

Background: ER stress associated with cerebral ischemia induces the expression of the transcription factor CHOP.
Results: Interaction with CHOP down-regulates cell surface GABAB receptors and, thus, GABAB receptor-mediated neuronal inhibition.
Conclusion: Interaction of CHOP with GABAB receptors in the ER prevents forward trafficking of the receptors.
Significance: This mechanism is expected to contribute to excitotoxicity in cerebral ischemia.
Keywords: Cell Surface, Endoplasmic Reticulum Stress, Endoplasmic Reticulum (ER), G Protein-coupled Receptors (GPCR), GABA Receptors, Neurons, Trafficking, CHOP, Cerebral Ischemia
Abstract
Cerebral ischemia frequently leads to long-term disability and death. Excitotoxicity is believed to be the main cause for ischemia-induced neuronal death. Although a role of glutamate receptors in this process has been firmly established, the contribution of metabotropic GABAB receptors, which control excitatory neurotransmission, is less clear. A prominent characteristic of ischemic insults is endoplasmic reticulum (ER) stress associated with the up-regulation of the transcription factor CCAAT/enhancer-binding protein-homologous protein (CHOP). After inducing ER stress in cultured cortical neurons by sustained Ca2+ release from intracellular stores or by a brief episode of oxygen and glucose deprivation (in vitro model of cerebral ischemia), we observed an increased expression of CHOP accompanied by a strong reduction of cell surface GABAB receptors. Our results indicate that down-regulation of cell surface GABAB receptors is caused by the interaction of the receptors with CHOP in the ER. Binding of CHOP prevented heterodimerization of the receptor subunits GABAB1 and GABAB2 and subsequent forward trafficking of the receptors to the cell surface. The reduced level of cell surface receptors diminished GABAB receptor signaling and, thus, neuronal inhibition. These findings indicate that ischemia-mediated up-regulation of CHOP down-regulates cell surface GABAB receptors by preventing their trafficking from the ER to the plasma membrane. This mechanism leads to diminished neuronal inhibition and may contribute to excitotoxicity in cerebral ischemia.
Introduction
GABAB receptors are Gi/o protein-coupled receptors composed of the two obligatory and functionally distinct subunits GABAB1 and GABAB2. GABAB1 harbors the binding site for orthosteric ligands, whereas GABAB2 contains a binding site for allosteric modulators, recruits the G protein, and is required for trafficking of the receptors from the endoplasmic reticulum (ER)2 to the plasma membrane (reviewed in Ref. 1). GABAB1 contains an ER retention signal in the C-terminal domain that retains unassembled GABAB1 in the ER. Heterodimerization with GABAB2 masks the ER retention signal and permits ER export of the receptor heterodimers (2–4). GABAB receptors are abundantly expressed throughout the mammalian central nervous system, where they mediate slow and persistent inhibition. According to their prominent role in regulating neuronal excitability, GABAB receptors have been implicated in a variety of neurological disorders, including cerebral ischemia.
In cerebral ischemia, excessive glutamatergic neurotransmission eventually leads to neuronal death (5). Decreased GABAergic activity appears to contribute to neuronal overexcitation (6), and there are indications that GABAB receptors are down-regulated under ischemic conditions (7–10). This suggests that impaired GABAB receptor signaling contributes to excitotoxicity. In line with these findings, enhancing GABAB receptor activity during ischemic insults by application of the GABAB receptor agonist baclofen has been reported to be neuroprotective in vitro and in vivo (10–19).
Cerebral ischemia induces ER stress, which is characterized by the accumulation of proteins in the ER, leading to the activation of several pathways to restore normal ER function (20). If, however, ER function is severely impaired and cellular homeostasis cannot be restored, apoptosis of the neuron is induced. ER stress triggers the activation and expression of a number of proteins, including up-regulation of the transcription factor CHOP (C/EBP-homologous protein, also known as C/EBPζ, growth arrest- and DNA damage-inducible gene 153 (GADD153) or DNA-damage inducible transcript 3 (DDIT3)). CHOP belongs to the C/EBP transcription factor family and has been shown to trigger apoptosis (21).
Besides its function as a transcription factor, there is evidence that CHOP interacts with GABAB receptors to regulate their cell surface expression. We showed previously that CHOP interacts with its C-terminal leucine zipper, with the leucine zipper present in the C-terminal domain of GABAB2 and with its N-terminal domain with an as yet unidentified site in GABAB1 (22). Upon coexpression in HEK 293 cells, CHOP induced the intracellular accumulation of GABAB receptors and a significant reduction of cell surface receptors (22). However, the mechanism behind this down-regulation of cell surface receptors as well as its physiological role remained unclear. Because ischemic conditions cause ER stress and up-regulate CHOP, we aimed, in this study, to elucidate the mechanism of CHOP-induced down-regulation of cell surface GABAB receptors in neurons and its putative role in cerebral ischemia. We found that ER stress-induced CHOP interacts with GABAB receptors in the ER to disrupt GABAB receptor heterodimerization. This prevents forward trafficking of the receptors to the plasma membrane and, thus, leads to the down-regulation of cell surface receptors and reduced GABAB receptor signaling. This mechanism is operative in an in vitro model of cerebral ischemia.
EXPERIMENTAL PROCEDURES
Antibodies
The following primary antibodies were used: rabbit GABAB1a,b directed against the C terminus of GABAB1 and rabbit GABAB1b directed against the N terminus of GABAB1b (affinity-purified, 1:200 for immunofluorescence, 1:40 for in situ PLA, custom-made by GenScript) (23) as well as rabbit GABAB2 directed against the N terminus of GABAB2 (24) (affinity-purified, 1:500 for immunofluorescence, 1:40 for in situ PLA, custom-made by GenScript), guinea pig GABAB2 (1:50, Chemicon International, catalog no. AB5394), rabbit GABAA receptor α1 subunit (25) (affinity-purified, 1:100 for in situ PLA), mouse GABAA receptor β2/3 subunits (25) (1:500 for in situ PLA), mouse GADD153/CHOP (1:100, Santa Cruz Biotechnology, catalog no. sc-7351), mouse GADD153/CHOP (1:100 for immunofluorescence, 1:30 for in situ PLA, Cell Signaling Technology, catalog no. 2895), rabbit Anti-MAP Kinase (ERK1, ERK2, 1:500, Sigma-Aldrich, catalog no. M5670), mouse anti-MAP kinase-activated (diphosphorylated ERK1 and ERK2, 1:250, Sigma-Aldrich, catalog no. M9692), rabbit NeuN (1:500, Millipore AG, catalog no. ABN78), goat PDI (1:150, Santa Cruz Biotechnology, catalog no. sc-17222), mouse GM130 (1:1000, Abcam, catalog no. ab32337), rabbit HA (1:200, Santa Cruz Biotechnology, catalog no. sc-805), and rabbit c-myc (1:500, Santa Cruz Biotechnology, catalog no. sc-789). Secondary antibodies were either coupled to Alexa Fluor 488 (1:1000, Invitrogen), Cy-3 (1:500, Jackson ImmunoResearch Laboratories), or Cy-5 (1:300, Jackson ImmunoResearch Laboratories).
Plasmids
Plasmids containing full-length cDNA of human CHOP and deletion mutants of CHOP (C-terminal deletion (ΔLZ) and N-terminal deletion (ΔN)) have been described previously (22). Plasmids containing wild-type and mutant rat GABAB1 and rat GABAB2 were provided by Bernhard Bettler (University of Basel, Basel, Switzerland) and are described in Ref. 3.
Drugs
The following drugs were used: thapsigargin (1 μm, Sigma-Aldrich), (R)-baclofen (100 μm, Tocris Bioscience), and CPG 56999A (10 μm, a gift from Novartis, Basel, Switzerland).
Cell Culture
Primary cortical neurons were prepared from E18 embryos of time-pregnant Wistar rats as described previously (26). Briefly, minced E8 cortex was incubated for 15 min with papain solution (0.5 mg/ml PBS, 1 mg/ml BSA, 10 mm glucose, and 10 μg/ml DNase I), washed with Dulbecco's modified Eagle's medium containing 10% fetal calf serum and titrated using a Pasteur pipette. Neurons were plated at a density of 120,000 cells onto poly-l-lysine coverslips (12 mm) in 24-well plates and kept in culture at 37 °C and 5% CO2 for 11–15 days.
Neurons in neuron-glia cocultures were transfected with plasmids using magnetofection, exactly as described in Ref. 27. 60,000 cells were plated on 18-mm coverslips and kept in culture for 11–15 days at 37 °C and 5% CO2. Magnetofection was performed for 30 min on a prewarmed magnetic plate.
