken with respect to the value under untreated conditions for each blot. Error bars, S.E.
α5β1 Is Recruited to Lipid Rafts through Interactions with uPAR
Because uPAR is a GPI-linked protein and Fyn is a doubly acylated protein, these would be expected to favor lipid raft microdomains. However, because α5β1 integrin is a non-acylated, transmembrane protein, we examined if α5β1 integrin is recruited to lipid rafts through specific and distinct protein-protein interactions. First, the disruptive effects of cholesterol depletion and the restorative effects of uPAR ligation on the lipid raft-associated proteins, flotillin and caveolin, paralleled that of the GPI-linked uPAR, acylated Fyn, and GM1. These findings suggest that uPAR ligation recruits general raft component proteins and lipids (Figs. 6 and 7 (A and B)). Second, only the caveolin-1-associated Fyn and α5β1 integrin were enhanced in the lipid rafts upon uPAR ligation (Fig. 7, E and F). Third, the integrin homologous peptide (α325) selectively blocks the recruitment of α5β1 and Fyn (but not flotillin, caveolin-1, or uPAR) into lipid rafts upon uPAR ligation (Fig. 8, A–C). This suggests that raft-localized uPAR recruits α5β1 integrin and Fyn in a ligation-dependent manner.
FIGURE 8.

uPAR-integrin interactions are required for α5β1 integrin and Fyn recruitment to lipid rafts. uPAR-integrin interactions were blocked by integrin homologous peptide (α325) or scrambled peptide followed by ligation of uPAR (with or without ATF, 10 nm, 60 min). A, uPAR ELISA from lipid raft fractions. *, p < 0.05 comparing −uPA versus +uPA conditions. B, representative Western blots from lipid raft fractions. Experiments were repeated in triplicate. C, average band densities from at least three independent experiments. Band densities were taken with respect to the value under untreated conditions for each blot. *, p < 0.05 comparing scrambled peptide alone versus scrambled peptide, +uPA conditions; #, p < 0.05 comparing α325 peptide versus α325, +uPA conditions. Error bars, S.E.
uPAR Ligation Induces a Raft-localized Migration Signaling Switch to an Acylated Fyn-dependent Pathway
Integrins are known to transmit intracellular signals though their β-cytoplasmic tail via Src-FAK-MAPK or via their α subunit through interactions with caveolin-Fyn-Shc (45). We demonstrate that uPAR ligation-induced migration predominantly signals via the caveolin-Fyn-Shc pathway (Fig. 9A). uPAR ligation-induced migration was selectively abrogated upon knockdown of caveolin-1, Fyn, and Shc (Fig. 9, A and B). In contrast, there was no effect of knocking down c-Src (Fig. 4B) or FAK (Fig. 9A) on uPAR ligation-induced migration. As expected, down-regulation of FAK, in the absence of uPAR ligation, attenuates the basal migration (41, 42).
FIGURE 9.

The caveolin-1-Fyn-Shc pathway mediates uPAR ligation-induced fibroblast motility. Fibroblasts were pretreated with siRNA for caveolin-1, Fyn, Shc, FAK, or scrambled siRNA. A, monolayer migration with or without ATF. *, p < 0.05 denotes loss of uPA-induced hypermotility with indicated siRNA versus scrambled siRNA. +, p < 0.05 denotes decrease in basal (−uPA) migration versus scrambled siRNA. B, validation of siRNA knockdown by Western blot. The individual proteins with their corresponding GAPDH loading controls are shown. Error bars, S.E.
We next aimed to determine the importance of lipid raft-localized (i.e. doubly acylated) Fyn to uPAR ligation-induced migration. Depletion of the palmitoylated form of Fyn using 2-bromopalmitate (46) (2-BP; 50 μm) selectively blocks the recruitment of Fyn but not of uPAR, caveolin-1, or α5β1 integrin (Fig. 10, A–C). At the functional level, both depalmitoylation (with 2-BP) or demyristoylation (with 2-hydroxymyristate (2-OHmyr); 50 μm) of Fyn blocks the uPAR ligation-induced migration enhancement, whereas the control treatment (palmitic acid; 50 μm) shows no effect (Fig. 10C). Taken together, these data indicate that the recruitment of Fyn to lipid rafts upon uPAR ligation requires acylation of Fyn and that raft-associated, acylated Fyn specifically initiates downstream migration signaling under these conditions. Furthermore, these data show that Fyn is not the primary driver of α5β1, caveolin, or uPAR to the rafts upon uPAR ligation.
FIGURE 10.

