

Background: The apoE4 mutant apoE4[L28P] is associated with an increased risk for Alzheimer disease (AD) in addition to the known effect of apoE4.
Results: ApoE4[L28P] displays folding defects and aberrant functions associated with AD pathogenesis.
Conclusion: The structural integrity of apoE4 is an important component of its role in AD pathogenesis.
Significance: We provide insights into the molecular basis for the added risk for AD in apoE4[L28P] carriers.
Keywords: Alzheimer Disease, ApoE, Apolipoproteins, Biophysics, Lipoprotein, Reactive Oxygen Species (ROS), Amyloid Peptide β, Mutation, Thermodynamic Stability, Toxicity
Abstract
The apolipoprotein (apo) E4 isoform has consistently emerged as a susceptibility factor for late-onset Alzheimer disease (AD), although the exact mechanism is not clear. A rare apoE4 mutant, apoE4[L28P] Pittsburgh, burdens carriers with an added risk for late-onset AD and may be a useful tool for gaining insights into the role of apoE4 in disease pathogenesis. Toward this end, we evaluated the effect of the L28P mutation on the structural and functional properties of apoE4. ApoE4[L28P] was found to have significantly perturbed thermodynamic properties, to have reduced helical content, and to expose a larger portion of the hydrophobic surface to the solvent. Furthermore, this mutant is thermodynamically destabilized and more prone to proteolysis. When interacting with lipids, apoE4[L28P] formed populations of lipoprotein particles with structural defects. The structural perturbations brought about by the mutation were accompanied by aberrant functions associated with the pathogenesis of AD. Specifically, apoE4[L28P] promoted the cellular uptake of extracellular amyloid β peptide 42 (Aβ42) by human neuroblastoma SK-N-SH cells as well as by primary mouse neuronal cells and led to increased formation of intracellular reactive oxygen species that persisted for at least 24 h. Furthermore, lipoprotein particles containing apoE4[L28P] induced intracellular reactive oxygen species formation and reduced SK-N-SH cell viability. Overall, our findings suggest that the L28P mutation leads to significant structural and conformational perturbations in apoE4 and can induce functional defects associated with neuronal Aβ42 accumulation and oxidative stress. We propose that these structural and functional changes underlie the observed added risk for AD development in carriers of apoE4[L28P].
Introduction
Apolipoprotein E (apoE)3 is a major protein of the human lipoprotein transport system in the circulation and brain (1, 2). The brain is second only to the liver with respect to apoE expression levels (3). ApoE contains 299 residues and has three common isoforms (apoE2, apoE3, and apoE4), each differing in amino acid positions 112 and 158 (1). ApoE3 has a cysteine at residue 112 and an arginine at residue 158, whereas apoE4 has an arginine at each position and apoE2 has cysteines. ApoE4 has been associated with a variety of neuropathological processes, including Alzheimer disease (AD) (2). ApoE4 is a major genetic risk factor for late-onset AD (LOAD) because 40% of all patients have at least one ϵ4 allele (4). Being homozygous or heterozygous for the ϵ4 allele increases the risk of LOAD 4-fold and lowers the age of onset of the disease (4, 5). Screening for apoE variants in patients with sporadic LOAD and related controls revealed a naturally occurring point mutation in apoE4, L28P (denoted as apoE4 Pittsburgh), in a heterozygous state (6, 7). Carriers of the apoE4[L28P] mutant are at a substantially higher risk of developing LOAD, and this risk remains significant even after adjusting for the known effect of apoE4 (6, 7).
A large number of studies have examined how apoE4 is associated with LOAD. It has been suggested that apoE4 is involved in the modulation of plaque formation and clearance of Aβ, promotes intraneuronal accumulation of Aβ, affects cholesterol homeostasis, alters phosphorylation of tau and formation of neurofibrillary tangles, disrupts cytoskeleton structure, impairs cholinergic signal transduction, and causes dysregulation of various signaling pathways (2, 8–11). Therefore, it appears that apoE4 has pleiotropic functions and that several parallel pathways may contribute to the pathogenic role of apoE4 in LOAD. It is also possible that some of the aforementioned pathological processes are early events, whereas others follow subsequently in LOAD pathogenesis.
Studies with humans and experimental animals suggested that amyloid β peptide accumulates inside neurons. Aβ accumulation occurs prior to extracellular amyloid formation, has been implicated in the onset of early cognitive alterations, and may contribute to the pathological cascade of events that leads to neuronal dysfunction and, eventually, to AD (12, 13). Furthermore, the 42-amino acid Aβ (Aβ42) constitutes the majority of intraneuronal Aβ (12). We have demonstrated previously that a specific apoE4 fragment can promote the uptake and intracellular accumulation of Aβ42 and lead to increased formation of reactive oxygen species in neuroblastoma cells, two events that are considered to be early events in the pathogenesis process of AD (10). These and other findings suggest that not all apoE4 forms found in the brains of AD patients are equally bioactive and that specific apoE4 forms may be involved in processes associated with AD pathogenesis (10, 14–17).
ApoE is highly helical, with a labile tertiary structure that can assume structures characteristic of a molten globule (18). It can undergo significant conformational changes during its physiological function, including lipid and receptor binding (19, 20). Lipid-free apoE is folded into two seemingly independent structural domains that can be separated after digestion with thrombin, generating an N-terminal, 22-kDa fragment (residues 1–191) and a C-terminal, 10-kDa fragment (residues 216–299) (21, 22). X-ray crystallographic analysis of the lipid-free apoE amino-terminal domain (residues 1–191) showed a 4 helix bundle spanning residues 24–164 that segregates the hydrophobic core of the four helices from the solvent (23). Biophysical and computational analyses suggest that unfolding of the N-terminal domain constitutes a necessary conformational change for lipid binding and apoE function (19). The C-terminal domain is also highly α-helical, but its tertiary structure is highly polymorphic and participates in interdomain interactions with the N-terminal domain as well as with lipids (19). ApoE4 has been described to have both structural and functional differences to apoE2 and apoE3 that stem from different interactions between the N-terminal and C-terminal domains of the molecule. These interactions are considered to be very important for the physiological function of apoE (24). ApoE variants with lower thermodynamic stability and changes in conformational plasticity have been shown to have altered interdomain interactions and defective physiological functions (19, 24–28).
In this study we evaluated the effects of the naturally occurring apoE4 mutation L28P, a hereditary mutation that is associated with an increased risk for LOAD, on the structural integrity and functional properties of the protein. Because the N-terminal domain of apoE is highly helical, we hypothesized that the substitution of leucine by proline, a structurally rigid amino acid that is often incompatible with helical segments, may cause perturbations on the local structure and adversely affect apoE4 structure and function. We produced recombinant apoE4[L28P] and characterized its structural, thermodynamic, and functional integrity. Our findings demonstrate that the mutation induced significant structural and conformational perturbations in the molecule of apoE4. Furthermore, lipid-free apoE4[L28P] promoted the neuronal accumulation of Aβ42 and led to increased formation of reactive oxygen species (ROS) inside cells. In addition, lipoprotein-associated apoE4[L28P] was found to promote the formation of ROS and reduce viability of SK-N-SH neuroblastoma cells. Overall, our data suggest that the L28P mutation significantly affects the structure of apoE4 both in solution and in lipoprotein particles and that these effects may underlie apoE4 functional disturbances in neurons that could be associated with AD pathogenesis.
EXPERIMENTAL PROCEDURES
Materials
Strain BL21-Gold (DE3) of Escherichia coli was purchased from Stratagene (Cedar Creek, TX). The Complete Mini EDTA-free protease inhibitor mixture was from Roche. Nickel-nitrilotriacetic acid (Ni-NTA) resin was purchased from Thermo Scientific (Rockford, IL). Chymotrypsin, trypsin, and elastase enzymes were obtained from Applichem (Darmstadt, Germany). Lyophilized Aβ(1–42), hexafluoroisopropanol-treated, was from JPT Peptide Technologies GmbH (Berlin, Germany). Culture media were purchased from Sigma-Aldrich (St. Louis, MO), Biochrom AG (Berlin, Germany), and Lonza (Verviers, Belgium). Egg phosphatidylcholine and cholesterol were from Sigma-Aldrich.
Site-directed Mutagenesis
The pET32-E43C vector containing a thioredoxin (Trx) tag, a His6 tag, and a 3C-protease site at the fusion junction with the human cDNA for full-length apoE4 has been described previously (29). The L28P mutation was introduced into the gene of apoE4 by site direct mutagenesis using the commercial available QuikChange II XL site-directed mutagenesis kit (Agilent, Santa Clara, CA) according to the instructions of the manufacturer. The sequences of the primers used for the mutagenesis were as follows: L28P, 5′-GCC AGC GCT GGG AAC CGG CAC TGG GTC GCT TTT G-3′ (forward) and 5′-CAA AAG CGA CCC AGT GCC GGT TCC CAG CGC TGG C-3′ (reverse). Successful mutagenesis was confirmed by DNA sequencing.