Immunocytochemistry and Confocal Laser-scanning Microscopy
Multiplex-labeling immunocytochemistry was performed as described previously (26). For the visualization of cell surface GABAB receptors, living neurons were incubated with primary antibodies for 2 h at 4 °C in ACSF (2 mm CaCl2, 2 mm MgCl2, 30 mm l-glucose, 5 mm KCl, 119 mm NaCl, and 25 mm HEPES (pH 7.4)) containing 10% normal goat serum. For staining of intracellularly localized proteins, neurons were subsequently fixed with 4% paraformaldehyde for 15 min at room temperature and permeabilized for 6 min with 0.2% Triton X-100. Neurons were then incubated with primary antibodies for 1 h (in PBS/10% normal goat serum) at room temperature, washed four times for 5 min with PBS, and incubated with secondary antibodies for 1 h. After four washes with PBS, neurons were mounted in fluorescence mounting medium and analyzed by confocal laser-scanning microscopy (LSM510 Meta, Zeiss). Images were acquired using a Zeiss ×100 plan apochromat oil differential interference contrast objective (1.4 numerical aperture) at 512 × 512 pixel resolution for fluorescence intensity measurements or 1024 × 1024 pixel resolution for colocalization studies. For each neuron, five optical sections spaced by 0.4 μm were taken.
Fluorescence intensity measurements were performed using the Mac Biophotonics ImageJ software (version 1.41n). For analysis of cell surface protein expression, cells were outlined carefully, and the mean fluorescence intensity of the soma was subtracted. For total protein expression analysis, somata of neurons were outlined carefully, and the mean intensity of the fluorescence signals was measured. An area of each image containing no specific signals was selected for determining background staining and was subtracted from the image.
Colocalization studies were performed using Imaris (version 7.1.1, Bitplane, Zurich, Switzerland). Images were smoothed using the median filter tool (filter size, 3 × 3 × 1) and processed further by setting threshold cutoffs for each channel to exclude background staining. Colocalization channels were built (colocalization intensity 255, constant value), and protein clusters (>15 pixels) as well as colocalized clusters were counted within a randomly selected, 30-μm2 area of the somata.
Gene Expression Assays
GABAB1, GABAB2, and CHOP RNA levels were determined in cortical primary neurons using real-time PCR. Total RNA was extracted from neurons using the GenElute mammalian total RNA miniprep kit (Sigma-Aldrich) according to the recommendations of the manufacturer. Reverse transcription was performed with the QuantiTec reverse transcription kit (Qiagen). Quantitative real-time PCR (7900HT fast real-time PCR system, Applied Biosciences) was done using the prepared cDNA and TaqMan gene expression assays (Applied Biosciences) for GABAB1 (Gabbr1, assay ID Rn00578911_m1), GABAB2 (Gabbr2, assay ID Rn00582550_m1), CHOP (Ddit3, assay ID Rn00492098_g1), and β-actin (Actb, assay ID Mm00607939_s1) as a control. Quantification of RNA levels was done using the ΔΔCt method.
Baclofen-induced ERK1/2 Phosphorylation
GABAB receptor activity was indirectly determined by measuring the levels of baclofen-induced ERK1/2 phosphorylation (28–31). Cortical neurons were incubated with the GABAB receptor agonist baclofen for 10 min at 37 °C, 5% CO2 or were left untreated for controls. Subsequently, the cultures were placed on ice, fixed with 4% paraformaldehyde for 15 min at 4 °C, and permeabilized with 0.2% Triton X-100 for 6 min. For determination of ERK1/2 phosphorylation, cultures were incubated overnight at 4 °C with antibodies directed against total ERK1/2 as well as with antibodies against diphosphorylated ERK1/2. After washing with PBS, secondary antibodies were added for 1 h at room temperature, and neurons were analyzed by measuring fluorescence intensities of total and diphosphorylated ERK1/2 levels using confocal laser-scanning microscopy. Levels of phosphorylated ERK1/2 were normalized to total ERK1/2 levels. The specificity of the baclofen-induced ERK1/2 phosphorylation was determined using the GABAB receptor antagonist CPG 56999A.
In Situ PLA
The in situ PLA is a highly sensitive antibody-based method for the visualization of protein-protein interactions and posttranslational modification in cultured cells and tissue sections (32, 33). This method employs two primary antibodies detecting the proteins of interest raised in different species and corresponding secondary antibodies (PLA probes) tagged with oligonucleotides. Only when the proteins of interest are in close proximity (<30 nm), specific connector oligonucleotides can be hybridized and ligated to the oligonucleotides attached to the secondary antibodies, forming a circular oligonucleotide. Rolling circle amplification then creates a large DNA strand to which numerous fluorophore-labeled oligonucleotides (detection probes) are hybridized. This generates a bright fluorescent spot that can be easily detected by microscopy. Quantification is done by counting the number of spots.
Here we used in situ PLA for analyzing the interaction of CHOP with GABAB receptors and the heterodimerization of GABAB1 and GABAB2. In situ PLA was performed using the Duolink kit (Olink Bioscience) according to the protocol of the manufacturer, as described previously (34). The specificity of the PLA signal was verified for both pairs of antibodies in HEK 293 cells expressing or not expressing one of the interaction partners. Furthermore, leaving out one of the primary antibodies completely prevented PLA signals. For in situ PLA, neurons were fixed and permeabilized as described above and incubated with primary antibodies (in PBS/10% normal goat serum) overnight at 4 °C. After in situ PLA, neurons were analyzed by confocal laser-scanning microscopy as described above. Protein-protein interactions were quantified by counting signal dots using the ImageJ software. Image stacks (five optical sections spaced by 0.4 μm) of individual neurons were merged, visualizing maximum intensities, and the number of maxima per area was determined.
Forward Trafficking Assay
For visualization of the amount of GABAB receptors inserted into the plasma membrane within a time period of 16 h, neurons were incubated in culture medium with antibodies directed against GABAB2 for 2 h at 37 °C. Following washes with ACSF to remove unbound primary antibody, cells were incubated for 2 h with a large excess of Alexa Fluor 488-conjugated secondary antibody to label (i.e. mask) the existing pool of cell surface receptors. After washing, the neurons were further incubated in culture medium for 16 h at 37 °C to allow neosynthesis of GABAB receptors and forward trafficking of receptors to the plasma membrane. Neurons were then placed on ice and stained at 4 °C for receptors newly inserted into the plasma membrane using antibodies directed against GABAB2 and Cy3-conjugated secondary antibodies. Neurons were then processed for confocal laser-scanning microscopy. Controls for judging the efficiency of labeling (i.e. masking) the pool of cell surface receptors were treated in exactly the same manner but kept at 4 °C for the 16-h incubation period.
Electrophysiology
Neurons of thapsigargin-treated or untreated control cultures were recorded in the whole-cell voltage clamp configuration at room temperature. Spontaneously occurring postsynaptic currents (sPSCs) were recorded before, during, and after the application of 50 μm baclofen at a holding potential of −60 mV. Patch electrodes were pulled from borosilicate glass and filled with 120 mm CsCl, 10 mm EGTA, 10 mm HEPES (pH 7.4), 4 mm MgCl2, 0.5 mm GTP, and 2 mm ATP. The external solution contained 140 mm NaCl, 10 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm HEPES (pH 7.4), and 10 mm glucose. Recordings were performed with a HEKA EPC-7 amplifier and Patch Master v2.11 software (HEKA Elektronik, Germany). Baclofen (50 μm) was applied locally using an outlet tube (inner diameter, 200 μm) of a custom-designed, gravity-fed microperfusion system positioned 50–100 μm of the recorded neuron. All synaptic events displaying amplitudes above the background noise (5–12 pA) were identified and analyzed offline using the MiniAnalysis 6.0.7 software (Synaptosoft). Mean amplitudes and frequency values were obtained from 1-min-long recordings for each experimental condition and normalized.
Oxygen and Glucose Deprivation (OGD) Model of Ischemia
Primary cortical neurons were washed twice with DMEM without glucose or with DMEM containing glucose for controls. For OGD, neurons were incubated for 10 min at 37 °C in 95% N2/5% CO2 prebubbled, glucose-free DMEM in an airtight box filled with 95% N2/5% CO2. Control cultures were incubated in DMEM containing glucose at 37 °C in a physiological oxygen-containing environment. Subsequently, cells were washed twice with DMEM containing glucose and incubated for 24 h in their original culture medium.
RESULTS
CHOP Interacts with GABAB Receptors and Down-regulates Cell Surface Receptors in Neurons
We have previously shown that the stress-induced transcription factor CHOP, which is only marginally expressed under normal physiological conditions, interacts with GABAB receptors by binding to the coiled coil motif in the C-terminal domain of GABAB2 and with a so far unidentified site in GABAB1 (22). Upon overexpression in HEK 293 cells, the interaction of CHOP with GABAB receptors resulted in the intracellular accumulation and a reduced cell surface expression of the receptors by an as yet unknown mechanism (22). To confirm an interaction of GABAB receptors with CHOP in neurons, we overexpressed CHOP in cultured cortical neurons and tested for an interaction with GABAB receptors using the in situ PLA (32, 33). In non-transfected neurons, only very few PLA dots were detected, in line with a low expression level of CHOP under normal physiological conditions. However, overexpression of CHOP dramatically increased PLA signals, indicating numerous GABAB receptor-CHOP interactions (Fig. 1A).