Lipid raft-associated Fyn is critical for the uPAR ligation enhancement of migration. Fibroblasts were treated with or without acylation inhibitors 2-BP (50 μm), 2-hydroxymyristolate (2-OHmyr; 50 μm), or palmitic acid control (50 μm) with or without 10 nm ATF. A, uPAR ELISA from lipid raft fractions. *, p < 0.05 comparing −uPA versus +uPA conditions. B, representative Western blots from lipid raft fractions. Experiments were repeated in triplicate. UT, untreated cells. C, average band densities from at least three independent experiments. Band densities were taken with respect to the value under untreated conditions for each blot. *, p < 0.05 comparing untreated versus +uPA conditions; #, p < 0.05 comparing +2-BP versus +2-BP, +uPA conditions. D, monolayer migration of fibroblasts with depleted levels of acylated specific Fyn. *, p < 0.05 denotes loss of uPA-induced hypermotility versus palmitic acid control; +, p < 0.05 denotes decrease in basal migration versus palmitic acid control. Error bars, S.E.
DISCUSSION
Our study, for the first time, demonstrates that uPAR ligation with uPA in physiological concentrations induces a motile phenotype in fibroblasts through a novel, integrin signaling switch mechanism (see proposed model in Fig. 11). This was demonstrable using fibroblasts migrating on fibronectin and occurred in a protease-independent but lipid raft-dependent manner. This switching function is dependent on uPAR-integrin coupling and results in a unique migration-inducing signal that localizes to lipid raft microdomains. This raft-constrained migration signal is dependent on integrin translocation into lipid rafts and acylation of Fyn and switches to an α5β1 integrin signaling pathway through caveolin-Fyn-Shc. Furthermore, the hypermotile phenotype, long noted in primary isolates of IPF patient-derived fibroblasts, is dependent on this uPAR ligation-generated Fyn signal, and uPAR ligation selectively enhances migration of fibroblasts on fibrotic lung areas. These data suggest that targeting uPAR or Fyn might lead to selective and novel therapies targeted to cell migration in the setting of uPAR ligation (i.e. cancer, IPF, angiogenesis, atherosclerosis) (1, 47, 48).
FIGURE 11.
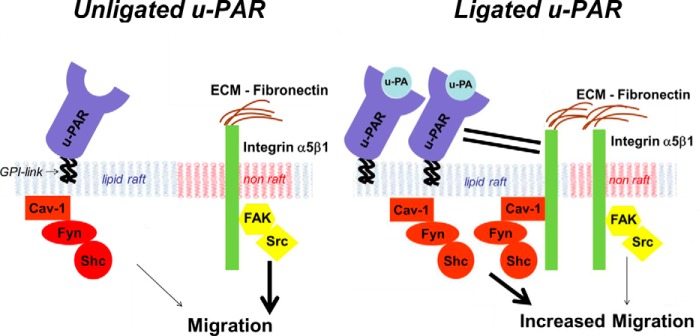
Proposed integrin signaling switch model. Under unligated uPAR conditions, migration is mainly initiated by non-raft (pink plasma membrane) signals through FAK-Src (yellow) and requires both α5β1 integrin and uPAR. In contrast, uPAR ligation enhances motility resulting from a recruitment of uPAR-α5β1 integrin complexes (solid black lines), along with Cav-1 and Fyn to lipid rafts (blue plasma membrane), and a corresponding increase in the dependence of migration on the lipid raft-localized Cav-1-Fyn-Shc (orange) pathway. The raft-localized Cav-1-Fyn-Shc pathway activation induces a hypermigratory phenotype.
Prior work has demonstrated that α5β1 and uPAR co-immunoprecipitate in a uPA-dependent manner in a cell-free system and that chemotaxis to uPA is dependent on integrins in uPAR-overexpressing CHO cells (19). Furthermore, uPAR, α5β1, and caveolin have been shown to interact but, upon doing so, inhibit α5β1 function (17, 18). Our work builds on these concepts by demonstrating a uPAR ligation-induced, integrin signaling switching mechanism that enhances integrin function. Further, our data indicate that this signaling switch is a consequence of the lipid raft localization of uPAR-α5β1 complexes. Furthermore, these effects were both present in untransformed human lung fibroblasts at their endogenous, low level uPAR expression levels and were up-regulated in a functional manner in diseased fibroblasts. In addition, we observed physiological effects of uPAR ligation on migration in physiologically relevant matrices using physiologically relevant concentrations of non-proteolytically active uPA (ATF or uPAR binding region). Our laboratory and others have shown that uPAR can interact with other integrins to influence cell behavior (10, 13–16, 18). This may extend the significance of our findings to the function of other integrins and potentially other diseases. However, to our knowledge, a uPAR ligation-dependent signaling pathway switch has not been described previously.
Our data indicate that uPAR-α5β1 interactions are the driving force for the translocation of the alternate caveolin-Fyn-Shc signaling pathway (see proposed model in Fig. 11). The α325 peptide, which disrupts uPAR-integrin interactions, not only abrogated the uPAR ligation-enhancing effect on migration but also selectively blocked the translocation of both α5β1 and Fyn (but not flotillin, caveolin-1, or uPAR) into lipid rafts upon uPAR ligation. However, deacylation of Fyn blocked only the recruitment of Fyn to lipid rafts upon uPAR ligation, with no effects on α5β1, caveolin-1, and uPAR. These data suggest that α5β1 is upstream and drives Fyn translocation into lipid rafts upon uPAR ligation.