Expression and Purification of WT ApoE4 and Mutant ApoE4[L28P]
The expression and purification of WT apoE4 were carried out as described previously (29). ApoE4[L28P] was expressed and purified following the same protocol with some modifications. Briefly, BL21-Gold(DE3) cells were transformed with the expression vector pET32-E43C carrying the sequence of the gene of mutant apoE4[L28P] and cultured in Luria broth medium containing 100 μg/ml ampicillin at 37 °C. Protein expression was induced with isopropyl 1-thio-β-d-galactopyranoside (final concentration 0.5 mm) for 2 h. Cells were lysed in 20 mm Tris-HCl buffer (pH 8.0) containing 0.5 m NaCl, Complete Mini EDTA-free protease inhibitor mixture, and 0.1 mg/ml lysozyme (40 ml/liter of original culture) by using a French press (SLM-AMINCO), and the lysate was centrifuged to remove cellular debris. The Trx-fused apoE4[L28P] in the supernatant was purified by Ni-NTA chromatography as follows. The supernatant was adjusted to contain 20 mm Tris-HCl (pH 8.0), 0.5 NaCl, and 5 mm imidazole and incubated with 2 ml (per 0.5 liter of original culture) Ni-NTA resin, under gentle stirring, at 4 °C overnight. The following day, the Ni-NTA suspension was loaded onto an empty chromatography column and washed with 180 ml 20 mm Tris-HCl (pH 8.0), 0.5 m NaCl, 5 mm imidazole. The Trx-fused apoE4[L28P] was eluted with 20 mm Tris-HCl (pH 8.0), 0.5 m NaCl containing 50–1000 mm imidazole. The collected fractions were dialyzed extensively against 20 mm Tris-HCl (pH 8.0), 0.5 m NaCl at 4 °C. Following dialysis, His-tagged 3C protease (prepared as described previously (29) using the vector pET-24/His-3C that was kindly provided by Dr. Arie Geerlof, EMBL, Heidelberg, Germany) was added at a ratio of 1/25 (3C-protease/Trx-apoE4, w/w), and cleavage of Trx from apoE4[L28P] was allowed to proceed for 18 h at 4 °C in 20 mm Tris-HCl buffer (pH 8.0) containing 0.5 m NaCl and 1 mm DTT. ApoE4[L28P] was separated by the cleaved tag by a second Ni-NTA resin affinity chromatography step. To facilitate the dissociation of noncovalent complexes among any uncut apoE4, cut apoE4, and cut Trx, the cleavage reaction protein solution was adjusted by adding 6 m urea (final concentration), and the solution was incubated with 1 ml of Ni-NTA resin (per 4 mg of protein) for 1 h at 4 °C. The suspension was loaded onto an empty chromatography column, and cut apoE4[L28P] was found in the column flow-through. The apoE4[L28P] solution was dialyzed extensively against 5 mm NH4HCO3, lyophilized, and stored at −80 °C.
Protein Refolding
Before all analyses, the lyophilized stocks of WT or mutant apoE4 were dissolved at a final concentration of 0.2 mg/ml in 6 m guanidine hydrochloride (GndHCl) in Dulbecco's phosphate-buffered saline (DPBS). The protein samples were incubated for 1 h at room temperature and then dialyzed extensively against DPBS (pH 7.4) (three changes over 48 h, two of which were overnight). The samples were then centrifuged at 10,000 × g for 10 min to remove any precipitated protein. The concentration of the proteins retrieved in the supernatant was calculated by measuring their absorbance at 280 nm and using an extinction coefficient of 1.285 ml/mg/cm. Proteins were kept at low concentrations (∼0.1 mg/ml) on ice to avoid aggregation. The refolded proteins were at least 96% pure, as estimated by SDS-PAGE. The purity and integrity of the protein was also confirmed by MALDI-TOF.
Preparation of Discoidal Reconstituted Lipoprotein Particles
Reconstituted lipoprotein particles containing WT or mutant apoE4 were prepared using a molar ratio of 100:10:1:100 of egg phosphatidylcholine:cholesterol:apoE:sodium cholate in DPBS (pH 7.4), as described previously (28). Lipoprotein particles containing WT apoE4 or apoE4[L28P] were prepared using the same phospholipid-cholesterol suspension, and the procedure was performed in parallel. Samples were stored at 4 °C under nitrogen to prevent the oxidation of lipids.
CD Measurements
Far-UV circular dichroism (CD) spectra were recorded using a Jasco 715 spectropolarimeter at 20 °C in a 1-mm path length quartz cuvette. The concentration of the protein samples was 0.1 mg/ml in DPBS (pH 7.4). Spectra were recorded from 190–260 nm by using the following measurement parameters: bandwidth 1 nm, response 8 s, step size 0.2 nm, and scan speed 50 nm/min. During the measurements, a Jasco PTC-348 WI Peltier temperature controller was connected to the instrument for thermostating the cuvette chamber. Each spectrum was the average of five accumulations. All spectra were obtained by subtracting the buffer baseline.
Helical content was calculated using the molecular ellipticity at 222 nm as described (30) using Equation 1:
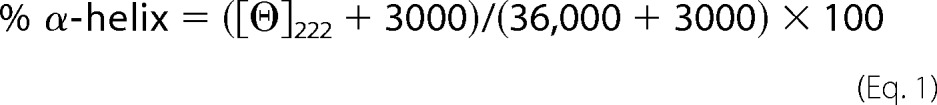 |
For thermal denaturation measurements, the change in molar ellipticity at 222 nm was monitored while varying the temperature in the range of 20–80 °C at a rate of 1 °C/min. The thermal denaturation curve was fitted to a Boltzman simple sigmoidal model curve using the GraphPad PrismTM software (GraphPad Software Inc., La Jolla, CA). The apparent melting temperature, Tm, was determined by the sigmoidal fit as midpoint of the thermal transition. The relative enthalpy change was calculated as described previously (31). For measurements using lipoprotein particles containing WT or mutant apoE4, the protein component of the particle was at 0.1 mg/ml, and all measuring parameters were identical to that of the free protein, with the exception of the temperature range for thermal denaturation, which varied from 20–100 °C.
Chemical Denaturation Experiments
To record the chemical denaturation profile of WT and mutant apoE4, we measured the changes in intrinsic tryptophan fluorescence (excitation 295, emission 340 nm) of the proteins in solution upon addition of increasing amounts of 8.0 m guanidine hydrochloride (GndHCl). Briefly, 0.05 mg/ml of freshly refolded protein was inserted into a 4-ml quartz fluorometer cuvette, small amounts of 8.0 m GndHCl were added gradually to the solution, and the contents were mixed by repeated pipetting for 5 s. Then, the mixture was incubated for 2 or 5 min in the dark, and the fluorescence signal of the sample was measured in a Quantamaster 4 fluorescence spectrometer (Photon Technology International). The experimental data were fitted to a three-state denaturation model as described previously (32).
ANS Fluorescence Measurement
1-anilinonaphthalene-8-sulfonic acid (1,8 ANS, Sigma-Aldrich) was dissolved into dimethyl sulfoxide to a final concentration of 50 mm (ANS stock solution) and stored at −20 °C. Freshly refolded WT or mutant apoE4 at 0.05 mg/ml in DPBS (pH 7.4) was inserted into a 4-ml quartz fluorometer cuvette. The fluorescence signal was measured in a Quantamaster 4 fluorescence spectrometer using scan rate of 1 nm/s, excitation wavelength at 395 nm, and emission range from 420–550 nm. After measuring the background fluorescence of the protein samples, 7.5 μl of 1,8 ANS stock solution was added into the cuvettes and mixed so that the final concentration of 1,8 ANS was 250 μm, and then the fluorescence signal was recorded. A control ANS spectrum in the absence of protein was also recorded to allow the calculation of ANS fluorescence enhancement in the presence of apoE4 forms.
DMPC Remodeling Kinetics
The assay of l-α-dimyristoylphosphatidylcholine (DMPC) vesicle remodeling by WT or mutant apoE4 was performed as described previously (33, 34) with some modifications. DMPC final concentration was 97 μg/ml, and apoE4 final concentration was 43 μg/ml. The reaction was performed in 1.1 ml of DPBS containing 3.5% KBr, 0.1 mm DTT, 0.1 m GndHCl, 0.1 mm EDTA. Reaction kinetics were followed by the change in absorbance at 325 nm using a Lambda 35 UV-visible spectrophotometer (PerkinElmer Life Sciences) for 60 min. The cuvette (1-cm path length) was thermostated at 24 ± 0.1 °C using the PerkinElmer Life Sciences PCB 1500 Peltier temperature controller. The contents were mixed by repeated pipetting for 3 s every 2 min. Experimental data were fitted to a two-phase exponential decay model using GraphPad PrismTM.
Protease Sensitivity Assay
The protease sensitivity of the lipid-free WT and mutant apoE4 was assessed by incubating 0.13 mg/ml of freshly refolded protein in DPBS (pH 7.4) with increasing concentrations of trypsin, chymotrypsin, or elastase (0.01, 0.1, 0.5, 1, 5, and 10 μg/ml final concentration) at room temperature for 1 h. The reactions were stopped with the addition of PMSF at a final concentration of 1.7 mm, and the samples were analyzed by SDS-PAGE.
Electron Microscopy
Reconstituted lipoprotein particles containing WT or mutant apoE4 were contrasted using the drop method of negative staining (35). A 4-μl aliquot of each preparation was applied to a freshly glow-discharged (36), Formvar carbon-coated, 300 mesh copper grid. After a 10-s incubation and 4-s filter paper blot, a 4-μl droplet of 1% sodium phosphotungstate (1% NaPT) (pH 7.4) stain was applied for 10 s. The grid was blotted and air-dried. Images were collected under low-dose conditions at a magnification of ×56,250 on a Philips CM-12 electron microscope (Philips Electron Optics, Eindhoven, The Netherlands) with a Teitz 1 K × 1 K charge-coupled device camera (TVIPS, Gauting, Germany). For analysis of the disc diameter distribution, two representative images from each sample were analyzed by measuring the diameter of 150 clearly formed discs using Adobe PhotoshopTM software (Adobe Inc., San Jose, CA). The data were plotted using the frequency distribution function of GraphPad PrismTM.
Cell Cultures
Human neuroblastoma SK-N-SH cells (ATCC) were cultured in minimal essential medium (MEM) Earle's (Biochrom AG, Berlin, Germany) supplemented with 2 mm l-glutamine, 0.1 mm nonessential amino acids, 1 mm sodium pyruvate, 1.5 g/liter sodium bicarbonate, 10% FBS (MEM-Earle's complete), and antibiotics.
Primary cultures of mouse cortical neurons were prepared from postnatal day 0 male pups of C57BL/6 mice as described previously (37, 38). Briefly, the cortices were dissected, neurons were dissociated by trypsin/DNase digestion, and then the cells were plated at a density of 6.6 × 104/cm2 on glass coverslips coated with poly-d-lysine (12 mm, no. 1 thickness, VitroCam, London, UK) and cultured in 0.5 ml Neurobasal medium supplemented with 2% B-27 serum-free supplement, 0.5 mm Glutamax, 100 units/ml penicillin G, and 100 mg/ml streptomycin sulfate.