FIGURE 1.

Up-regulated CHOP interacts with GABAB receptors in neurons. A, CHOP overexpressed in neurons interact with GABAB receptors. Cortical neurons were left non-transfected (−CHOP) or were transfected with CHOP (+CHOP) and analyzed for interaction with GABAB receptors by in situ PLA using antibodies directed against CHOP and GABAB2. Because under normal physiological conditions CHOP expression is low, only few interactions (white dots in representative images) were detected. However, overexpression of CHOP resulted in numerous CHOP-GABAB receptor interactions. The images represent merged Z-stack reconstructions of five optical section spaced by 0.4 μm. Scale bar = 5 μm. B, thapsigargin-induced CHOP interacts with GABAB receptors. Cortical neurons were incubated with 2 μm thapsigargin (TG) for 2 h and allowed to recover in their original culture medium for 16 h. The interaction of CHOP with GABAB receptors was determined by in situ PLA using antibodies directed against CHOP and GABAB2 (white dots in representative images, left panel, scale bar = 5 μm). Right panel, quantification of in situ PLA signals. Data are means ± S.E., 30 neurons from four experiments. ***, p < 0.001, Student's t test. Ctrl, control. C, CHOP does not interact with GABAA receptors. Cortical neurons were treated (TG) or not treated (Ctrl) with 2 μg of thapsigargin for 2 h and allowed to recover in their original culture medium for 16 h. The interaction of CHOP with GABAA receptors was determined by in situ PLA using antibodies directed against CHOP and the α1 subunit of GABAA receptors. No interactions were observed (right panel). As a positive control, the interaction of the GABAA receptor subunits α1 and β2/3 was tested. Numerous interactions were detected in untreated neurons (white dots in representative image, left panel). The images represent merged Z-stack reconstructions of five optical section spaced by 0.4 μm. Scale bar = 5 μm.
Because ER stress-induced up-regulation of CHOP has been shown to play an important role in ischemia-induced neuronal death (35–42), we then analyzed the mechanism of CHOP-induced down-regulation of cell surface GABAB receptors by exposing cultured cortical neurons to ER stress. ER stress was induced by treating neurons for 2 h with the sarco/endoplasmic reticulum Ca2+-ATPase blocker thapsigargin. After a 16-h recovery period, neurons were analyzed for CHOP and GABAB receptor interaction using an in situ PLA. Under control conditions, i.e. in untreated neurons, only few interactions were observed (Fig. 1B). However, upon up-regulation of CHOP with thapsigargin, numerous interactions with GABAB receptors were detected (454 ± 53% of control, n = 30, p < 0.001, Fig. 1B). Thus, up-regulation of CHOP by ER stress resulted in an interaction with GABAB receptors. Under these conditions, no in situ PLA signals were generated using CHOP and GABAA receptor antibodies, documenting the specificity of the CHOP-GABAB receptor interaction (Fig. 1C).
An analysis of the protein expression levels revealed that CHOP was vastly up-regulated in thapsigargin-treated neurons (706 ± 65% of control, n = 57, p < 0.001; Fig. 2A), whereas the cell surface staining of GABAB1 and GABAB2 was decreased significantly (GABAB1, 67 ± 4%; GABAB2, 50 ± 3% of control, n = 50–55, p < 0.001, Fig. 2A). No colocalization of CHOP with cell surface GABAB receptors was detected. In contrast to cell surface receptors, total GABAB1 and GABAB2 expression levels remained unchanged in thapsigargin-treated neurons (Fig. 2B). These findings suggest that the interaction of CHOP with GABAB receptors caused a down-regulation of the receptors from the cell surface and their intracellular accumulation.
FIGURE 2.

Up-regulated CHOP in neurons mediates down-regulation of cell surface GABAB receptors. A, up-regulation of CHOP down-regulates cell surface GABAB receptors. Neurons were treated with thapsigargin and stained for surface GABAB1 and GABAB2 (green). Subsequently, cells were fixed, permeabilized, and stained for CHOP (red). Neurons not treated with thapsigargin served as controls. Left panel, representative images. Scale bar = 7 μm. Right panel, quantification of cell surface GABAB1, GABAB2, and CHOP. Data are means ± S.E., 50–55 neurons from five experiments. ***, p < 0.001, Student's t test. B, up-regulation of CHOP does not affect total GABAB receptor expression levels. Neurons were treated with thapsigargin (TG) and stained for total GABAB1 and GABAB2 (green) and CHOP (red). Neurons not treated with thapsigargin served as controls (Ctrl). Scale bar = 10 μm. Bar graphs show the quantification of total expression levels of GABAB1 and GABAB2. Data are means ± S.E., 30 neurons from three experiments. n.s., not significant, p > 0.05, Student's t test. C, mutant forms of CHOP that cannot bind to GABAB receptors do not mediate down-regulation of cell surface receptors. Neurons were cotransfected with EGFP and CHOP or mutant forms of CHOP lacking its interaction site with GABAB2 (CHOPΔLZ) or its interaction site with GABAB1 (CHOPΔN). After 48 h, transfected neurons were stained for cell surface GABAB2 (green) and CHOP (red). Left panel, representative images. Scale bar = 10 μm. Right panel, quantification of cell surface GABAB2 levels in neurons expressing either CHOP, CHOPΔLZ, or CHOPΔN. Data are means ± S.E.; 82 (EGFP and CHOP), 79 (CHOPΔLZ), and 101 (CHOPΔN) neurons from three experiments. ***, p < 0.001; one-way analysis of variance, Bonferroni's multiple comparison test. D, CHOP does not regulate GABAB receptor mRNA levels. Total mRNA was isolated from neuronal cultures treated or not treated (controls) with thapsigargin. CHOP, GABAB1, and GABAB2 mRNA was quantified using real-time PCR. Data are means ± S.E., four (CHOP) and five (GABAB1 and GABAB2) individual cultures were analyzed. ***, p < 0.001; n.s., not significant (p > 0.05); Student's t test.
To verify that the loss of cell surface receptors was caused by the interaction of CHOP with GABAB receptors, neurons were transfected with mutant forms of CHOP that are unable to interact with GABAB1 (CHOPΔN, N-terminal deletion (22)) or GABAB2 (CHOPΔLZ, deletion of C-terminal leucine zipper motif (22)). Neurons overexpressing wild-type CHOP displayed a significantly reduced level of cell surface GABAB2 compared with control neurons transfected with EGFP (63 ± 4%, n = 82, p < 0.001, Fig. 2C). However, overexpressing either of the CHOP mutants did not affect cell surface expression of GABAB receptors (CHOPΔLZ, 92 ± 5%, p > 0.05, n = 79; CHOPΔN, 103 ± 5%, n = 101, p > 0.05), suggesting that down-regulation of cell surface receptors was mediated by its interaction with CHOP.
To rule out that up-regulation of CHOP affects transcription of GABAB1 and GABAB2, their mRNA levels were quantified in neurons treated with thapsigargin using real-time PCR (Fig. 2D). CHOP mRNA levels were increased drastically in thapsigargin-treated neurons (432 ± 54% of control, n = 4, p < 0.001), but GABAB1 and GABAB2 mRNA levels remained unchanged (GABAB1, 113 ± 6% of control, n = 5, p > 0.05; GABAB2, 94 ± 12% of control, n = 5, p > 0.05). Thus, CHOP does not regulate GABAB receptor expression at the transcriptional level.
CHOP-mediated Down-regulation of Cell Surface Receptors Reduces GABAB Receptor Signaling
It is well established that GABAB receptors activate extracellular-signal-regulated kinase 1/2 (ERK1/2) in neurons (28–31). To analyze the functional consequences of CHOP-mediated down-regulation of GABAB receptors, receptor activity was assayed on the basis of baclofen-induced ERK1/2 phosphorylation. Activation of GABAB receptors with 100 μm baclofen in control neurons significantly increased ERK1/2 phosphorylation (159 ± 10%, n = 26, p < 0.001, Fig. 3A), which was abolished by preincubation of neurons with the GABAB receptor antagonist CPG 56999A (106 ± 10%, n = 26, p > 0.05; Fig. 3A). However, in thapsigargin-treated neurons, no increase of ERK1/2 phosphorylation was observed (108 ± 8% of control, n = 28, p > 0.05, Fig. 3A), indicating that CHOP-mediated down-regulation of cell surface GABAB receptors inhibits downstream signaling.
FIGURE 3.