Although overexpression of uPAR has been shown to stimulate α5β1 activation (49), prior work shows that the percentage of total cellular uPAR associated with integrins varies (32, 35). In our study, less than 1% of the α5β1 was located in lipid rafts under basal conditions, and this value increased to just 2.4% upon uPAR ligation. It is surprising that such a small increase in lipid raft-associated α5β1 has a dominant effect on uPAR ligation-induced signaling.
Our data demonstrate that uPAR ligation induces a raft with unique molecular composition, enriched in α5β1 integrin and caveolin-Fyn-Shc. Furthermore, we show that uPAR ligation trumps cholesterol depletion in supporting raft-localized signaling/assembly and downstream physiological effects. Prior work has shown similar cholesterol-independent effects through cross-linking of raft proteins by antibodies or raft lipids with cholera toxin (50, 51). We speculate that the binding of ATF influences protein-protein interactions (α5β1 and uPAR), much like antibody cross-linking. The α integrin peptide results (Fig. 8) suggest that uPAR-α5β1 interactions are critical protein-protein interactions for the recruitment of α5β1 integrin and Fyn to rafts. Prior work suggests that uPAR directly binds α5β1, thereby inducing a conformational change in the integrin extracellular domain, with distinct physiological consequences (49, 52). We speculate that this conformational change might result in an integrin with increased avidity for rafts. However, recruitment of other raft-localized proteins (i.e. caveolin and flotillin), although increased by uPAR ligation, is less dependent on uPAR- integrin interactions, suggesting that a second distinct mechanism is operative.
Our data suggest that uPAR expression and function may be important to the pathogenesis of IPF. We and others have shown that primary fibroblasts from IPF patients overexpress uPAR as compared with those from normal controls (10, 24). Also, bronchoalveolar lavage fluid from patients with rapidly progressing IPF has been reported to induce a higher fibroblast migration rate than that from controls or from slower progressors (26). Although several putative mediators/modulators of this hypermotile/invasive phenotype have been identified, including PGE2, LPA, Thy-1, and hyaluronan (20, 28, 53–56), we have shown, for the first time, that IPF fibroblasts are hyperresponsive to the promigratory effects of uPAR ligation, as compared with those from normal controls. The response of the primary fibroblast isolates from normal controls to uPAR ligation is identical to that of the human lung fibroblasts (19Lu) described throughout this work. Furthermore, this response is dependent on uPAR signaling through Fyn. This finding indicates a switch from the predominant non-raft, integrin-mediated migration signal (i.e. to PDGF) that we have characterized through FAK in the absence of uPAR ligation (25, 41, 42) to a raft-dominant, hypermigratory signal through caveolin-Fyn-Shc when uPAR is ligated with uPA. Because the down-regulation of Fyn with siRNA only significantly affected the migration of uPA-ligated cells and not those that were untreated, it is possible that selective targeting of these uPA-ligated cells with a Fyn inhibitor could be a potential therapeutic inhibitor of fibrosis.
Our observation regarding the selectivity of the hypermigratory effect of uPAR ligation to fibroblasts interacting with fibrotic lung matrix further supports the possibility for selective targeting of fibrotic lesional fibroblasts (Fig. 1D). In addition, it extends the uPAR-α5β1 interaction to the tissue level because we have shown that α5β1 is the predominant integrin mediating migration in our cells (10).
Plasma soluble uPAR levels have been associated with fibrotic disease of the kidney glomeruli and other acute illnesses, although the precise cellular source of the uPAR in plasma has yet to be identified (31, 57–60). Our data demonstrate the novel finding that plasma uPAR levels are elevated in IPF and correlate with disease severity, using the standard measure of the DLCO. DLCO is also an important prognosticator in IPF patients, opening up the possibility that plasma uPAR might act as a surrogate marker for prognosis in IPF (61–65).
uPAR ligation has been demonstrated to have protean effects on tissue repair and disease pathobiology. For example, ligated uPAR can mediate fibronectin matrix assembly by activating Src, epidermal growth factor receptor, and β1 integrin; and ligated uPAR has also been shown to mediate cancer cell migration/transformation and angiogenesis (1, 47, 48, 66). Similarly, Shc-initiated signals have been shown to be involved in multiple illnesses related to aging, including cardiovascular and neurological diseases and cancer (67). Therefore, it is possible that the uPAR ligation-initiated caveolin-Fyn-Shc signaling described herein could be important not only to IPF but to these other diseases of aging as well.