Cell Viability Assay
Cell viability was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma) assay (39). SK-N-SH cells were plated on 96-well plates at a density of 2 × 104 cells/well in MEM-Earle's complete medium and then incubated with lipid-free or lipoprotein particle-associated WT or mutant apoE4 (at concentrations of 0.375 or 1 μm) for 24 h at 37 °C. The cells were then incubated for 3 h at 37 °C in serum-free cell medium containing 0.65 mg/ml MTT. After incubation, the medium was removed, the dark blue formazan crystals, formed by metabolically active cells containing functional mitochondria, were dissolved in dimethyl sulfoxide, and absorbance was measured at 550 nm in an Infinite M200 plate reader (Tecan Group Ltd., Mannedorf, Switzerland).
Confocal Microscopy
SK-N-SH cells or primary mouse cortical neurons seeded on coverslips were incubated with 25 ng/ml Aβ42 in the presence or absence of 0.375 μm lipid-free or lipoprotein particle-associated WT or mutant apoE4 for 24 h at 37 °C or after removal of apoE4 and Aβ42 from the cell medium for another 24 h at 37 °C. At the end of incubation, cells were washed with PBS, fixed with 4% paraformaldehyde at room temperature for 15 min, washed again with PBS, and incubated in blocking and permeabilization buffer (PBS/5% FBS/0.05% Tween 20) for 30 min at 25 °C. Aβ42 was stained by using primary mouse anti-Aβ monoclonal antibody (6E10, 1:200, Chemicon, Temecula, CA) and secondary Alexa Fluor 488-conjugated goat anti-mouse IgG antibody (1:1000, Invitrogen). For F-actin staining, rhodamine phalloidin (1:200, Cytoskelton Inc., Denver, CO) was used. Coverslips were mounted onto slides and viewed with an MRC 1024ES laser-scanning confocal microscope (Bio-Rad) equipped with Lasersharp software (Bio-Rad) and a krypton-argon laser with a motor step of 0.5 μm.
Measurement of ROS Generation
Intracellular ROS generation was measured by following a method described previously (40) modified for fluorescent microscopy. SK-N-SH cells were plated on 24-well plates at a density of 1 × 105 cells/well in MEM-Earle's complete cell medium and cultured until 90% confluent. Cells were then incubated with 0.375 μm lipid-free or lipoprotein particle-associated WT or mutant apoE4 in the presence or absence of 25 ng/ml Aβ42 in serum-free cell medium for 24 h at 37 °C or, after removal of apoE4 and Aβ42 from the cell medium, for another 24 h at 37 °C. At the end of the incubation period, the cells were washed with DMEM and incubated in the dark for 45min at 37 °C in DMEM containing 25μΜ 2′,7′-dichlorofluorescein diacetate (Molecular Probes/Invitrogen). Subsequently, the cells were washed with preheated at 37 °C PBS, and the production of ROS was detected by recording the fluorescence of 2′,7′-dichlorofluorescein (DCF) with an Axiovert 25 (Zeiss, Göttingen, Germany) inverted microscope equipped for fluorescence microscopy (excitation 450–490 nm, emission 520 nm). DCF fluorescence intensity was measured for at least 40 cells from the fluorescent images of each sample using ImageJ image analysis software (National Institutes of Health, Bethesda, MD) (41), and the relative fluorescence intensity was taken as the average of the values of at least five images for each experiment.
RESULTS
The L28P Mutation Destabilizes the Structure of ApoE4
To investigate whether the L28P mutation (Fig. 1, A and B) affects the structure of apoE4, we produced and purified recombinant apoE4 carrying the mutation, as well as WT apoE4, using an E. coli expression system established previously (29). Both proteins were at least 96% pure, as judged by SDS-PAGE analysis (Fig. 1C). MALDI-TOF analysis of the samples confirmed the purity of our preparations as well as the expected molecular mass (34585.5 ± 30.9 Da for WT apoE4 and 34570.3 ± 30.4 Da for apoE4[L28P]) (Fig. 1, D and E).
FIGURE 1.

Schematic of the crystal structure of the N-terminal domain of human apoE4, indicating the location of the L28P mutation and SDS-PAGE and MALDI-TOF analyses of purified WT apoE4 and mutant apoE4[L28P]. A and B, the position of the mutated leucine residue on the N-terminal helical segment of apoE4 was mapped on the crystal structure of apoE4 with PDB code 1GS9. C, SDS-PAGE analysis of refolded WT and mutant apoE4, which were produced and purified as described under “Experimental Procedures.” D and E, MALDI-TOF spectra of refolded WT and mutant apoE4. a.u., arbitrary units.
The circular dichroism spectrum of lipid-free apoE4[L28P] was found to overall have a similar shape to the spectrum of the WT apoE4, which is identical to spectra reported previously for that molecule (25, 29, 42, 43). The calculated α-helical content, on the basis of the CD signal at 222 nm, for WT apoE4 was 53.1%, in the middle of the range of 44–62% that has been reported for the helical content of apoE4 and apoE3, produced by bacterial or mammalian expression systems (22, 25, 29, 43–45). ApoE4[L28P] displayed a loss of about 10% of helical content compared with the WT apoE4 (Fig. 2A). This amount of helicity loss greatly exceeds the expected loss of helicity because of local structure disruption as a result of the mutation and suggests a more generalized unfolding. Furthermore, apoE4[L28P] was less stable toward thermal denaturation, having a lower melting temperature midpoint (Tm) and a calculated enthalpy for the transition of 9.1 kcal/mol less than WT apoE4 (Fig. 2B).
FIGURE 2.

Physicochemical properties of WT apoE4 and apoE4[L28P]. A, far UV CD spectra of WT and mutant apoE4. Spectra are averages of three separate experiments. The percentage of helical content was calculated on the basis of the molar ellipticity at 222 nm, as described under “Experimental Procedure.” B, thermal denaturation profiles of WT and mutant apoE4. The y axis has been normalized to correspond to the fraction of the protein in the unfolded state. Experimental data were fit to a simple two-state Boltzman transition (solid line). Gray lines indicate the portion of the apoE4[L28P] mutant that is in an unfolded state at physiological temperatures (37 °C). Apparent Tm and ΔH values were calculated as described under “Experimental Procedures.” C, chemical denaturation profiles of WT and mutant apoE4. The titration of WT apoE4 with GndHCl was performed using a 2-min incubation between each measurement, whereas that of apoE4[L28P] was performed using a 2- or 5-min incubation. The y axis has been normalized to correspond to the fraction of the protein remaining in the folded state. Experimental data were fitted to a three-state denaturation model as described under “Experimental Procedures” (solid line). D, time course of the fluorescence signal change of the WT or mutant apoE4 in the absence or presence of GndHCl at a final concentration of 0.5 m. E, ANS fluorescence spectra in the presence or absence of WT and mutant apoE4. Spectra are the average of three separate measurements. The fold increase is the increase in ANS fluorescence in the presence of the protein relative to free ANS in the same buffer. *, p < 0.05 versus WT; **, p < 0.01 versus WT; ***, p < 0.005 versus WT.
To better understand the possible changes in the thermodynamic stability of apoE4 because of the presence of the L28P mutation, we studied the chemical denaturation profile of the apoE4 variant. The chemical denaturation experiments were performed by serial additions of GndHCl in the cuvette containing the WT or mutant apoE4, incubation for 2 min, and then measurement of the fluorescence signal, as described under “Experimental Procedures.” This approach yields a chemical denaturation profile (including the intermediate phase) for WT apoE4 that is identical to what reported in the literature using longer (up to overnight) incubation times of protein with GndHCl (43, 46). When treated with GndHCl using the same approach, apoE4[L28P] demonstrated a single transition with a loss of the unfolding intermediate and a shift to lower GndHCl concentrations (Fig. 2C). To examine whether the loss of the unfolding intermediate for apoE4[L28P] is due to changes in equilibrium thermodynamics or kinetics, we measured the time course of the fluorescence signal change after the addition of GndHCl at a final concentration of 0.5 m to both the WT and mutant apoE4. The same measurement was also performed in the absence of GndHCl. We detected a small decrease in the fluorescence signal during the time course that plateaued after about 2–5 min (Fig. 2D). This decrease, however, was present even in the absence of GndHCl (Fig. 2D) and is more likely to be due to some photobleaching of the protein because of the illumination. Furthermore, the loss of fluorescence signal was similar for both WT and mutant apoE4 (Fig. 2D). Regardless, we repeated the chemical denaturation of the mutant protein using the longer, 5-min incubation per data point (Fig. 2C). The overall shape of the denaturation curve for apoE4[L28P] was unchanged, indicating that the loss of the intermediate state is not due to incomplete equilibration after each addition of GndHCl but due to a destabilized structure. This behavior has been correlated in the past with the misfolding of the N-terminal domain of the protein, where this mutation is located (21, 25, 46).
Finally, to evaluate whether the introduction of the mutation interferes with the hydrophobic profile of apoE4, we used the hydrophobic reporter probe ANS. Binding of ANS onto apoE has been used previously to probe the solvent-exposed hydrophobic sites of the molecule and as a measure of possible conformational rearrangements because of mutation (25, 44). The ANS binding analysis suggested that the L28P mutation resulted in an increase of the solvent-accessible hydrophobic surface of apoE4, a finding that could be consistent with exposure of the N-terminal four-helix bundle hydrophobic core because of misfolding (Fig. 2E). Overall, biophysical analysis indicates that introduction of the L28P mutation into apoE4 has serious repercussions for the structure and folding of the protein and leads to thermodynamic destabilization and, possibly, local or generalized misfolding.
ApoE4 is prone to proteolysis both in vivo and in vitro, something that has been suggested to be associated with Alzheimer disease pathogenesis (14). Because thermodynamic destabilization can enhance the susceptibility of a protein to proteolysis, we investigated whether the introduction of the L28P mutation can affect apoE4 sensitivity to proteolysis in vitro (Fig. 3). Limited proteolysis with elastase, chymotrypsin, and trypsin revealed that the introduction of the L28P mutation further sensitizes apoE4 toward proteolysis. Specific proteolysis-resistant apoE4 fragments (outlined in rectangles in Fig. 3) that are generated after limited proteolysis of WT apoE4 failed to form when apoE4[L28P] was subjected to the same treatment. Because proline is not the preferred residue for any of the proteases used, this finding is consistent with local or even generalized unfolding that exposes protease-sensitive sites in the variant apoE4.