CHOP-mediated down-regulation of cell surface GABAB receptors impairs GABAB receptor-mediated downstream signaling. A, GABAB receptor-induced phosphorylation of ERK1/2 was prevented by the GABAB receptor antagonist CPG 56999A (CPG) and by thapsigargin-induced up-regulation of CHOP. Neurons were treated with thapsigargin (+TG) to up-regulate CHOP, whereas control cultures remained untreated. Subsequently, neurons were incubated for 10 min with 100 μm baclofen (Bac) in the presence or absence of 10 μm CPG 56999A (+CGP, GABAB receptor antagonist). After fixation and permeabilization, neurons were stained for total ERK1/2 (ERK, green) and diphosphorylated ERK1/2 (pERK, red). Left panel, representative images. Scale bar = 5 μm. Right panel, quantification of pERK levels. Data are means ± S.E., 26 (Baclofen, Baclofen + CPG) and 28 (Baclofen + TG) neurons from three experiments. ***, p < 0.001, one-way analysis of variance, Bonferroni's multiple comparison test. B, up-regulation of CHOP diminished baclofen-induced inhibition of sPSCs. Left panel, representative current traces depicting spontaneous sPSCs recorded from untreated control neurons (n = 9) or from neurons treated with thapsigargin for up-regulation of CHOP (n = 5). Right panel, normalized amplitude and frequency values of the sPSCs. Mean amplitudes and frequency values were normalized to the control condition of the individual neuron. Con., control. Data are mean ± S.E. *, p < 0.05; Student's t test.
To test whether CHOP-induced down-regulation of GABAB receptors affects GABAB receptor-mediated neuronal inhibition, spontaneous synaptic activity was measured in electrophysiological experiments using the whole-cell voltage clamp configuration. In untreated control neurons, application of baclofen considerably decreased the amplitude (66 ± 6% of control, n = 9) and the frequency (38 ± 8% of control, n = 5) of sPSCs (Fig. 3B). However, after up-regulation of CHOP by treating cultures with thapsigargin, baclofen-induced inhibition of sPSCs was strongly reduced (amplitude, 87 ± 6% of control, n = 9, p < 0.05; frequency, 65 ± 9% of control, n = 5, p < 0.05; Fig. 3B). This finding is consistent with an up-regulation of CHOP and, subsequently, reduced levels of functional GABAB receptors available for neuronal inhibition.
CHOP Interacts with GABAB Receptors in the ER and Interferes with Receptor Heterodimerization
Next, we investigated the mechanism of CHOP-mediated down-regulation of cell surface GABAB receptors. We envisioned two scenarios. 1) CHOP disrupts functional GABAB receptor heterodimers on the cell surface, which would lead to their internalization and degradation, or 2) CHOP interacts with GABAB receptors in the ER and inhibits receptor heterodimerization and, consequently, forward transport to the cell surface. To test these two scenarios, we analyzed the colocalization of CHOP and GABAB receptor subunits in different cellular compartments in untreated control neurons and neurons treated with thapsigargin to up-regulate CHOP. In thapsigargin-treated neurons, GABAB receptors accumulated in the ER, as indicated by an increased colocalization of GABAB1 (control, 10 ± 0.7 clusters, n = 20; thapsigargin, 15 ± 0.8 clusters, n = 21, p < 0.001) and GABAB2 (control, 7 ± 0.7 clusters, n = 32; thapsigargin, 13 ± 0.7 clusters, n = 32, p < 0.001) with the ER marker PDI (Fig. 4, A and B). Hardly any CHOP was observed in the ER under control conditions, whereas CHOP accumulated in the ER after thapsigargin treatment (GABAB1-stained neurons, control, 2 ± 0.4 clusters, n = 20; thapsigargin, 9 ± 0.9 clusters, n = 21, p < 0.001; Fig. 4A; GABAB2-stained neurons, control, 1.5 ± 0.4 clusters, n = 32; thapsigargin, 9 ± 0.6 clusters, n = 32, p < 0.001; Fig. 4B). Triple colocalization of GABAB-CHOP-PDI was basically absent under control conditions but increased significantly after up-regulation of CHOP by thapsigargin (Fig. 4, A and B; GABAB1-CHOP-PDI, control, 1.4 ± 0.3 clusters, n = 20; thapsigargin, 6 ± 0.7 clusters, n = 21, p < 0.01; GABAB2-CHOP-PDI, control, 0.7 ± 0.3 clusters, n = 32; thapsigargin, 6 ± 0.5 clusters, n = 32, p < 0.001). These findings indicate that GABAB receptors interact with CHOP in the ER, which leads to the accumulation of GABAB1 and GABAB2 in the ER.
FIGURE 4.

Up-regulated CHOP colocalizes with GABAB receptors in the ER and induces their intracellular accumulation. A and B, GABAB receptors accumulate in the ER and colocalize with CHOP. Neurons were treated with thapsigargin (TG) to up-regulate CHOP and stained for GABAB1 (A) or GABAB2 (B) (green), CHOP (red), and the ER marker PDI (blue). Cultures not treated with thapsigargin served as controls. Left panel, representative images depicting GABAB1/B2-CHOP-PDI colocalization (white dots marked with arrows, scale bar = 5 μm). Right panel, quantification of colocalization. Data are means ± S.D., 20–32 neurons/colocalization from three experiments. ***, p < 0.001, Student's t test. C, GABAB receptors do not accumulate or colocalize with CHOP in the Golgi apparatus. Neurons were treated with thapsigargin to up-regulate CHOP and stained for GABAB2 (green), CHOP (red), and the Golgi marker GM130 (blue). Cultures not treated with thapsigargin served as controls. Left panel, representative images. Scale bar = 5 μm. Right panel, quantification of colocalization. Data are means ± S.D., 19 neurons/colocalization from three experiments. n.s., not significant (p > 0.05), Student's t test. D, GABAB receptors do not colocalize with CHOP on the cell surface. Neurons were treated with thapsigargin to up-regulate CHOP and then stained for cell surface GABAB2 (green) as well as for CHOP (red). Cultures not treated with thapsigargin served as controls. Left panel, representative images. Scale bar = 5 μm. Right panel, quantification of colocalization. Data are means ± S.D., 44 (control) and 45 (thapsigargin) neurons from four experiments. n.s., not significant (p > 0.05), Student's t test.
In contrast, GABAB receptors did not display increased colocalization with the Golgi apparatus marker (GM) GM130 in thapsigargin-treated neurons (control, 27 ± 3 clusters, n = 19; thapsigargin, 30 ± 3 clusters, p > 0.05, n = 19; Fig. 4C), indicating no accumulation of GABAB receptors in the Golgi after up-regulation of CHOP. In line with this finding, there was no statistically significant increase in CHOP-GM colocalization control (3 ± 1 clusters, n = 19; thapsigargin, 7 ± 2 clusters, p > 0.05, n = 19, p > 0.05; Fig. 4C) nor in GABAB2-CHOP-GM (control, 2 ± 0.7 clusters, n = 19; thapsigargin, 6 ± 2 clusters, n = 19, p > 0.05; Fig. 4C), indicating marginal interactions between CHOP and GABAB receptors in the Golgi after thapsigargin-induced up-regulation of CHOP. In addition, there was no significant colocalization of CHOP with GABAB receptors at the cell surface (control, 1.3 ± 0.2 clusters, n = 45; thapsigargin, 2.3 ± 0.5 clusters, n = 44, p > 0.05; Fig. 4D), indicating that GABAB receptors do not interact with CHOP in the plasma membrane.
To demonstrate that the accumulation of GABAB receptors in the ER is mediated by direct interaction with CHOP, we expressed in neurons a mutant form of GABAB2 lacking its leucine zipper (GABAB2ΔLZ) and, thus, its CHOP interaction site, together with GABAB1 containing a mutated ER retention signal (GABAB1ASA) to ensure forward trafficking of the receptors. GABAB1ASA was required because deletion of the GABAB2 leucine zipper unmasks the ER retention signal in the C terminus of GABAB1 and, therefore, prevents forward trafficking of the receptors. To verify normal trafficking behavior of the GABAB1ASA/GABAB2ΔLZ heterodimers, we analyzed the colocalization of GABAB1ASA and GABAB2ΔLZ with PDI and CHOP in thapsigargin-treated and untreated neurons. In thapsigargin-treated neurons, GABAB1ASA (control, 10 ± 3 clusters, n = 12; thapsigargin, 15 ± 5 clusters, n = 19, p < 0.01, Fig. 5A), GABAB2 (control, 13 ± 5 clusters, n = 17; thapsigargin, 17 ± 4 clusters, n = 15, p < 0.01, Fig. 5B), and CHOP (Fig. 5A, control, 1.3 ± 1.4 clusters, n = 13; thapsigargin, 11 ± 5 clusters, n = 20, p < 0.001; Fig. 5B, control, 1.4 ± 1.8 clusters, n = 17; thapsigargin, 11 ± 5 clusters, n = 16, p < 0.001) showed increased colocalization with the ER marker PDI. Also, increased colocalization of GABAB1 or GABAB2 with CHOP and PDI (GABAB1/CHOP/PDI and GABAB2/CHOP/PDI) was detected in thapsigargin-treated neurons (GABAB1ASA, control, 0.7 ± 1.1 clusters, n = 13; thapsigargin, 7 ± 4 clusters, n = 20, p < 0.001; GABAB2, control, 0.8 ± 1.1 clusters, n = 17; thapsigargin, 6 ± 3 clusters, n = 16, p < 0.001; Fig. 5, A and B). The identical colocalization patterns of GABAB1ASA/GABAB2 receptors and endogenous GABAB receptors indicate normal trafficking capabilities of the mutant receptors.