Although we have shown that lipid raft-localized, ligated uPAR signals through α5 and Fyn to increase migration and that uPAR may be related to IPF pathogenesis, there are limitations to our work. Although lipid raft isolation results can vary depending on methodology (4, 68), we used an identical, established protocol under all conditions (37) and, importantly, verified the isolation methods and the effects of methyl-β-cyclodextrin with several known lipid raft and non-raft markers. In addition, our described mechanism of uPAR ligation modulating migration is distinct from the integrin-independent mechanism described previously (69–71). In that prior work, direct binding of raft-localized, dimeric cell surface uPAR to the somatomedin B region of vitronectin mediates cell adhesion and lamellipodia formation through the MAPK pathway in epithelial cells (69–71). Also, other work supports the hypothesis that the ameliorative effects of exogenous uPA on lung fibrosis in animal models of human IPF are a consequence of the proteolytic effects of uPA (72–75).
In summary, uPAR ligation leads to the enhanced attachment and migration of human lung fibroblasts through recruitment of α5β1 integrin and caveolin-Fyn-Shc signaling to lipid rafts. Upon doing so, the predominant integrin-initiated signal for migration switches from a FAK-based signal to a lipid raft-localized caveolin-acylated Fyn-dependent signal (see proposed model in Fig. 11). Further, we have shown the importance of uPAR signaling to the hypermotile phenotype in IPF fibroblasts, whether using a system with matrix protein-coated plastic wells or physiologically relevant matrix from actual fibrotic lung. Taken together, these observations suggest that the unique lipid raft signaling platforms that are formed under conditions of uPAR ligation mediate the migratory processes in the fibrotic lung. In doing so, they may provide a novel selective target for strategies designed to ameliorate IPF and other devastating fibrotic diseases. In addition, results of this study may be applicable to other diseases in which uPAR signaling has been reported to play a role, including severe sepsis, angiogenesis, and many cancers (1, 30, 32, 43, 57).
Acknowledgments
We thank Dr. Edward Plow for critical reading of the manuscript. This study utilized biological specimens and data provided by the Lung Tissue Research Consortium supported by NHLBI, National Institutes of Health. We thank Dr. Patricia Sime (University of Rochester) for providing primary isolates of human lung fibroblasts from IPF patients and normal controls, and we thank Dr. Stanley Hazen (Cleveland Clinic) for providing heparinized plasma from healthy controls.
This work was supported, in whole or in part, by National Institutes of Health (NIH) Grants HL-58655 and HL-103553 (to M. A. O.), HL-085324 (to Q. D. and M. A. O.), and HL-44712 (to H. A. C.) and NIH, NCI, Grants R01 CA152883 and CA127620 (to C. L. G.). This work was also supported by grants from the Veterans Administration MERIT Review (to M. A. O.) and Department of Defense Idea Development Award PC 111318 (to C. S. C.).
- uPAR
- urokinase-type plasminogen activator receptor
- uPA
- urokinase-type plasminogen activator
- GPI
- glycosylphosphatidylinositol
- SFK
- Src family kinase
- IPF
- idiopathic pulmonary fibrosis
- HLF
- human lung fibroblasts
- FN
- fibronectin
- ATF
- amino-terminal fragment
- scuPA
- single chain human urokinase-type plasminogen activator
- CD
- methyl-β-cyclodextrin
- DLCO
- diffusion capacity for carbon monoxide
- 2-BP
- 2-bromopalmitate
- FAK
- focal adhesion kinase
- GM1
- monosialotetrahexosylganglioside.
REFERENCES
- 1. Blasi F., Carmeliet P. (2002) UPAR: a versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 3, 932–943 [DOI] [PubMed] [Google Scholar]
- 2. Brown D. A., London E. (1998) Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14, 111–136 [DOI] [PubMed] [Google Scholar]
- 3. Brown D. A. (2006) Lipid rafts, detergent-resistant membranes, and raft targeting signals. Physiology 21, 430–439 [DOI] [PubMed] [Google Scholar]
- 4. Munro S. (2003) Lipid rafts: elusive or illusive? Cell 115, 377–388 [DOI] [PubMed] [Google Scholar]
- 5. Maxfield F. R. (2002) Plasma membrane microdomains. Curr. Opin. Cell Biol. 14, 483–487 [DOI] [PubMed] [Google Scholar]
- 6. Lingwood D., Simons K. (2010) Lipid rafts as a membrane-organizing principle. Science 327, 46–50 [DOI] [PubMed] [Google Scholar]
- 7. Simons K., Toomre D. (2000) Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1, 31–39 [DOI] [PubMed] [Google Scholar]