FIGURE 3.
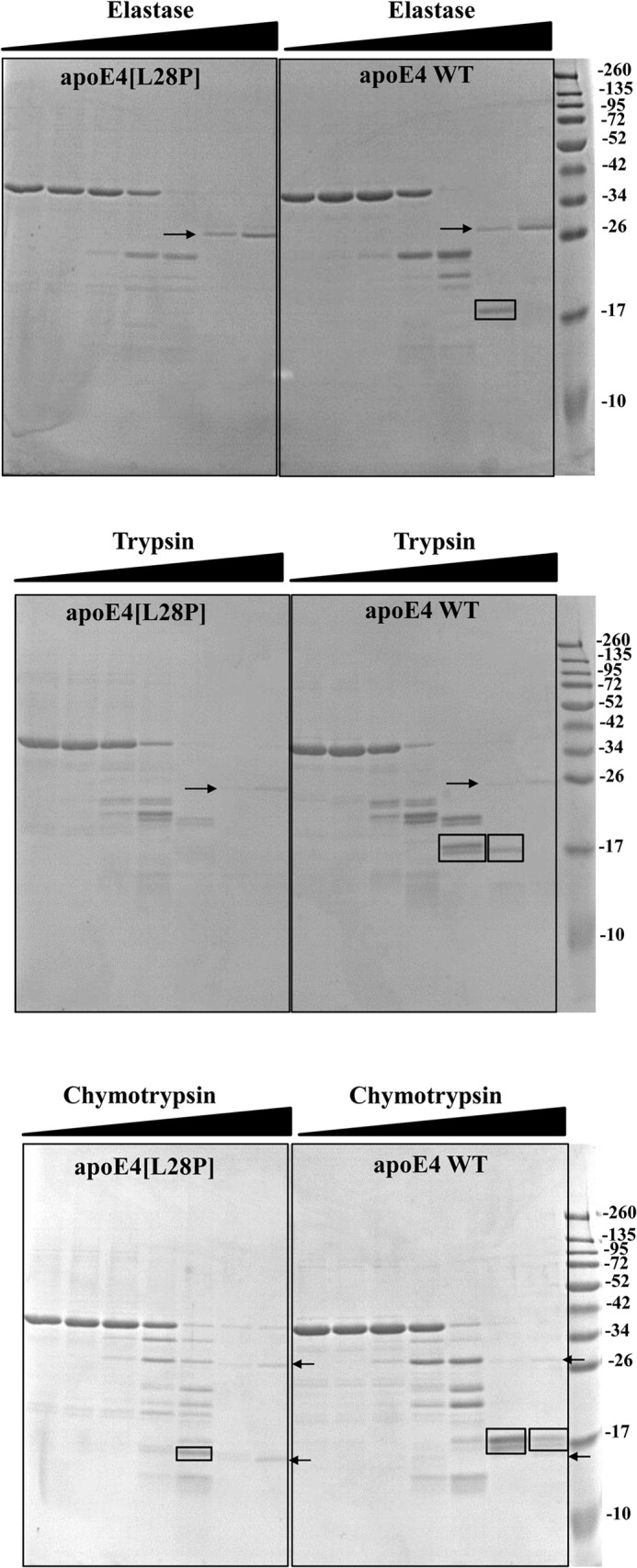
Protease digestion sensitivity of WT apoE4 and apoE4[L28P]. WT and mutant apoE4 were incubated for 1 h at room temperature with increasing amounts of elastase (top panel), trypsin (center panel), and chymotrypsin (bottom panel) as described under “Experimental Procedures.” Reactions were stopped by the addition of PMSF and analyzed on SDS-PAGE. Arrows indicate the bands that correspond to the protease (only visible for the highest concentrations used). Rectangles highlight different apoE4 fragments that accumulate in WT apoE4 but are degraded in the mutant apoE4 or apoE4 fragments that are observed for chymotrypsin proteolysis of mutant apoE4 but not of WT apoE4.
The L28P Mutation Affects the Interaction of ApoE4 with Lipids
ApoE4 can interact with lipids, forming lipoprotein particles as part of its normal function (19, 26). To achieve this, it has to undergo a series of conformational changes that include the unfolding of its N-terminal four-helix bundle where the L28P mutation resides. To investigate possible effects of the mutation to the ability of apoE4 to interact with lipids, we measured the rate of remodeling of DMPC vesicles after addition of lipid-free WT and mutant apoE4. Similar to results published previously, WT apoE4 remodeled DMPC vesicles following biphasic kinetics that include an initial “burst” phase and a slower major transition (28, 33, 34). Although apoE4[L28P] was also able to remodel DMCP vesicles, it did so with slower kinetics compared with the WT protein, suggesting possible defects in the ability of the protein to undergo the necessary conformational changes to bind lipids (Fig. 4).
FIGURE 4.
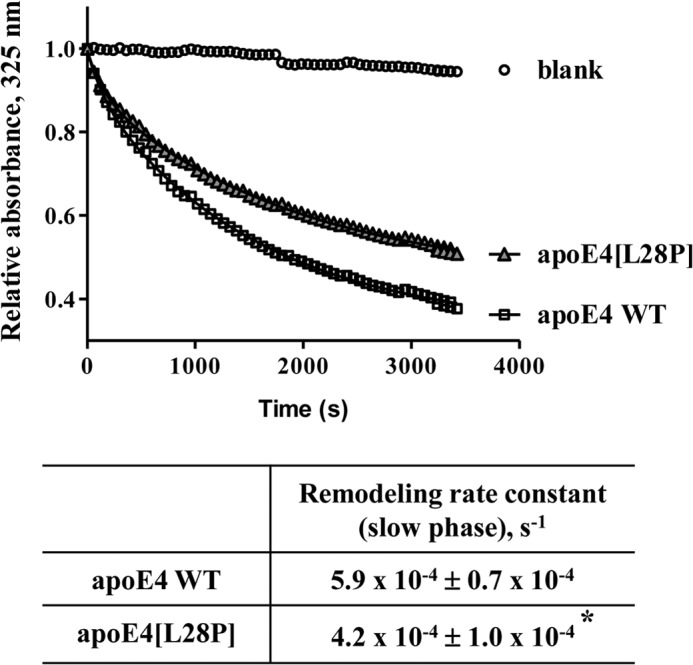
Time course of remodeling of multilamellar DMPC vesicles by WT apoE4 and apoE4[L28P]. Absorbance at 325 nm was followed for 1 h after addition of WT or mutant apoE4 to DMPC vesicles, as described under “Experimental Procedures.” Experimental data (□ or ▵) were fit to a two-phase exponential decay model (solid lines). *, p < 0.05 versus WT.
We then examined the structural integrity of lipoprotein particles made with apoE4[L28P]. We prepared reconstituted lipoprotein particles consisting of WT apoE4 or apoE4[L28P], phosphatidylcholine, and cholesterol (PC/C-apoE4) and analyzed them by electron microscopy (Fig. 5, A and B). Both WT and mutant protein were able to form lipoprotein particles with the expected discoidal geometry that appear as rouleaux because they tend to stack on edge during the negative staining procedure (28). Statistical analysis of the diameter distribution of those particles revealed that the mutant protein resulted in particles that had, on average, larger diameters (13.8 ± 0.2 nm versus 17.8 ± 0.3 nm) (Fig. 5C). This, however, was primarily due to the presence of several larger-diameter outliers (Fig. 5C, insets). Such larger-sized malformed lipoprotein particles may indicate aggregation tendencies because of structural defects brought about by the mutation. CD analysis of the lipoprotein particles revealed that the lipoprotein particles that contain the mutant apoE4 had a statistically significant but minor reduction in helical content (Fig. 6A) and also a slightly perturbed thermal denaturation profile (Fig. 6B), which may indicate some minor perturbation because of mutation. These effects are significantly less pronounced than the effects observed for the lipid-free protein and may be due to the presence of a subpopulation of aberrant particles, such as the ones seen during electron microscopy analysis.
FIGURE 5.

Characteristic electron microscopy pictures and size distribution of reconstituted discoidal lipoprotein particles consisting of WT apoE4 or apoE4[L28P]. A and B, reconstituted discoidal lipoprotein particles consisting of WT or mutant apoE4, phosphatidylcholine, and cholesterol (PC/C-apoE4) were prepared and analyzed by electron microscopy as described under “Experimental Procedures.” C, the calculation of the disc diameter distribution of PC/C-apoE4 particles was performed as described under “Experimental Procedures.” The mutant protein resulted in particles that had, on average, larger diameters (p < 0.0001 versus WT). The insets show lipoprotein particle structures for WT apoE4 or apoE4[L28P].
FIGURE 6.

Biophysical characterization of lipoprotein particles containing WT apoE4 or apoE4[L28P]. Reconstituted discoidal lipoprotein particles consisting of WT or mutant apoE4, phosphatidylcholine, and cholesterol (PC/C-apoE4) were prepared as described under “Experimental Procedures.” A, far UV CD spectra of the protein component (WT or mutant apoE4) of PC/C-apoE4 particles. Spectra are averages of three separate experiments. The percentage of helical content was calculated on the basis of the molar ellipticity at 222 nm, as described under “Experimental Procedures.” *, p < 0.05 versus WT. B, thermal denaturation profiles of WT and mutant apoE4 in lipoprotein particles. The y axis has been normalized to correspond to the fraction of the protein in the unfolded state.