FIGURE 5.

Accumulation of GABAB receptors in the ER is mediated by interaction with CHOP. A and B, neurons were transfected with EGFP in combination with GABAB1ASA/GABAB2 or GABAB1ASA/GABAB2ΔLZ. After 48 h, neurons were treated with thapsigargin (TG) to up-regulate CHOP and then stained for GABAB1ASA (A) or GABAB2ΔLZ (B), CHOP, and the ER marker PDI. Cultures not treated with thapsigargin served as controls. Left panels, representative images depicting colocalization of GABAB1-CHOP-PDI and GABAB2-CHOP-PDI (white dots). Scale bars = 5 μm). Right panels, quantification of colocalization. Data are means ± S.D., 11–22 neurons/colocalization from two experiments. **, p < 0.01;***, p < 0.001, n.s., not significant (p > 0.05); Student's t test. C, Up-regulation of CHOP interferes with GABAB receptor heterodimerization. Neurons were treated with thapsigargin to up-regulate CHOP and tested for GABAB1/GABAB2 heterodimerization by in situ PLA using antibodies directed against GABAB1 and GABAB2 (white dots in representative images, left panel, scale bar = 5 μm). Cultures not treated with thapsigargin served as controls (Ctrl). The images represent merged Z-stack reconstructions of five optical section spaced by 0.4 μm. Right panel, quantification of in situ PLA signals. Data are means ± S.E., 32 neurons from three experiments. ***, p < 0.001; Student's t test.
However, transfection of neurons with GABAB1ASA/GABAB2ΔLZ receptors, which lack the CHOP interaction site in GABAB2, fully abolished the increased colocalization of GABAB1-PDI (control, 13 ± 4 clusters, n = 14; thapsigargin, 10 ± 4 clusters, n = 20, p > 0.05), GABAB2-PDI (control, 12 ± 5 clusters, n = 14; thapsigargin, 9 ± 3 clusters, n = 11, p > 0.05), GABAB1-CHOP-PDI (control, 0.5 ± 1.1 clusters, n = 15; thapsigargin, 1.5 ± 1.5 clusters, n = 22, p > 0.05), and GABAB2-CHOP-PDI (control, 0.3 ± 0.8 clusters, n = 14; thapsigargin, 1.3 ± 1.2 clusters, n = 12, p > 0.05; Fig. 5, A and B) after induction of CHOP with thapsigargin. This result verified that CHOP directly interacts with GABAB receptors to induce their accumulation in the ER.
Because GABAB1 and GABAB2 interact via the leucine zippers in their C-terminal domains, binding of CHOP to GABAB2 may prevent GABAB receptor heterodimerization and, thus, inhibit forward trafficking of the receptors. Therefore, we tested whether up-regulation of CHOP interferes with heterodimerization of GABAB1 and GABAB2 using in situ PLA. In thapsigargin-treated neurons, we observed a significant reduction of in situ PLA signals (64 ± 5% of control, n = 32, p < 0.001, Fig. 5C), indicating the presence of considerably less GABAB1/GABAB2 heterodimers compared with untreated control neurons. This result suggests that the interaction of CHOP with GABAB receptors disrupts or prevents the heterodimerization of GABAB1 and GABAB2.
These findings suggest a mechanism in which CHOP interacts with GABAB receptors in the ER after cellular stress to prevent heterodimerization of GABAB1 and GABAB2. Because only heterodimerized GABAB receptors can leave the ER (2–4), this mechanism is expected to impede forward trafficking of newly synthesized receptors to the plasma membrane.
Up-regulation of CHOP Prevents Forward Trafficking of GABAB Receptors to the Cell Surface
To test whether up-regulation of CHOP impairs forward trafficking of GABAB receptors to the cell surface, we transfected neurons with HA-tagged GABAB2 and CHOP. Twenty-four hours after transfection, newly synthetized HA-tagged receptors inserted into the plasma membrane were detected using an anti-HA antibody. In control neurons only transfected with HA-GABAB2, strong cell surface expression of HA-tagged GABAB2 was detected (Fig. 6A). However, in neurons transfected with CHOP, staining for cell surface HA-GABAB2 was strongly reduced (36 ± 4% of control, n = 25, p < 0.001, Fig. 6A). This reduction of cell surface HA-GABAB2 was not observed in neurons expressing a mutant of CHOP (CHOPΔLZ) that is unable to bind to GABAB2 (80 ± 7% of control, n = 25, p > 0.05, Fig. 6A). These results suggest that the interaction of CHOP with GABAB2 impairs forward trafficking of GABAB receptors to the cell surface.
FIGURE 6.
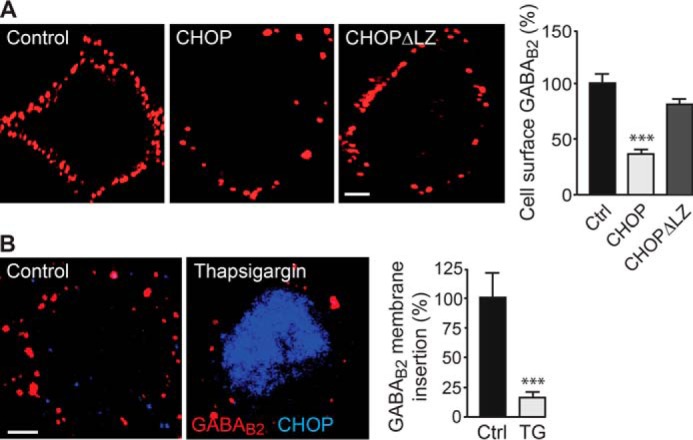
Up-regulated CHOP impairs forward trafficking of GABAB receptors. A, neurons were transfected with either EGFP and HA-tagged GABAB2 (Control), EGFP, HA-GABAB2 and CHOP, or EGFP HA-GABAB2 and CHOPΔLZ (CHOPΔLZ). Twenty-four hours after transfection, GABAB receptors newly inserted into the plasma membrane were visualized by staining with HA-antibodies. Left panel, representative images. Scale bar = 5 μm. Right panel, quantification of GABAB receptor membrane insertion. Data are means ± S.E., 25 neurons/condition from three experiments. ***, p < 0.001; one-way analysis of variance, Dunnett's post-hoc test. B, neurons were treated with thapsigargin (TG) to up-regulate CHOP und subjected to an immunofluorescence-based forward trafficking assay to determine the amounts of GABAB receptors inserted into the plasma membrane within a time period of 16 h (red clusters in representative images, scale bar = 5 μm). CHOP expression is depicted in blue. Cultures not treated with thapsigargin served as controls. Right panel, quantification of GABAB receptor membrane insertion. Data are means ± S.E., 38 (control) and 33 (thapsigargin) neurons from three experiments. ***, p < 0.001; Student's t test.
This result was confirmed for native GABAB receptors with an immunofluorescence-based forward trafficking assay using untreated control neurons and thapsigargin-treated neurons. After masking the existing receptor pool on the cell surface with primary and secondary antibodies, the insertion of new receptors into the plasma membrane was tested after 16 h. Membrane insertion of GABAB receptors was reduced drastically in neurons expressing high levels of CHOP (16 ± 6% of control, n = 33, p < 0.001, Fig. 6B).
Down-regulation of Cell Surface GABAB Receptors by CHOP in the OGD Model of Ischemia
Next, we tested whether up-regulation of CHOP under ischemic conditions mediates down-regulation of cell surface GABAB receptors. We used the OGD in vitro model of ischemia, in which cultured cortical neurons were deprived of oxygen and glucose for 10 min, followed by a recovery period of 24 h. 24 h after OGD, CHOP expression in neurons was increased significantly (157 ± 5% of control, n = 311, p < 0.001, Fig. 7A). As expected, cell surface GABAB receptors decreased considerably 24 h after OGD in CHOP-expressing neurons (44 ± 3% of control, n = 49, p < 0.001, Fig. 7B).
FIGURE 7.