- 8. Davy A., Gale N. W., Murray E. W., Klinghoffer R. A., Soriano P., Feuerstein C., Robbins S. M. (1999) Compartmentalized signaling by GPI-anchored ephrin-A5 requires the fyn tyrosine kinase to regulate cellular adhesion. Genes Dev. 13, 3125–3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lagares D., Busnadiego O., García-Fernández R. A., Kapoor M., Liu S., Carter D. E., Abraham D., Shi-Wen X., Carreira P., Fontaine B. A., Shea B. S., Tager A. M., Leask A., Lamas S., Rodríguez-Pascual F. (2012) Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum. 64, 1653–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhu S., Gladson C. L., White K. E., Ding Q., Stewart J. E., Jr., Jin T. H., Chapman H. A., Jr., Olman M. A. (2009) Urokinase receptor mediates lung fibroblast attachement and migration towards provisional matrix proteins through interaction with multiple integrins. Am. J. Physiol. Lung Cell Mol. Physiol. 297, L97–L108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. D'Alessio S., Blasi F. (2009) The urokinase receptor as an entertainer of signal transduction. Front. Biosci. 14, 4575–4587 [DOI] [PubMed] [Google Scholar]
- 12. Goel H. L., Sayeed A., Breen M., Zarif M. J., Garlick D. S., Leav I., Davis R. J., Fitzgerald T. J., Morrione A., Hsieh C. C., Liu Q., Dicker A. P., Altieri D. C., Languino L. R. (2013) β1 integrins mediate resistance to ionizing radiation in vivo by inhibiting c-Jun amino terminal kinase 1. J. Cell Physiol. 228, 1601–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ossowski L., Aguirre-Ghiso J. A. (2000) Urokinase receptor and integrin partnership: coordination of signaling for cell adhesion, migration and growth. Curr. Opin. Cell Biol. 12, 613–620 [DOI] [PubMed] [Google Scholar]
- 14. Pluskota E., Soloviev D. A., Plow E. F. (2003) Convergence of the adhesive and fibrinolytic systems: recognition of urokinase by integrin αMβ2 as well as by the urokinase receptor regulates cell adhesion and migration. Blood 101, 1582–1590 [DOI] [PubMed] [Google Scholar]
- 15. Wei Y., Lukashev M., Simon D. I., Bodary S. C., Rosenberg S., Doyle M. V., Chapman H. A. (1996) Regulation of integrin function by the urokinase receptor. Science 273, 1551–1555 [DOI] [PubMed] [Google Scholar]
- 16. Simon D. I., Rao N. K., Xu H., Wei Y., Majdic O., Ronne E., Kobzik L., Chapman H. A. (1996) Mac-1 (CD11b/CD18) and the urokinase receptor (CD87) form a functional unit on monocytic cells. Blood 88, 3185–3194 [PubMed] [Google Scholar]
- 17. Wei Y., Yang X., Liu Q., Wilkins J. A., Chapman H. A. (1999) A role for caveolin and the urokinase receptor in integrin-mediated adhesion and signaling. J. Cell Biol. 144, 1285–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chapman H. A., Wei Y., Simon D. I., Waltz D. A. (1999) Role of urokinase receptor and caveolin in regulation of integrin signaling. Thromb. Haemost. 82, 291–297 [PubMed] [Google Scholar]
- 19. Tarui T., Andronicos N., Czekay R.-P., Mazar A. P., Bdeir K., Parry G. C., Kuo A., Loskutoff D. J., Cines D. B., Takada Y. (2003) Critical role of integrin α5β1 in urokinase (uPA)/urokinase receptor (uPAR, CD87) signaling. J. Biol. Chem. 278, 29863–29872 [DOI] [PubMed] [Google Scholar]
- 20. Li Y., Jiang D., Liang J., Meltzer E. B., Gray A., Miura R., Wogensen L., Yamaguchi Y., Noble P. W. (2011) Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J. Exp. Med. 208, 1459–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hinz B., Phan S. H., Thannickal V. J., Prunotto M., Desmoulière A., Varga J., De Wever O., Mareel M., Gabbiani G. (2012) Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am. J. Pathol. 180, 1340–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. King T. E., Jr., Schwarz M. I., Brown K., Tooze J. A., Colby T. V., Waldron J. A., Jr., Flint A., Thurlbeck W., Cherniack R. M. (2001) Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am. J. Respir. Crit. Care Med. 164, 1025–1032 [DOI] [PubMed] [Google Scholar]
- 23. Suganuma H., Sato A., Tamura R., Chida K. (1995) Enhanced migration of fibroblasts derived from lungs with fibrotic lesions. Thorax 50, 984–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shetty S., Kumar A., Johnson A. R., Pueblitz S., Holiday D., Raghu G., Idell S. (1996) Differential expression of the urokinase receptor in fibroblasts from normal and fibrotic human lungs. Am. J. Respir. Cell Mol. Biol. 15, 78–87 [DOI] [PubMed] [Google Scholar]