Effect of Lipid-free or Lipoprotein Particle-associated WT ApoE4 and ApoE4[L28P] on SK-N-SH Cell Viability
The structural defects brought about by the introduction of the L28P mutation on apoE4 prompted us to investigate the presence of functional alterations, especially in the context of possible roles of apoE4 in the pathogenesis of AD. We first evaluated the effects of lipid-free or lipidated apoE4 on the viability of human neuroblastoma SK-N-SH cells. Cells were incubated for 24 h with two concentrations of either WT apoE4 or apoE4[L28P] in lipid-free form or in lipoprotein particle form. Cell viability was determined post-incubation using an MTT assay (Fig. 7). Both WT and mutant apoE4 in their lipid-free form displayed no apparent toxicity to the cells at the low concentration used but resulted in a similar reduced cell viability when used at the higher concentration (Fig. 7A). However, when using the lipidated forms of the proteins and at both concentrations assayed, lipidated apoE4[L28P] resulted in significantly less cell viability compared with lipidated WT apoE4 (Fig. 7B). More importantly, toxicity (28% decrease in cell viability) was evident even when lipidated apoE4[L28P] was used at the lower concentration of 0.375 μm, which is similar to the reported concentration of apoE in human cerebrospinal fluid (0.365–0.395 μm) (47, 48). No toxicity was evident for lipidated WT apoE4 at that concentration. These findings suggest that lipoproteins containing apoE4[L28P] may negatively affect neuronal cell viability.
FIGURE 7.
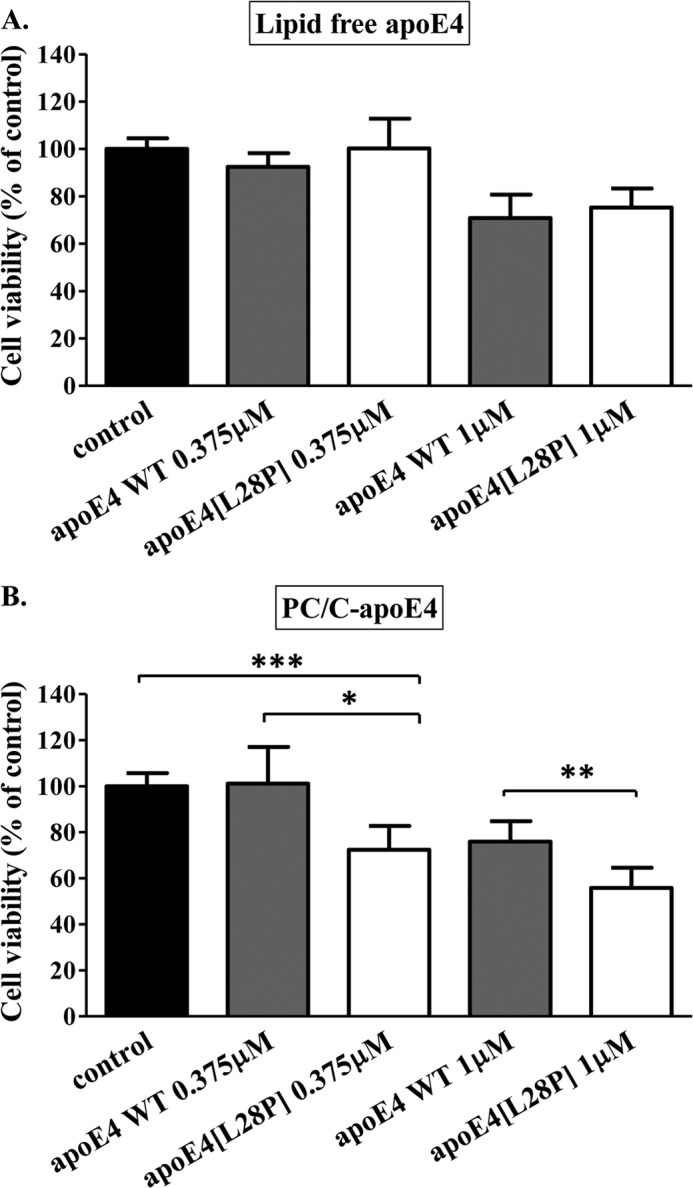
Effect of lipid free or lipoprotein particle-associated WT apoE4 and apoE4[L28P] on SK-N-SH cell viability. Shown is the survival of SK-N-SH cells incubated with lipid-free (A) or lipoprotein particle-associated (PC/C) (B) WT apoE4 or apoE4[L28P] (0.375 or 1 μm) for 24 h, as determined by a MTT assay. Cell viability is expressed as percent relative to the viability of control untreated cells (incubation without apoE4 forms) set to100%. Data are the means ± S.D. of three experiments performed in triplicate. *, p < 0.05 versus WT; **, p < 0.01 versus WT; ***, p = 0.0003 versus control.
Effect of Lipid-free ApoE4[L28P] on Cellular Uptake of Aβ42 by SK-N-SH and Primary Mouse Neurons
In a previous study, we discovered that a specific fragment of apoE4, in particular apoE4[(Δ(166–299)], can induce Aβ42 peptide uptake by SK-N-SH cells and result in persistent ROS formation inside the cells (10). Using this experimental setup, we now show that the lipid-free, full-length apoE4[L28P] variant also shares this property. SK-N-SH cells incubated with Aβ42 and physiological concentrations of apoE4[L28P] for 24 h resulted in internalization of the Aβ42 peptide (Fig. 8c). Use of the cytoskeletal F-actin marker rhodamine-phalloidin, which traces the outline of individual cells, indicated that Aβ42 accumulates primarily in the cytosol of the cells (Fig. 8, d–f). No Aβ42 uptake was evident when WT apoE4 was used or in the absence of apoE4 (Fig. 8, a and b). Furthermore, Aβ42 accumulation inside the cells was persistent even 24 h post-removal of both Aβ42 and apoE4 from the medium (Fig. 8, g–i). This finding links the functional properties of this apoE4 variant with a property reported previously (intracellular accumulation of Aβ42) of proteolytic fragments of apoE4 that may be associated with AD pathogenesis.
FIGURE 8.
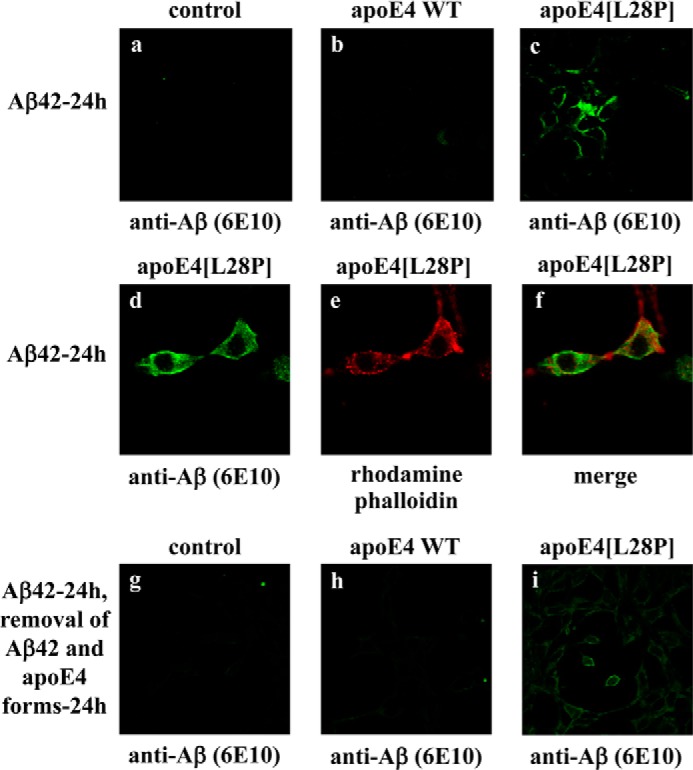
Fluorescence confocal laser-scanning microscopy of SK-N-SH cells incubated in the presence of Aβ42 and WT apoE4 or apoE4[L28P]. SK-N-SH cells were incubated with 25 ng/ml Aβ42 in the absence (control) or presence of 375 nm lipid-free WT apoE4 or apoE4[L28P] for 24 h, as indicated (a–f). SK-N-SH cells were incubated with 25 ng/ml Aβ42 in the absence (control) or presence of 375 nm lipid-free WT apoE4 or apoE4[L28P] for 24 h and then washed and incubated in fresh medium without Aβ42 and apoE4 forms for another 24 h, as indicated (g–i). Aβ immunostaining of cells was detected with the antibody 6E10, followed by an FITC-conjugated secondary antibody (a–d, g–i, green). F-actin was stained with rhodamine phalloidin (e, red). The merger of images d and e is shown in f.
Incubation of SK-N-SH cells with Aβ42 and lipoprotein particles containing the apoE4[L28P] variant did not promote the cellular uptake of Aβ42. Similarly, there was no Aβ42 internalization in the presence of lipoprotein particles containing WT apoE4 (Fig. 9).
FIGURE 9.
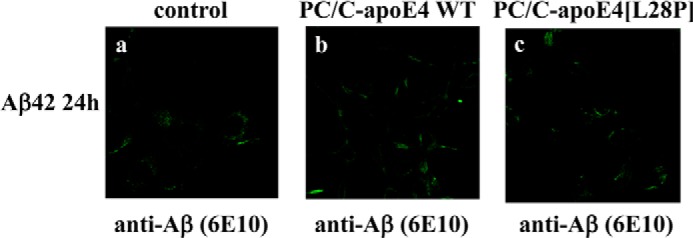
Fluorescence confocal laser-scanning microscopy of SK-N-SH cells incubated in the presence of Aβ42 and lipoprotein particle-associated WT apoE4 or apoE4[L28P]. SK-N-SH cells were incubated with 25 ng/ml Aβ42 in the absence (control) or presence of 375 nm lipoprotein particle-associated (PC/C) WT apoE4 or apoE4[L28P] for 24 h, as indicated (a–c). Aβ immunostaining of cells was detected with the antibody 6E10, followed by an FITC-conjugated secondary antibody.
To test whether the functional properties of the lipid-free apoE4[L28P] variant extend to a more physiologically relevant cellular system, we incubated primary mouse cortical neurons with Aβ42 and either WT apoE4 or apoE4[L28P] (Fig. 10). Similar to the SK-N-SH cells, the apoE4[L28P] variant, but not WT apoE4, induced the intracellular accumulation of Aβ42 peptide (Fig. 10, a–c). Use of the actin marker rhodamine-phalloidin indicated some cytosolic localization of Aβ42, with the majority of the Aβ42 to localize on the neurites, as described previously for primary mouse cortical neurons incubated with Aβ42 (38) (Fig. 10, d–f).