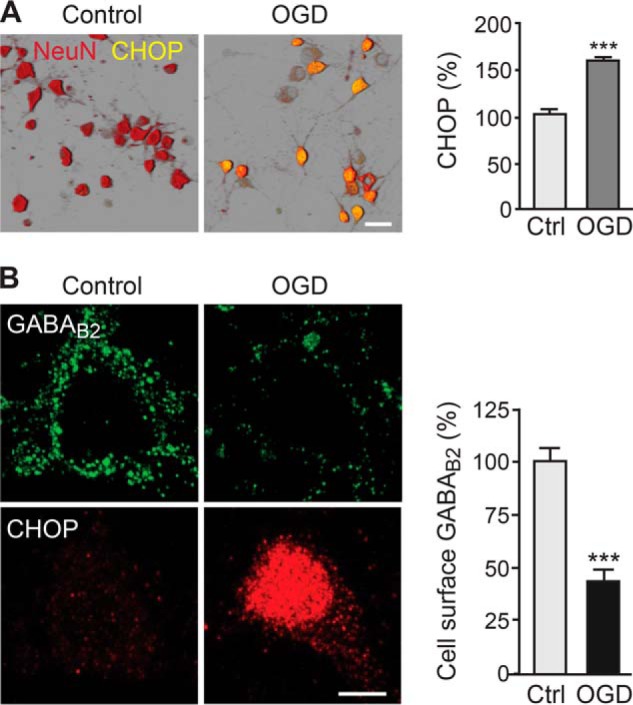
CHOP is up-regulated and GABAB receptors are down-regulated in the OGD model of cerebral ischemia. A, OGD in neurons up-regulates CHOP. Neurons were subjected to OGD for 10 min, followed by a recovery period of 24 h. Subsequently, neurons were stained for CHOP (green) and for NeuN to visualize the neurons (red). CHOP expression in neurons is depicted in yellow. Cultures not subjected to OGD served as controls (Ctrl). Left panel, representative images. Scale bar = 30 μm. Right panel, quantification of CHOP expression in neurons. Data are means ± S.E., 271 (control) and 311 (thapsigargin) neurons from three experiments. ***, p < 0.001; Student's t test. B, cell surface GABAB receptors are down-regulated following OGD. Neurons were subjected to OGD for 10 min, followed by a recovery period of 24 h, and were then stained for cell surface GABAB2 (green) and CHOP (red). Cultures not subjected to OGD served as controls. Left panel, representative images. Scale bar = 5 μm. Right panel, quantification of cell surface GABAB receptor expression. Data are means ± S.E., 30 (control) and 49 (thapsigargin) neurons from three experiments. ***, p < 0.001; Student's t test.
To test whether the interaction of CHOP with GABAB receptors is responsible for the down-regulation of cell surface receptors after OGD, we overexpressed CHOPΔLZ in cortical neurons. CHOPΔLZ is a mutant form of CHOP that is not able to interact with GABAB2 and does not affect cell surface numbers of GABAB receptors (Fig. 1C). Endogenous CHOP was significantly up-regulated after OGD in transfected (154 ± 8% of control, n = 20, p < 0.001) and in non-transfected neurons (167 ± 12% of control, n = 22, p < 0.001, Fig. 8). In non-transfected neurons, GABAB2 receptors were down-regulated after OGD (60 ± 7% of control, n = 27, p < 0.001, Fig. 8), whereas in neurons overexpressing CHOPΔLZ, despite similar up-regulation of endogenous CHOP, the down-regulation of GABAB receptors was completely prevented (113 ± 7% of control, n = 45, p > 0.05,; Fig. 8). These results verify that down-regulation of cell surface GABAB receptors after ischemic conditions is mediated by their interaction with CHOP.
FIGURE 8.
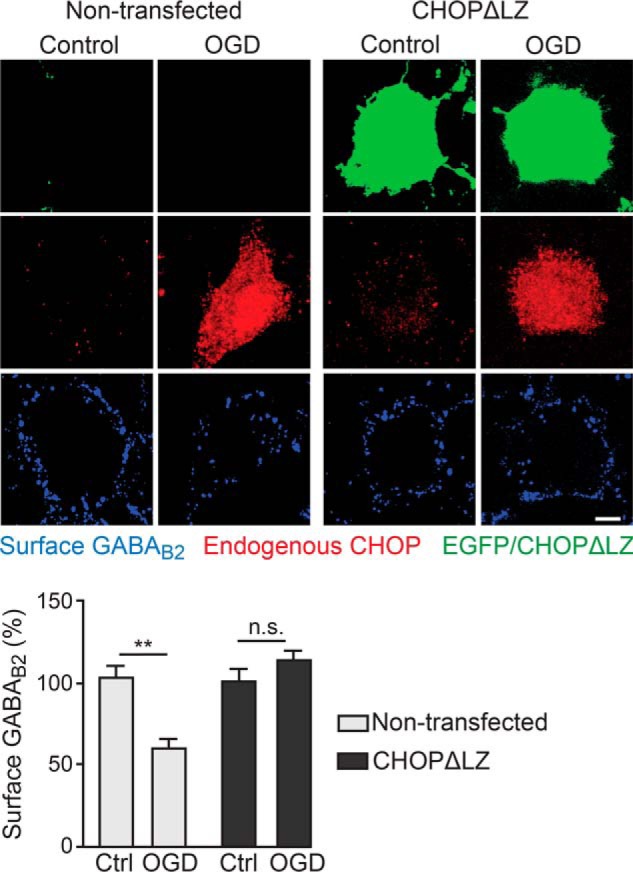
Interaction of CHOP with GABAB receptors is responsible for OGD-induced down-regulation of cell surface receptors. Neurons were transfected with a CHOP mutant that is not able to interact with GABAB2 (CHOPΔLZ) and EGFP. After 24 h, neurons were subjected to OGD for 10 min, followed by a recovery period of 24 h. Subsequently, neurons were stained for cell surface GABAB receptors (blue) and endogenous CHOP (red). CHOPΔLZ-overexpressing neurons were selected by means of EGFP expression. Cultures not subjected to OGD served as controls. Top panel, representative images. Scale bar = 5 μm. Bottom panel, quantification of cell surface GABAB receptors. Data are means ± S.E.; 38 (control, non-transfected), 27 (OGD, non-transfected), 36 (control, transfected), and 45 (OGD, transfected) neurons from three experiments. **, p < 0.01; n.s., not significant (p > 0.05); Student's t test.
DISCUSSION
GABAB receptor-mediated neuronal inhibition critically depends on the availability of receptors in the plasma membrane. Receptor numbers might be altered under pathological conditions and a loss of receptors resulting in diminished neuronal inhibition is expected to contribute to the disease state. We showed previously that the ER stress-induced transcription factor CHOP interacts with GABAB receptors, causing their down-regulation from the cell surface upon coexpression in HEK 293 cells (22). This finding suggests that CHOP, besides its function as transcription factor, may regulate GABAB receptor-mediated neuronal inhibition by affecting the availability of cell surface receptors. Because CHOP is expressed at very low levels under normal physiological conditions but is highly up-regulated after induction of ER stress (21, 43–45), CHOP-induced down-regulation of cell surface GABAB receptors may be a contributing factor to neurological disease states associated with ER stress, such as stroke, Alzheimer and Parkinson disease, or bipolar disorders (20).
In this study, we verified that ER stress-induced CHOP expression also down-regulates cell surface GABAB receptors in neurons and disclosed the mechanism underlying this effect. An in vitro model of ischemia suggests that this mechanism is operative in cerebral ischemia.
We induced up-regulation of CHOP in cultured neurons by inhibition of the sarco/endoplasmic reticulum Ca2+-ATPase with thapsigargin, which leads to the depletion of Ca2+ from the ER, thereby causing ER stress (46). Thapsigargin up-regulated CHOP in neurons and considerably down-regulated GABAB receptors from the cell surface. The reduced level of cell surface receptors consequently affected downstream signaling of GABAB receptors, as shown by impaired ERK1/2 phosphorylation and reduced baclofen-induced inhibition of spontaneous neuronal activity.
Down-regulation of cell surface GABAB receptors was mediated by the interaction of the receptors with CHOP because overexpression in neurons of mutant forms either lacking the site binding to GABAB1 or the site binding to GABAB2 prevented down-regulation of the receptors. Most importantly, up-regulation of CHOP did not affect the mRNA levels of GABAB1 and GABAB2, ruling out a contribution of impaired subunit transcription (Fig. 2D).