- 25. Cai G. Q., Zheng A., Tang Q., White E. S., Chou C. F., Gladson C. L., Olman M. A., Ding Q. (2010) Downregulation of FAK-related non-kinase mediates the migratory phenotype of human fibrotic lung fibroblasts. Exp. Cell Res. 316, 1600–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Selman M., Carrillo G., Estrada A., Mejia M., Becerril C., Cisneros J., Gaxiola M., Pérez-Padilla R., Navarro C., Richards T., Dauber J., King T. E., Jr., Pardo A., Kaminski N. (2007) Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern. PLoS One 2, e482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. White E. S., Thannickal V. J., Carskadon S. L., Dickie E. G., Livant D. L., Markwart S., Toews G. B., Arenberg D. A. (2003) Integrin α4β1 regulates migration across basement membranes by lung fibroblasts: a role for phosphatase and tensin homologue deleted on chromosome 10. Am. J. Respir. Crit. Care Med. 168, 436–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. White E. S., Atrasz R. G., Dickie E. G., Aronoff D. M., Stambolic V., Mak T. W., Moore B. B., Peters-Golden M. (2005) Prostaglandin E2 inhibits fibroblast migration by E-prostanoid 2 receptor-mediated increase in PTEN activity. Am. J. Respir. Cell Mol. Biol. 32, 135–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Raghu H., Sodadasu P. K., Malla R. R., Gondi C. S., Estes N., Rao J. S. (2010) Localization of uPAR and MMP-9 in lipid rafts is critical for migration, invasion, and angiogenesis in human breast cancer cells. BMC Cancer 10, 647–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Veeravalli K. K., Rao J. S. (2012) MMP-9 and uPAR regulated glioma cell migration. Cell Adh. Migr. 6, 509–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wei C., El Hindi S., Li J., Fornoni A., Goes N., Sageshima J., Maiguel D., Karumanchi S. A., Yap H. K., Saleem M., Zhang Q., Nikolic B., Chaudhuri A., Daftarian P., Salido E., Torres A., Salifu M., Sarwal M. M., Schaefer F., Morath C., Schwenger V., Zeier M., Gupta V., Roth D., Rastaldi M. P., Burke G., Ruiz P., Reiser J. (2011) Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat. Med. 17, 952–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chaurasia P., Aguirre-Ghiso J. A., Liang O. D., Gardsvoll H., Ploug M., Ossowski L. (2006) A region in urokinase plasminogen receptor domain III controlling a functional association with α5β1 integrin and tumor growth. J. Biol. Chem. 281, 14852–14863 [DOI] [PubMed] [Google Scholar]
- 33. Busso N., Masur S. K., Lazega D., Waxman S., Ossowski L. (1994) Induction of cell migration by pro-urokinase binding to its receptor, possible mechanism for signal transduction in human epithelial cells. J. Cell Biol. 126, 259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rasch M. G., Lund I. K., Almasi C. E., Hoyer-Hansen G. (2008) Intact and cleaved uPAR forms: diagnostic and prognostic value in cancer. Front. Biosci. 13, 6752–6762 [DOI] [PubMed] [Google Scholar]
- 35. Wei Y., Eble J. A., Wang Z., Kreidberg J. A., Chapman H. A. (2001) Urokinase receptors promote β1 integrin function through interactions with integrin α3β1. Mol. Biol. Cell 12, 2975–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Duriseti S., Goetz D. H., Hostetter D. R., LeBeau A. M., Wei Y., Craik C. S. (2010) Antagonistic anti-urokinase plasminogen activator receptor (uPAR) antibodies significantly inhibit uPAR-mediated cellular signaling and migration. J. Biol. Chem. 285, 26878–26888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lingwood D., Simons K. (2007) Detergent resistance as a tool in membrane research. Nat. Protoc. 2, 2159–2165 [DOI] [PubMed] [Google Scholar]
- 38. Olman M. A., Simmons W. L., Pollman D. J., Loftis A. Y., Bini A., Miller E. J., Fuller G. M., Rivera K. (1996) Polymerization of fibrinogen in murine bleomycin-induced lung injury. Am. J. Physiol. 271, L519–L526 [DOI] [PubMed] [Google Scholar]
- 39. Pörn M. I., Slotte J. P. (1995) Localization of cholesterol in sphingomyelinase-treated fibroblasts. Biochem. J. 308, 269–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. del Pozo M. A., Alderson N. B., Kiosses W. B., Chiang H. H., Anderson R. G., Schwartz M. A. (2004) Integrins regulate Rac targeting by internalization of membrane domains. Science 303, 839–842 [DOI] [PubMed] [Google Scholar]