FIGURE 10.
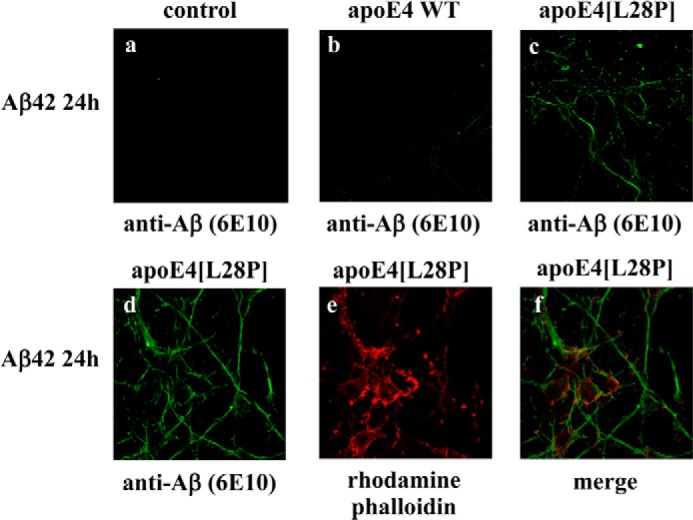
Fluorescence confocal laser-scanning microscopy of primary mouse cortical neurons incubated in the presence of Aβ42 and WT apoE4 or apoE4[L28P]. Primary mouse cortical neurons were incubated with 25 ng/ml Aβ42 in the absence (control) or presence of 375 nm lipid-free WT apoE4 or apoE4[L28P] for 24 h, as indicated. Aβ immunostaining of cells was detected with the antibody 6E10, followed by an FITC-conjugated secondary antibody (a–d, green). F-actin was stained with rhodamine phalloidin (e, red). The merger of images d and e is shown in f.
Effect of Lipid-free or Lipidated ApoE4[L28P] on ROS Formation by SK-N-SH in the Presence or Absence of Aβ42
To evaluate the effect of apoE4[L28P]-induced Aβ42 peptide uptake by SK-N-SH cells, we measured intracellular formation of ROS, which are markers of oxidative stress, using a specialized fluorescent probe as described under “Experimental Procedures.” Incubation of SK-N-SH cells with Aβ42 and physiological concentrations of apoE4[L28P] for 24 h resulted in a significant increase of intracellular ROS, whereas WT apoE4 had no such effect (Fig. 11A). This phenomenon was persistent for at least 24 h after removal of the medium that contained Aβ42 and apoE4[L28P] (Fig. 11B). The increase in ROS formation was completely dependent on the uptake of Aβ42 by cells because there was no enhanced ROS formation in SK-N-SH cells incubated with lipid-free WT apoE4 or apoE4[L28P] in the absence of Aβ42 (Fig. 11C).
FIGURE 11.

Effect of lipid free or lipoprotein particle-associated WT apoE4 and apoE4[L28P] in the presence or absence of Aβ42 on ROS formation by SK-N-SH cells. A, SK-N-SH cells were incubated with 25 ng/ml Aβ42 in the absence (control) or presence of 375 nm lipid-free WT apoE4 or apoE4[L28P] for 24 h. B, SK-N-SH cells were incubated with 25 ng/ml Aβ42 in the absence (control) or presence of 375 nm lipid-free WT apoE4 or apoE4[L28P] for 24 h and then washed and incubated in fresh medium without Aβ42 and apoE4 forms for another 24 h. C, SK-N-SH cells were incubated in the absence (control) or presence of 375 nm lipid-free WT apoE4 or apoE4[L28P] for 24 h (without Aβ42). D, SK-N-SH cells were incubated in the absence (control) or presence of 375 nm lipoprotein particle-associated (PC/C) WT apoE4 or apoE4[L28P] for 24 h (without Aβ42). At the end of each incubation period, the cells were incubated with 2′,7′-dichlorofluorescein diacetate for 45 min. The formation of ROS was measured by detection of fluorescent DCF emitted from cells using a fluorescence microscope, as described under “Experimental Procedures.” The DCF fluorescence of cells incubated with lipid-free or lipoprotein particle-associated apoE4 forms in the presence or absence of Aβ42 is shown relative to DCF fluorescence of control cells set as 100%. DCF fluorescence intensity was measured for at least 40 cells from the fluorescent images of each sample, as described under “Experimental Procedures,” and the relative fluorescent intensity was taken as the average of the values of at least five images for each experiment. Values are the means ± S.D. (n = 20) of four experiments. **, p = 0.053 versus control; ***, p < 0.0001 versus control; ***1, p = 0.0001 versus WT.
Furthermore, to examine whether the effect of lipidated apoE4[L28P] on SK-N-SH viability is induced by increased cellular oxidative stress, we measured the formation of ROS. Our analysis showed that lipoprotein particle-associated apoE4[L28P] was capable to induce ROS formation inside SK-N-SH cells to a much higher degree than WT apoE4 (Fig. 11D). Overall, our findings suggest that the apoE4[L28P] variant can induce an increased ROS burden inside SK-N-SH cells in lipid-free form after induction of cellular Aβ42 uptake or in lipidated form independently of Aβ42.
DISCUSSION
Although the association of the polymorphic variation of apoE with the risk of LOAD is very well established, the mechanism by which a particular apoE isoform, apoE4, can promote LOAD pathogenesis is not clear. A rare hereditary mutation in apoE4, L28P, has been associated with an increased risk for LOAD in patients (6, 7).
LOAD is a complex disease, and the most recent genetic studies of LOAD have focused on the identification of common variants associated with a risk for LOAD through genome-wide association studies. These studies have identified several common variants of several new genes to be associated with LOAD, but, with the exception of variants of triggering receptor expressed on myeloid cells 2, no rare variants were identified (49, 50). Rare variants, like apoE4[L28P], although equally important as common variants for disease pathogenesis, are seldom found by initial genome-wide association studies, primarily because of the low representation in the cohort, and require resequencing of large populations or targeted family-based studies and linkage analysis to be identified (51, 52). A characteristic example involves a mutation screening study that showed that rare variants in amyloid β precursor protein, presenilin 1, and presenilin 2 increased the risk for AD in LOAD families, suggesting that rare variants in genes associated with AD pathogenesis could explain a significant proportion of genetic heritability of AD, which is not detected by genome-wide association studies (53). Furthermore, functional analyses of rare variants, like the one presented in this study, can provide additional validation of their association with LOAD, even in the absence of their identification from genome-wide association studies.
In this study, we pursued the structural and functional characterization of apoE4[L28P], aiming both to understand its possible role in the pathogenesis of AD but to also gain insights into the role of WT apoE4 in the development of this disease. The study of disease-associated rare mutations can be a powerful tool for understanding the involvement of particular gene products on disease pathogenesis because the mutation may often exacerbate pre-existing functional roles in the pathophysiological process of disease progression. This may be very relevant in the case of apoE because the differences between the three common apoE isoforms have been hypothesized extensively to be structural, and these exact differences in structural properties are presumed to underlie changes in function that affect disease predisposition.
The L28P apoE4 mutation is characterized by the fact that it introduces a proline residue into a highly helical segment of the protein. Although proline residues can occasionally be found in helical segments of proteins, the structural rigidity of this amino acid generally makes it less than favorable for α helices. According to this rationale, this substitution would be expected to be tolerated poorly in the local region of the protein. Our findings, however, suggest that the structural perturbations because of the mutation are not only restricted to the local area but are, instead, transmitted to the folding integrity of the whole N-terminal domain of apoE4, suggesting that the mutation interferes with key folding pathways of the protein. These structural perturbations appear to lead to important functional changes. Although the mutant protein can still interact with lipids, both the kinetics and final products are altered. More importantly this single-amino acid mutation gives apoE4 the ability to promote the intracellular accumulation of Aβ42, a function that has also been found previously in a heavily truncated apoE4 variant, apoE4[(Δ(166–299)] (10), which has a similar molecular weight as carboxyl-terminal truncated apoE4 fragments found in the brain of AD patients (54, 55). We have proposed previously an association between two molecular events that are considered to be early events in the pathogenesis of AD, namely the proteolysis of apoE4 and the intraneuronal accumulation of Aβ42 that leads to persistent ROS formation and, therefore, increased oxidative stress, which is also an early event in AD (10, 12, 13, 56). Interestingly, both the hereditary mutant apoE4[L28P] and apoE4[(Δ(166–299)] have significant structural perturbations that could affect function, although the one variant only has a single amino acid substitution, whereas the other is missing the entire C-terminal domain. The structural perturbations of these two variants are not, however, identical. ApoE4[(Δ(166–299)] is largely thermodynamically stabilized (25), whereas apoE4[L28P] is destabilized. This apparent paradox, that two opposite thermodynamic stability changes are associated with the same gain of function, can be reconciled in the context that apoE4 is a thermodynamically metastable and structural plastic protein that relies, for its function, on its ability to change conformational states, an ability that necessitates an optimized thermodynamic stability. Furthermore, both variants display one common characteristic, namely the loss of a key unfolding intermediate during chemical denaturation (25). This intermediate has been proposed to correspond to the unfolding of the N-terminal domain where the L28P mutation lies, a conformational change that is key to apoE4 interaction with hydrophobic surfaces and membranes.
We show here that the lipid-free apoE4[L28P] induces the intracellular accumulation of Aβ42 in SK-N-SH human neuroblastoma cells and mouse primary neurons as well as ROS formation, whereas the lipoprotein-associated apoE4[L28P] increases ROS formation and reduces the viability of SK-N-SH cells. Studies with humans and experimental animals have proposed that Aβ accumulates inside neurons. Accumulation of Aβ, the majority of which is Aβ42, occurs prior to extracellular amyloid formation, has been implicated in the onset of early cognitive alterations, and may contribute to the pathological cascade of events that lead to neuronal dysfunction and, eventually, to AD (12, 13). Oxidative stress has also been proposed to be one of the earliest events in AD that plays important roles in the onset and progression of the disease. This stress has been suggested to be chronic in neurons (56). Aβ has been shown to be one of the factors that contribute to oxidative stress in neurons (57, 58). It has been proposed that ROS, which are considered markers of oxidative stress, are generated in the cytoplasm of neurons and can cause damage to key cellular components, including DNA, proteins, and lipids, leading to neuronal dysfunction and death (56, 59, 60). Therefore, it is possible that apoE4[L28P], regardless of its lipidation status, affects processes that lead to chronic oxidative stress and, in that manner, contributes to the pathogenesis of AD.