Up-regulated CHOP selectively accumulates together with GABAB receptors in the ER, suggesting that interaction with CHOP retains GABAB receptors in the ER and prevents their forward trafficking to the cell surface. This conclusion is supported by the finding that mutant GABAB receptors not containing the CHOP binding site in GABAB2 do not accumulate in the ER and that the insertion of new receptors into the plasma membrane is strongly reduced upon up-regulation of CHOP. The mechanism that interferes with forward trafficking of the receptors appears to involve prevention or disruption of receptor heterodimerization, as indicated by our in situ PLA experiments. It is currently not clear whether CHOP prevents heterodimerization of newly synthesized GABAB1 and GABAB2 by binding to GABAB1 and GABAB2 or directly disrupts already existing heterodimers. In either case, preventing heterodimerization exposes the ER retention signal in the C-terminal domain of GABAB1, which prohibits ER exit of GABAB1 (2–4). In addition, GABAB2 contains a C-terminal sequence (amino acids 841–862) that is important for forward trafficking (47). Binding of CHOP to the leucine zipper domain of GABAB2, upstream of this motif, might sterically interfere with the function of this motif and prevent ER export of GABAB2.
The mechanism of CHOP-induced down-regulation of cell surface receptors appears to be operative under ischemic conditions. This is not surprising because cerebral ischemia has been shown to be associated with ER stress and the profound up-regulation of CHOP (35–42). Using the OGD in vitro model of ischemia, we found that up-regulated CHOP mediates down-regulation cell surface GABAB receptors, which depended on the interaction of CHOP with the receptors.
So far, the effect of cerebral ischemia on GABAB receptor expression levels has been rarely investigated, and the results are difficult to correlate because different animal species and experimental conditions were used. In vivo models of ischemia demonstrated a loss of GABAB receptors 1–4 days after the insult (7–9). Because considerable ischemia-induced neuronal death occurs during this time period, the loss of GABAB receptors might be due, at least in part, to a loss of GABAB receptor-expressing neurons. A recent in vitro study on cultured hippocampal slices using a similar OGD paradigm as in this study detected down-regulation of total GABAB2 but no effect on the expression of total GABAB1 (10). In this study, we detected down-regulation of both GABAB1 and GABAB2 selectively from the cell surface but did not observe changes in total receptor expression. The reason for this discrepancy is not clear yet, but it may be caused by different experimental conditions used (organotypic hippocampal slices, 45-min OGD versus cultured cortical neurons, 10-min OGD).
Cerebral ischemia is associated with excessive glutamate release, and overstimulation of glutamate receptors triggers a signaling cascade leading to neuronal death (48). The enhanced neuronal activity also increases extracellular levels of GABA (6), which, in turn, should activate GABAB receptors located at glutamatergic synapses to counteract, at least partially, the increased neuronal excitation to reduce excitotoxicity. The down-regulation of functional GABAB receptors from the cell surface might, therefore, be one factor that fosters excitotoxicity. It is well established that sustained activation of GABAB receptors (by application of baclofen) under ischemic conditions reduces neuronal cell death (10–19). In this regard, it is conceivable that restoring normal cell surface expression of GABAB receptors under ischemic conditions would also reduce excitotoxicity. This view is supported by a recent study showing that mild hypothermia reverses down-regulation of total GABAB1 after cerebral ischemia by an unknown mechanism and reduces neuronal death (8).
One opportunity to restore normal cell surface GABAB receptor levels would be the application of small synthetic peptides interfering with the CHOP-GABAB receptor interaction. Further experiments are required to identify small peptide sequences that inhibit the interaction of CHOP with GABAB receptors but do not interfere with receptor heterodimerization. This approach provides the opportunity to unambiguously test the hypothesis whether CHOP-induced down-regulation of cell surface GABAB receptors promotes excitotoxicity. If this is the case, small interfering peptides may be of potential therapeutic use to limit neuronal death under ischemic conditions. Such an intervention would be highly specific because CHOP is significantly expressed only upon ER stress, and it targets a specific protein-protein interaction in response to the ischemic insult and is, thus, expected to be associated with a low risk of side effects.
Acknowledgments
We thank Dr. J.-M. Fritschy for support with confocal microscopy and for providing E18 rat cortex, C. Sidler and G. Bosshard for preparation of E18 rat cortex, and T. Grampp for technical assistance. We also thank L. Scheurer for help with real-time PCR.
This study was supported by Swiss National Science Foundation grants 31003A_121963 and 31003A_138382 (to D. B.).
- ER
- endoplasmic reticulum
- CHOP
- CCAAT/enhancer-binding protein-homologous protein
- C/EBP
- CCAAT/enhancer-binding protein
- PLA
- proximity ligation assay
- PDI
- protein disulfide isomerase
- sPSC
- spontaneous postsynaptic current
- OGD
- oxygen and glucose deprivation
- GM
- Golgi apparatus marker
- EGFP
- enhanced GFP
- E18
- embryonic day 18.
REFERENCES
- 1. Bettler B., Kaupmann K., Mosbacher J., Gassmann M. (2004) Molecular structure and physiological functions of GABAB receptors. Physiol. Rev. 84, 835–867 [DOI] [PubMed] [Google Scholar]
- 2. Margeta-Mitrovic M., Jan Y. N., Jan L. Y. (2000) A trafficking checkpoint controls GABAB receptor heterodimerization. Neuron 27, 97–106 [DOI] [PubMed] [Google Scholar]
- 3. Pagano A., Rovelli G., Mosbacher J., Lohmann T., Duthey B., Stauffer D., Ristig D., Schuler V., Meigel I., Lampert C., Stein T., Prezeau L., Blahos J., Pin J., Froestl W., Kuhn R., Heid J., Kaupmann K., Bettler B. (2001) C-terminal interaction is essential for surface trafficking but not for heteromeric assembly of GABAB receptors. J. Neurosci. 21, 1189–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Calver A. R., Robbins M. J., Cosio C., Rice S. Q., Babbs A. J., Hirst W. D., Boyfield I., Wood M. D., Russell R. B., Price G. W., Couve A., Moss S. J., Pangalos M. N. (2001) The C-terminal domains of the GABAB receptor subunits mediate intracellular trafficking but are not required for receptor signaling. J. Neurosci. 21, 1203–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lipton P. (1999) Ischemic cell death in brain neurons. Physiol. Rev. 79, 1431–1568 [DOI] [PubMed] [Google Scholar]
- 6. Schwartz-Bloom R. D., Sah R. (2001) γ-Aminobutyric acid(A) neurotransmission and cerebral ischemia. J. Neurochem. 77, 353–371 [DOI] [PubMed] [Google Scholar]
- 7. Vollenweider F., Bendfeldt K., Maetzler W., Otten U., Nitsch C. (2006) GABAB receptor expression and cellular localization in gerbil hippocampus after transient global ischemia. Neurosci. Lett. 395, 118–123 [DOI] [PubMed] [Google Scholar]
- 8. Kim J. Y., Kim N., Yenari M. A., Chang W. (2011) Mild hypothermia suppresses calcium-sensing receptor (CaSR) induction following forebrain ischemia while increasing GABAB receptor 1 (GABAB-R1) expression. Transl. Stroke Res. 2, 195–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Francis J., Zhang Y., Ho W., Wallace M. C., Zhang L., Eubanks J. H. (1999) Decreased hippocampal expression, but not functionality, of GABAB receptors after transient cerebral ischemia in rats. J. Neurochem. 72, 87–94 [DOI] [PubMed] [Google Scholar]
- 10. Cimarosti H., Kantamneni S., Henley J. M. (2009) Ischaemia differentially regulates GABAB receptor subunits in organotypic hippocampal slice cultures. Neuropharmacology 56, 1088–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Babcock A. M., Everingham A., Paden C. M., Kimura M. (2002) Baclofen is neuroprotective and prevents loss of calcium/calmodulin-dependent protein kinase II immunoreactivity in the ischemic gerbil hippocampus. J. Neurosci. Res. 67, 804–811 [DOI] [PubMed] [Google Scholar]