- 41. Ding Q., Gladson C. L., Wu H., Hayasaka H., Olman M. A. (2008) Focal adhesion kinase (FAK)-related non-kinase inhibits myofibroblast differentiation through differential MAPK activation in a FAK-dependent manner. J. Biol. Chem. 283, 26839–26849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ding Q., Cai G. Q., Hu M., Yang Y., Zheng A., Tang Q., Gladson C. L., Hayasaka H., Wu H., You Z., Southern B. D., Grove L. M., Rahaman S. O., Fang H., Olman M. A. (2013) FAK-related non-kinase is a multifunctional negative regulator of pulmonary fibrosis. Am. J. Pathol. 182, 1572–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hu J., Jo M., Cavenee W. K., Furnari F., VandenBerg S. R., Gonias S. L. (2011) Crosstalk between the urokinase-type plasminogen activator receptor and EGF receptor variant III supports survival and growth of glioblastoma cells. Proc. Natl. Acad. Sci. U.S.A. 108, 15984–15989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ding Q., Stewart J., Jr., Olman M. A., Klobe M. R., Gladson C. L. (2003) The pattern of enhancement of SRC kinase activity on PDGF stimulation of glioblastoma cells is affected by the integrin engaged. J. Biol. Chem. 278, 39882–39891 [DOI] [PubMed] [Google Scholar]
- 45. Wary K. K., Mariotti A., Zurzolo C., Giancotti F. G. (1998) A requirement for caveolin-1 and associated kinase fyn in integrin signaling and anchorage-dependent cell growth. Cell 94, 625–634 [DOI] [PubMed] [Google Scholar]
- 46. Webb Y., Hermida-Matsumoto L., Resh M. D. (2000) Inhibition of protein palmitoylation, raft localization, and T cell signaling by 2-bromopalmitate and polyunsaturated fatty acids. J. Biol. Chem. 275, 261–270 [DOI] [PubMed] [Google Scholar]
- 47. Aguirre Ghiso J. A., Kovalski K., Ossowski L. (1999) Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J. Cell Biol. 147, 89–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nguyen D. H., Hussaini I. M., Gonias S. L. (1998) Binding of urokinase-type plasminogen activator to its receptor in MCF-7 cells activates extracellular signal-regulated kinase 1 and 2 which is required for increased cellular motility. J. Biol. Chem. 273, 8502–8507 [DOI] [PubMed] [Google Scholar]
- 49. Wei Y., Czekay R.-P., Robillard L., Kugler M. C., Zhang F., Kim K. K., Xiong J. P., Humphries M. J., Chapman H. A. (2005) Regulation of alpha5beta1 integrin conformation and function by urokinase receptor binding. J. Cell Biol. 168, 501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Harder T., Scheiffele P., Verkade P., Simons K. (1998) Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 141, 929–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Janes P. W., Ley S. C., Magee A. I. (1999) Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J. Cell Biol. 147, 447–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wei Y., Tang C. H., Kim Y., Robillard L., Zhang F., Kugler M. C., Chapman H. A. (2007) Urokinase receptors are required for α5β1 integrin-mediated signaling in tumor cells. J. Biol. Chem. 282, 3929–3939 [DOI] [PubMed] [Google Scholar]
- 53. Rege T. A., Hagood J. S. (2006) Thy-1 as a regulator of cell-cell and cell-matrix interactions in axon regeneration, apoptosis, adhesion, migration, cancer, and fibrosis. FASEB J. 20, 1045–1054 [DOI] [PubMed] [Google Scholar]
- 54. Rege T. A., Pallero M. A., Gomez C., Grenett H. E., Murphy-Ullrich J. E., Hagood J. S. (2006) Thy-1, via its GPI anchor, modulates Src family kinase and focal adhesion kinase phosphorylation and subcellular localization, and fibroblast migration, in response to thrombospondin-1/hep I. Exp. Cell Res. 312, 3752–3767 [DOI] [PubMed] [Google Scholar]
- 55. Tager A. M., LaCamera P., Shea B. S., Campanella G. S., Selman M., Zhao Z., Polosukhin V., Wain J., Karimi-Shah B. A., Kim N. D., Hart W. K., Pardo A., Blackwell T. S., Xu Y., Chung J., Luster A. D. (2008) The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 14, 45–54 [DOI] [PubMed] [Google Scholar]
- 56. Zhou Y., Hagood J. S., Lu B., Merryman W. D., Murphy-Ullrich J. E. (2010) Thy-1-integrin αvβ5 interactions inhibit lung fibroblast contraction-induced latent TGF-b1 activiation and myofibroblast differentiation. J. Biol. Chem. 285, 22382–22393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Donadello K., Scolletta S., Covajes C., Vincent J.-L. (2012) suPAR as a prognostic biomarker in sepsis. BMC Med. 10, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Koch A., Voigt S., Kruschinski C., Sanson E., Dückers H., Horn A., Yagmur E., Zimmermann H., Trautwein C., Tacke F. (2011) Circulating soluble urokinase plasminogen activator receptor is stably elevated during the first week of treatment in the intensive care unit and predicts mortality in critically ill patients. Crit. Care 15, R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ostrowski S. R., Ullum H., Goka B. Q., Høyer-Hansen G., Obeng-Adjei G., Pedersen B. K., Akanmori B. D., Kurtzhals J. A. (2005) Plasma concentrations of soluble urokinase-type plasminogen activator receptor are increased in patients with malaria and are associated with a poor clinical or a fatal outcome. J. Infect. Dis. 191, 1331–1341 [DOI] [PubMed] [Google Scholar]