Although the genetic association of apoE4 and, more specifically, apoE4[L28P] with LOAD has been established, previous attempts to model this effect in experimental animals have yielded conflicting results. In a previous study, Huber et al. (61) generated transgenic mice on a background of endogenous mouse apoE for apoE4[L28P] and WT apoE4 and analyzed these mice for the presence of an AD phenotype. These apoE4 transgenes were controlled by the glial fibrillary acidic protein promoter that led to major transgene expression in glial cells and to limited apoE production in neurons. Neither transgenic mouse differed from non-transgenic mice in cognitive tests, nor did they show any change in immunohistological and biochemical brain protein analyses, a finding that is in conflict with the established association of apoE4 and AD in humans (61). In contrast, in another study, transgenic WT apoE4 mice lacking endogenous mouse apoE and expressing human apoE transgenes in neurons, under the control of the neuron-specific enolase promoter, showed impairments in learning a water maze task and in vertical exploratory behavior that increased with age compared with neuron-specific enolase-apoE3 transgenic mice and control non-transgenic mice (62). In addition, neuron-specific enolase-apoE4 mice and, to a significantly lesser extent, neuron-specific enolase-apoE3 mice, but not glial fibrillary acidic protein-apoE3 or glial fibrillary acidic protein-apoE4 mice, are characterized by the accumulation of C-terminal-truncated fragments of apoE4 and phosphorylated tau in an age-dependent manner (15). Therefore, the number of important differences among the study of Huber et al. (61) and other studies of apoE4 transgenic mice, including the presence or absence of endogenous mouse apoE, which could compensate or mask certain biological effects as well as the different cellular expression sites for the apoE transgenes (glia versus neurons), seem to be critical for the development of an AD phenotype in mice. Future studies in mice expressing the apoE4[L28P] transgene in neurons and in the absence of endogenous mouse apoE may be necessary for elucidating the role of apoE4 and apoE4[L28P] in AD pathogenesis.
Our findings suggest a molecular basis that may underlie the genetic association of a naturally occurring mutation in apoE4 with the development of LOAD. The mutation induces serious folding defects on the protein, leading to a functional phenotype demonstrated previously with a heavily truncated apoE4 proteolytic fragment similar to apoE4 fragments proposed to appear early in the pathogenetic process of AD (15). The common functional properties of apoE4[L28P] and the C-terminal truncated apoE4 further support the idea that Aβ internalization by neuronal cells is an early event in AD pathogenesis that is exacerbated in the case of the apoE4[L28P] variant. The structural integrity of apoE4 is almost certainly an important component of its role in the pathogenesis of LOAD, and analysis of structurally defective apoE4 variants may hold clues regarding the pathogenic mechanisms behind this disease.
Acknowledgments
We thank Drs. Antonia Vlahou and Manousos Makridakis for performing the MALDI-TOF analysis.
This work was supported by the ARISTEIA II program (project code 4839), which is cofunded by the European Social Fund, European Union and Greek National Resources, under the Operational Program “Education and Lifelong Learning.”
- ApoE
- apolipoprotein E
- AD
- Alzheimer disease
- LOAD
- late-onset Alzheimer disease
- Aβ
- amyloid β peptide
- ROS
- reactive oxygen species
- Ni-NTA
- nickel-nitrilotriacetic acid
- Trx
- thioredoxin
- GndHCl
- guanidine hydrochloride
- DPBS
- Dulbecco's phosphate-buffered saline
- ANS
- 1-anilinonaphthalene-8-sulfonic acid
- DMPC
- l-α-dimyristoylphosphatidylcholine
- MEM
- minimal essential medium
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- DCF
- 2′,7′-dichlorofluorescein
- PC/C
- phosphatidylcholine and cholesterol.
REFERENCES
- 1. Zannis V. I., Kypreos K. E., Chroni A., Kardassis D., Zanni E. E. (2004) in Molecular Mechanisms of Atherosclerosis (Loscalzo J., ed) pp. 111–174, Taylor & Francis, Abington, UK [Google Scholar]
- 2. Mahley R. W., Weisgraber K. H., Huang Y. (2006) Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 103, 5644–5651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zannis V. I., Cole F. S., Jackson C. L., Kurnit D. M., Karathanasis S. K. (1985) Distribution of apolipoprotein A-I, C-II, C-III, and E mRNA in fetal human tissues: time-dependent induction of apolipoprotein E mRNA by cultures of human monocyte-macrophages. Biochemistry 24, 4450–4455 [DOI] [PubMed] [Google Scholar]
- 4. Corder E. H., Saunders A. M., Strittmatter W. J., Schmechel D. E., Gaskell P. C., Small G. W., Roses A. D., Haines J. L., Pericak-Vance M. A. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261, 921–923 [DOI] [PubMed] [Google Scholar]
- 5. Myers R. H., Schaefer E. J., Wilson P. W., D'Agostino R., Ordovas J. M., Espino A., Au R., White R. F., Knoefel J. E., Cobb J. L., McNulty K. A., Beiser A., Wolf P. A. (1996) Apolipoprotein E ϵ4 association with dementia in a population-based study: The Framingham study. Neurology 46, 673–677 [DOI] [PubMed] [Google Scholar]
- 6. Kamboh M. I., Aston C. E., Perez-Tur J., Kokmen E., Ferrell R. E., Hardy J., DeKosky S. T. (1999) A novel mutation in the apolipoprotein E gene (APOE*4 Pittsburgh) is associated with the risk of late-onset Alzheimer's disease. Neurosci. Lett. 263, 129–132 [DOI] [PubMed] [Google Scholar]
- 7. Scacchi R., Gambina G., Ferrari G., Corbo R. M. (2003) Screening of two mutations at exon 3 of the apolipoprotein E gene (sites 28 and 42) in a sample of patients with sporadic late-onset Alzheimer's disease. Neurobiol. Aging 24, 339–343 [DOI] [PubMed] [Google Scholar]
- 8. Cedazo-Mínguez A. (2007) Apolipoprotein E and Alzheimer's disease: molecular mechanisms and therapeutic opportunities. J. Cell Mol. Med. 11, 1227–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim J., Basak J. M., Holtzman D. M. (2009) The role of apolipoprotein E in Alzheimer's disease. Neuron 63, 287–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dafnis I., Stratikos E., Tzinia A., Tsilibary E. C., Zannis V. I., Chroni A. (2010) An apolipoprotein E4 fragment can promote intracellular accumulation of amyloid peptide β 42. J. Neurochem. 115, 873–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao W., Dumanis S. B., Tamboli I. Y., Rodriguez G. A., Jo L. M., Moussa C. E., William R. G. (2013) Human APOE genotype affects intraneuronal Aβ1–42 accumulation in a lentiviral gene transfer model. Hum. Mol. Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. LaFerla F. M., Green K. N., Oddo S. (2007) Intracellular amyloid-β in Alzheimer's disease. Nat. Rev. Neurosci. 8, 499–509 [DOI] [PubMed] [Google Scholar]
- 13. Bayer T. A., Wirths O. (2008) Review on the APP/PS1KI mouse model: intraneuronal Aβ accumulation triggers axonopathy, neuron loss and working memory impairment. Genes Brain Behav. 7, 6–11 [DOI] [PubMed] [Google Scholar]
- 14. Harris F. M., Brecht W. J., Xu Q., Tesseur I., Kekonius L., Wyss-Coray T., Fish J. D., Masliah E., Hopkins P. C., Scearce-Levie K., Weisgraber K. H., Mucke L., Mahley R. W., Huang Y. (2003) Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer's disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 100, 10966–10971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brecht W. J., Harris F. M., Chang S., Tesseur I., Yu G. Q., Xu Q., Dee Fish J., Wyss-Coray T., Buttini M., Mucke L., Mahley R. W., Huang Y. (2004) Neuron-specific apolipoprotein e4 proteolysis is associated with increased Tau phosphorylation in brains of transgenic mice. J. Neurosci. 24, 2527–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chang S., ran Ma T., Miranda R. D., Balestra M. E., Mahley R. W., Huang Y. (2005) Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 102, 18694–18699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dafnis I., Tzinia A. K., Tsilibary E. C., Zannis V. I., Chroni A. (2012) An apolipoprotein E4 fragment affects matrix metalloproteinase 9, tissue inhibitor of metalloproteinase 1 and cytokine levels in brain cell lines. Neuroscience 210, 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Morrow J. A., Hatters D. M., Lu B., Hochtl P., Oberg K. A., Rupp B., Weisgraber K. H. (2002) Apolipoprotein E4 forms a molten globule: a potential basis for its association with disease. J. Biol. Chem. 277, 50380–50385 [DOI] [PubMed] [Google Scholar]
- 19. Hatters D. M., Peters-Libeu C. A., Weisgraber K. H. (2006) Apolipoprotein E structure: insights into function. Trends Biochem. Sci. 31, 445–454 [DOI] [PubMed] [Google Scholar]
- 20. Chen J., Li Q., Wang J. (2011) Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc. Natl. Acad. Sci. U.S.A. 108, 14813–14818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wetterau J. R., Aggerbeck L. P., Rall S. C., Jr., Weisgraber K. H. (1988) Human apolipoprotein E3 in aqueous solution: I: evidence for two structural domains. J. Biol. Chem. 263, 6240–6248 [PubMed] [Google Scholar]
- 22. Aggerbeck L. P., Wetterau J. R., Weisgraber K. H., Wu C. S., Lindgren F. T. (1988) Human apolipoprotein E3 in aqueous solution: II: properties of the amino- and carboxyl-terminal domains. J. Biol. Chem. 263, 6249–6258 [PubMed] [Google Scholar]