- 12. Dave K. R., Lange-Asschenfeldt C., Raval A. P., Prado R., Busto R., Saul I., Pérez-Pinzón M. A. (2005) Ischemic preconditioning ameliorates excitotoxicity by shifting glutamate/γ-aminobutyric acid release and biosynthesis. J. Neurosci. Res. 82, 665–673 [DOI] [PubMed] [Google Scholar]
- 13. Jackson-Friedman C., Lyden P. D., Nunez S., Jin A., Zweifler R. (1997) High dose baclofen is neuroprotective but also causes intracerebral hemorrhage: a quantal bioassay study using the intraluminal suture occlusion method. Exp. Neurol. 147, 346–352 [DOI] [PubMed] [Google Scholar]
- 14. Kulinskii V. I., Mikhel'son G. V. (2000) Additivity and independence of neuroprotective effects of GABAA and GABAB receptor agonists in complete global cerebral ischemia. Bull. Exp. Biol. Med. 130, 772–774 [DOI] [PubMed] [Google Scholar]
- 15. Han D., Zhang Q. G., Yong-Liu Li C., Zong Y. Y., Yu C. Z., Wang W., Yan J. Z., Zhang G. Y. (2008) Co-activation of GABA receptors inhibits the JNK3 apoptotic pathway via the disassembly of the GluR6-PSD95-MLK3 signaling module in cerebral ischemic-reperfusion. FEBS Lett. 582, 1298–1306 [DOI] [PubMed] [Google Scholar]
- 16. Lal S., Shuaib A., Ijaz S. (1995) Baclofen is cytoprotective to cerebral ischemia in gerbils. Neurochem. Res. 20, 115–119 [DOI] [PubMed] [Google Scholar]
- 17. Xu J., Li C., Yin X. H., Zhang G. Y. (2008) Additive neuroprotection of GABAA and GABAB receptor agonists in cerebral ischemic injury via PI-3K/Akt pathway inhibiting the ASK1-JNK cascade. Neuropharmacology 54, 1029–1040 [DOI] [PubMed] [Google Scholar]
- 18. Zhou C., Li C., Yu H. M., Zhang F., Han D., Zhang G. Y. (2008) Neuroprotection of γ-aminobutyric acid receptor agonists via enhancing neuronal nitric oxide synthase (Ser847) phosphorylation through increased neuronal nitric oxide synthase and PSD95 interaction and inhibited protein phosphatase activity in cerebral ischemia. J. Neurosci. Res. 86, 2973–2983 [DOI] [PubMed] [Google Scholar]
- 19. Zhang F., Li C., Wang R., Han D., Zhang Q. G., Zhou C., Yu H. M., Zhang G. Y. (2007) Activation of GABA receptors attenuates neuronal apoptosis through inhibiting the tyrosine phosphorylation of NR2A by Src after cerebral ischemia and reperfusion. Neuroscience 150, 938–949 [DOI] [PubMed] [Google Scholar]
- 20. Kim I., Xu W., Reed J. C. (2008) Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat. Rev. Drug. Discov. 7, 1013–1030 [DOI] [PubMed] [Google Scholar]
- 21. Oyadomari S., Mori M. (2004) Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11, 381–389 [DOI] [PubMed] [Google Scholar]
- 22. Sauter K., Grampp T., Fritschy J. M., Kaupmann K., Bettler B., Mohler H., Benke D. (2005) Subtype-selective interaction with the transcription factor CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP) regulates cell surface expression of GABAB receptors. J. Biol. Chem. 280, 33566–33572 [DOI] [PubMed] [Google Scholar]
- 23. Benke D., Honer M., Michel C., Bettler B., Mohler H. (1999) γ-Aminobutyric acid type B receptor splice variant proteins GBR1a and GBR1b are both associated with GBR2 in situ and display differential regional and subcellular distribution. J. Biol. Chem. 274, 27323–27330 [DOI] [PubMed] [Google Scholar]
- 24. Benke D., Michel C., Mohler H. (2002) Structure of GABAB receptors in rat retina. J. Recept. Signal Transduct. Res. 22, 253–266 [DOI] [PubMed] [Google Scholar]
- 25. Benke D., Mertens S., Trzeciak A., Gillessen D., Mohler H. (1991) GABAA receptors display association of γ 2-subunit with α1- and β2/3-subunits. J. Biol. Chem. 266, 4478–4483 [PubMed] [Google Scholar]
- 26. Grampp T., Notz V., Broll I., Fischer N., Benke D. (2008) Constitutive, agonist-accelerated, recycling and lysosomal degradation of GABAB receptors in cortical neurons. Mol. Cell. Neurosci. 39, 628–637 [DOI] [PubMed] [Google Scholar]
- 27. Buerli T., Pellegrino C., Baer K., Lardi-Studler B., Chudotvorova I., Fritschy J. M., Medina I., Fuhrer C. (2007) Efficient transfection of DNA or shRNA vectors into neurons using magnetofection. Nat. Protoc. 2, 3090–3101 [DOI] [PubMed] [Google Scholar]
- 28. Vanhoose A. M., Emery M., Jimenez L., Winder D. G. (2002) ERK activation by G-protein-coupled receptors in mouse brain is receptor identity-specific. J. Biol. Chem. 277, 9049–9053 [DOI] [PubMed] [Google Scholar]
- 29. Ren X., Mody I. (2003) γ-Hydroxybutyrate reduces mitogen-activated protein kinase phosphorylation via GABAB receptor activation in mouse frontal cortex and hippocampus. J. Biol. Chem. 278, 42006–42011 [DOI] [PubMed] [Google Scholar]
- 30. Tu H., Rondard P., Xu C., Bertaso F., Cao F., Zhang X., Pin J. P., Liu J. (2007) Dominant role of GABAB2 and Gβγ for GABAB receptor-mediated-ERK1/2/CREB pathway in cerebellar neurons. Cell Signal. 19, 1996–2002 [DOI] [PubMed] [Google Scholar]
- 31. Im B. H., Rhim H. (2012) GABAB receptor-mediated ERK1/2 phosphorylation via a direct interaction with CaV1.3 channels. Neurosci. Lett. 513, 89–94 [DOI] [PubMed] [Google Scholar]
- 32. Leuchowius K. J., Jarvius M., Wickström M., Rickardson L., Landegren U., Larsson R., Söderberg O., Fryknäs M., Jarvius J. (2010) High content screening for inhibitors of protein interactions and post-translational modifications in primary cells by proximity ligation. Mol. Cell Proteomics 9, 178–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
- 34. Zemoura K., Benke D. (2014) Proteasomal degradation of γ-aminobutyric acidB receptors is mediated by the interaction of the GABAB2 C terminus with the proteasomal ATPase Rtp6 and regulated by neuronal activity. J. Biol. Chem. 289, 7738–7746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hossmann K. A. (1993) Disturbances of cerebral protein synthesis and ischemic cell death. Prog. Brain Res. 96, 161–177 [DOI] [PubMed] [Google Scholar]
- 36. Paschen W., Doutheil J. (1999) Disturbance of endoplasmic reticulum functions: a key mechanism underlying cell damage? Acta Neurochir. Suppl. 73, 1–5 [DOI] [PubMed] [Google Scholar]
- 37. Osada N., Kosuge Y., Kihara T., Ishige K., Ito Y. (2009) Apolipoprotein E-deficient mice are more vulnerable to ER stress after transient forebrain ischemia. Neurochem. Int. 54, 403–409 [DOI] [PubMed] [Google Scholar]
- 38. Osada N., Kosuge Y., Ishige K., Ito Y. (2010) Characterization of neuronal and astroglial responses to ER stress in the hippocampal CA1 area in mice following transient forebrain ischemia. Neurochem. Int. 57, 1–7 [DOI] [PubMed] [Google Scholar]
- 39. Tajiri S., Oyadomari S., Yano S., Morioka M., Gotoh T., Hamada J.-I., Ushio Y., Mori M. (2004) Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 11, 403–415 [DOI] [PubMed] [Google Scholar]
- 40. Paschen W., Gissel C., Linden T., Althausen S., Doutheil J. (1998) Activation of Gadd153 expression through transient cerebral ischemia: evidence that ischemia causes endoplasmic reticulum dysfunction. Mol. Brain Res. 60, 115–122 [DOI] [PubMed] [Google Scholar]
- 41. Oida Y., Shimazawa M., Imaizumi K., Hara H. (2008) Involvement of endoplasmic reticulum stress in the neuronal death induced by transient forebrain ischemia in gerbil. Neuroscience 151, 111–119 [DOI] [PubMed] [Google Scholar]
- 42. Morimoto N., Oida Y., Shimazawa M., Miura M., Kudo T., Imaizumi K., Hara H. (2007) Involvement of endoplasmic reticulum stress after middle cerebral artery occlusion in mice. Neuroscience 147, 957–967 [DOI] [PubMed] [Google Scholar]
- 43. Fornace A. J., Jr., Alamo I., Jr., Hollander M. C. (1988) DNA damage-inducible transcripts in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 85, 8800–8804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oyadomari S., Araki E., Mori M. (2002) Endoplasmic reticulum stress-mediated apoptosis in pancreatic β-cells. Apoptosis 7, 335–345 [DOI] [PubMed] [Google Scholar]
- 45. Zinszner H., Kuroda M., Wang X., Batchvarova N., Lightfoot R. T., Remotti H., Stevens J. L., Ron D. (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lytton J., Westlin M., Hanley M. R. (1991) Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 266, 17067–17071 [PubMed] [Google Scholar]
- 47. Pooler A. M., Gray A. G., McIlhinney R. A. (2009) Identification of a novel region of the GABAB2 C-terminus that regulates surface expression and neuronal targeting of the GABAB receptor. Eur. J. Neurosci. 29, 869–878 [DOI] [PubMed] [Google Scholar]
- 48. Kostandy B. B. (2012) The role of glutamate in neuronal ischemic injury: the role of spark in fire. Neurol. Sci. 33, 223–237 [DOI] [PubMed] [Google Scholar]