- 60. Lawn S. D., Myer L., Bangani N., Vogt M., Wood R. (2007) Plasma levels of soluble urokinase-type plasminogen activator receptor (suPAR) and early mortality risk among patients enrolling for antiretroviral treatment in South Africa. BMC Infect. Dis. 7, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hamada K., Nagai S., Tanaka S., Handa T., Shigematsu M., Nagao T., Mishima M., Kitaichi M., Izumi T. (2007) Significance of pulmonary arterial pressure and diffusion capacity of the lung as prognosticator in patients with idiopathic pulmonary fibrosis. Chest 131, 650–656 [DOI] [PubMed] [Google Scholar]
- 62. Martinez F. J., Safrin S., Weycker D., Starko K. M., Bradford W. Z., King T. E., Jr., Flaherty K. R., Schwartz D. A., Noble P. W., Raghu G., Brown K. K., and IPF Study Group (2005) The clinical course of patients with idiopathic pulmonary fibrosis. Ann. Intern. Med. 142, 963–967 [DOI] [PubMed] [Google Scholar]
- 63. Schwartz D. A., Van Fossen D. S., Davis C. S., Helmers R. A., Dayton C. S., Burmeister L. F., Hunninghake G. W. (1994) Determinants of progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 149, 444–449 [DOI] [PubMed] [Google Scholar]
- 64. Fell C. D., Martinez F. J. (2007) The impact of pulmonary arterial hypertension on idiopathic pulmonary fibrosis. Chest 131, 641–643 [DOI] [PubMed] [Google Scholar]
- 65. Dunn T. L., Watters L. C., Hendrix C., Cherniack R. M., Schwarz M. I., King T. E., Jr. (1988) Gas exchange at a given degree of volume restriction is different in sarcoidosis and idiopathic pulmonary fibrosis. Am. J. Med. 85, 221–224 [DOI] [PubMed] [Google Scholar]
- 66. Monaghan-Benson E., McKeown-Longo P. J. (2006) Urokinase-type plasminogen activator receptor regulates a novel pathway of fibronectin matrix assembly requiring src-dependent transactivation of epidermal growth factor receptor. J. Biol. Chem. 281, 9450–9459 [DOI] [PubMed] [Google Scholar]
- 67. Wills M. K., Jones N. (2012) Teaching an old dogma new tricks: twenty years of Shc adaptor signalling. Biochem. J. 447, 1–16 [DOI] [PubMed] [Google Scholar]
- 68. Schuck S., Honsho M., Ekroos K., Shevchenko A., Simons K. (2003) Resistance of cell membranes to different detergents. Proc. Natl. Acad. Sci. U.S.A. 100, 5795–5800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cunningham O., Andolfo A., Santovito M. L., Iuzzolino L., Blasi F., Sidenius N. (2003) Dimerization controls the lipid raft partitioning of uPAR/CD87 and regulates its biological functions. EMBO J. 22, 5994–6003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Madsen C. D., Ferraris G. M., Andolfo A., Cunningham O., Sidenius N. (2007) uPAR-induced cell adhesion and migration: vitronectin provides the key. J. Cell Biol. 177, 927–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Madsen C. D., Sidenius N. (2008) The interaction between urokinase receptor and vitronectin in cell adhesion and signaling. Eur. J. Cell Biol. 87, 617–629 [DOI] [PubMed] [Google Scholar]
- 72. Sisson T. H., Hanson K. E., Subbotina N., Patwardhan A., Hattori N., Simon R. H. (2002) Inducible lung-specific urokinase expression reduces fibrosis and mortality after lung injury in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 283, L1023–L1032 [DOI] [PubMed] [Google Scholar]
- 73. Bauman K. A., Wettlaufer S. H., Okunishi K., Vannella K. M., Stoolman J. S., Huang S. K., Courey A. J., White E. S., Hogaboam C. M., Simon R. H., Toews G. B., Sisson T. H., Moore B. B., Peters-Golden M. (2010) The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J. Clin. Invest. 120, 1950–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sisson T. H., Hattori N., Xu Y., Simon R. H. (1999) Treatment of bleomycin-induced pulmonary fibrosis by transfer of urokinase-type plasminogen activator genes. Hum. Gene Ther. 10, 2315–2323 [DOI] [PubMed] [Google Scholar]
- 75. Günther A., Lübke N., Ermert M., Schermuly R. T., Weissmann N., Breithecker A., Markart P., Ruppert C., Quanz K., Ermert L., Grimminger F., Seeger W. (2003) Prevention of bleomycin-induced lung fibrosis by aerosolization of heparin or urokinase in rabbits. Am. J. Respir. Crit. Care Med. 168, 1358–1365 [DOI] [PubMed] [Google Scholar]