- 23. Wilson C., Wardell M. R., Weisgraber K. H., Mahley R. W., Agard D. A. (1991) Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science 252, 1817–1822 [DOI] [PubMed] [Google Scholar]
- 24. Mahley R. W., Weisgraber K. H., Huang Y. (2009) Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer's disease to AIDS. J. Lipid Res. 50, S183–S188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chroni A., Pyrpassopoulos S., Thanassoulas A., Nounesis G., Zannis V. I., Stratikos E. (2008) Biophysical analysis of progressive C-terminal truncations of human apolipoprotein E4: insights into secondary structure and unfolding properties. Biochemistry 47, 9071–9080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saito H., Dhanasekaran P., Baldwin F., Weisgraber K. H., Lund-Katz S., Phillips M. C. (2001) Lipid binding-induced conformational change in human apolipoprotein E: evidence for two lipid-bound states on spherical particles. J. Biol. Chem. 276, 40949–40954 [DOI] [PubMed] [Google Scholar]
- 27. Georgiadou D., Chroni A., Vezeridis A., Zannis V. I., Stratikos E. (2011) Biophysical analysis of apolipoprotein E3 variants linked with development of type III hyperlipoproteinemia. PLoS ONE. 6, e27037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Georgiadou D., Stamatakis K., Efthimiadou E. K., Kordas G., Gantz D., Chroni A., Stratikos E. (2013) Thermodynamic and structural destabilization of apoE3 by hereditary mutations associated with the development of lipoprotein glomerulopathy. J. Lipid Res. 54, 164–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Argyri L., Skamnaki V., Stratikos E., Chroni A. (2011) A simple approach for human recombinant apolipoprotein E4 expression and purification. Protein Expr. Purif. 79, 251–257 [DOI] [PubMed] [Google Scholar]
- 30. Greenfield N., Fasman G. D. (1969) Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry 8, 4108–4116 [DOI] [PubMed] [Google Scholar]
- 31. Gorshkova I. N., Liadaki K., Gursky O., Atkinson D., Zannis V. I. (2000) Probing the lipid-free structure and stability of apolipoprotein A-I by mutation. Biochemistry 39, 15910–15919 [DOI] [PubMed] [Google Scholar]
- 32. Clément-Collin V., Barbier A., Dergunov A. D., Visvikis A., Siest G., Desmadril M., Takahashi M., Aggerbeck L. P. (2006) The structure of human apolipoprotein E2, E3 and E4 in solution. 2. Multidomain organization correlates with the stability of apoE structure. Biophys. Chem. 119, 170–185 [DOI] [PubMed] [Google Scholar]
- 33. Segall M. L., Dhanasekaran P., Baldwin F., Anantharamaiah G. M., Weisgraber K. H., Phillips M. C., Lund-Katz S. (2002) Influence of apoE domain structure and polymorphism on the kinetics of phospholipid vesicle solubilization. J. Lipid Res. 43, 1688–1700 [DOI] [PubMed] [Google Scholar]
- 34. Chou C. Y., Jen W. P., Hsieh Y. H., Shiao M. S., Chang G. G. (2006) Structural and functional variations in human apolipoprotein E3 and E4. J. Biol. Chem. 281, 13333–13344 [DOI] [PubMed] [Google Scholar]
- 35. Harris J. R., Horne R. W. (1991) in Electron Microscopy in Biology (Harris J. R., ed) pp. 203–238, IRL Press, Oxford, UK [Google Scholar]
- 36. Dubochet J., Groom M., Mueller-Neuteboom S. (1982) The mounting of macromolecules for electron microscopy with particular reference to surface phenomena and the treatment of support films by glow discharge. Adv. Opt. Electron Microsc. 8, 107–135 [Google Scholar]
- 37. Lesuisse C., Martin L. J. (2002) Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J. Neurobiol. 51, 9–23 [DOI] [PubMed] [Google Scholar]
- 38. Li J., Kanekiyo T., Shinohara M., Zhang Y., LaDu M. J., Xu H., Bu G. (2012) Differential regulation of amyloid-β endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J. Biol. Chem. 287, 44593–44601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Berridge M. V., Tan A. S. (1993) Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys. 303, 474–482 [DOI] [PubMed] [Google Scholar]
- 40. Bae Y. S., Kang S. W., Seo M. S., Baines I. C., Tekle E., Chock P. B., Rhee S. G. (1997) Epidermal growth factor (EGF)-induced generation of hydrogen peroxide: role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem. 272, 217–221 [PubMed] [Google Scholar]
- 41. Abramoff M. D., Magelhaes P. J., Ram S. J. (2004) Image processing with ImageJ. Biophotonics International 11, 36–42 [Google Scholar]
- 42. Chou C. Y., Lin Y. L., Huang Y. C., Sheu S. Y., Lin T. H., Tsay H. J., Chang G. G., Shiao M. S. (2005) Structural variation in human apolipoprotein E3 and E4: secondary structure, tertiary structure, and size distribution. Biophys. J. 88, 455–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tanaka M., Vedhachalam C., Sakamoto T., Dhanasekaran P., Phillips M. C., Lund-Katz S., Saito H. (2006) Effect of carboxyl-terminal truncation on structure and lipid interaction of human apolipoprotein E4. Biochemistry 45, 4240–4247 [DOI] [PubMed] [Google Scholar]
- 44. Gorshkova I. N., Kypreos K. E., Gantz D. L., Zannis V. I., Atkinson D. (2008) Biophysical properties of apolipoprotein E4 variants: implications in molecular mechanisms of correction of hypertriglyceridemia. Biochemistry 47, 12644–12654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sakamoto T., Tanaka M., Vedhachalam C., Nickel M., Nguyen D., Dhanasekaran P., Phillips M. C., Lund-Katz S., Saito H. (2008) Contributions of the carboxyl-terminal helical segment to the self-association and lipoprotein preferences of human apolipoprotein E3 and E4 isoforms. Biochemistry 47, 2968–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Morrow J. A., Segall M. L., Lund-Katz S., Phillips M. C., Knapp M., Rupp B., Weisgraber K. H. (2000) Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry 39, 11657–11666 [DOI] [PubMed] [Google Scholar]
- 47. Kay A. D., Petzold A., Kerr M., Keir G., Thompson E. J., Nicoll J. A. (2003) Cerebrospinal fluid apolipoprotein E concentration decreases after traumatic brain injury. J. Neurotrauma 20, 243–250 [DOI] [PubMed] [Google Scholar]
- 48. Wang L., Han Y., Chen D., Xiao Z., Xi Z., Xiao F., Wang X. (2010) Cerebrospinal fluid apolipoprotein E concentration decreases after seizure. Seizure 19, 79–83 [DOI] [PubMed] [Google Scholar]
- 49. European Alzheimer's Disease Initiative (EADI), Genetic and Environmental Risk in Alzheimer's Disease, Alzheimer's Disease Genetic Consortium, and Cohorts for Heart and Aging Research in Genomic Epidemiology. (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat. Genet. 45, 1452–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Guerreiro R., Wojtas A., Bras J., Carrasquillo M., Rogaeva E., Majounie E., Cruchaga C., Sassi C., Kauwe J. S., Younkin S., Hazrati L., Collinge J., Pocock J., Lashley T., Williams J., Lambert J. C., Amouyel P., Goate A., Rademakers R., Morgan K., Powell J., St George-Hyslop P., Singleton A., Hardy J. (2013) TREM2 variants in Alzheimer's disease. N. Engl. J. Med. 368, 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bush W. S., Moore J. H. (2012) Chapter 11: Genome-wide association studies. PLoS Comput. Biol. 8, e1002822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gibson G. (2011) Rare and common variants: twenty arguments. Nat. Rev. Genet. 13, 135–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cruchaga C., Haller G., Chakraverty S., Mayo K., Vallania F. L., Mitra R. D., Faber K., Williamson J., Bird T., Diaz-Arrastia R., Foroud T. M., Boeve B. F., Graff-Radford N. R., St Jean P., Lawson M., Ehm M. G., Mayeux R., Goate A. M. (2012) Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer's disease families. PLoS ONE 7, e31039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Huang Y., Liu X. Q., Wyss-Coray T., Brecht W. J., Sanan D. A., Mahley R. W. (2001) Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc. Natl. Acad. Sci. U.S.A. 98, 8838–8843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jones P. B., Adams K. W., Rozkalne A., Spires-Jones T. L., Hshieh T. T., Hashimoto T., von Armin C. A., Mielke M., Bacskai B. J., Hyman B. T. (2011) Apolipoprotein E: isoform specific differences in tertiary structure and interaction with amyloid-β in human Alzheimer brain. PLoS ONE. 6, e14586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nunomura A., Castellani R. J., Zhu X., Moreira P. I., Perry G., Smith M. A. (2006) Involvement of oxidative stress in Alzheimer disease. J. Neuropathol. Exp. Neurol. 65, 631–641 [DOI] [PubMed] [Google Scholar]
- 57. Smith D. G., Cappai R., Barnham K. J. (2007) The redox chemistry of the Alzheimer's disease amyloid β peptide. Biochim. Biophys. Acta 1768, 1976–1990 [DOI] [PubMed] [Google Scholar]
- 58. Butterfield D. A., Swomley A. M., Sultana R. (2013) Amyloid β-peptide (1–42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid. Redox Signal. 19, 823–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dasuri K., Zhang L., Keller J. N. (2013) Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free. Radic. Biol. Med. 62, 170–185 [DOI] [PubMed] [Google Scholar]
- 60. Yan M. H., Wang X., Zhu X. (2013) Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free. Radic. Biol. Med. 62, 90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Huber G., März W., Martin J. R., Malherbe P., Richards J. G., Sueoka N., Ohm T., Hoffmann M. M. (2000) Characterization of transgenic mice expressing apolipoprotein E4(C112R) and apolipoprotein E4(L28P; C112R). Neuroscience. 101, 211–218 [DOI] [PubMed] [Google Scholar]
- 62. Raber J., Wong D., Buttini M., Orth M., Bellosta S., Pitas R. E., Mahley R. W., Mucke L. (1998) Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc. Natl. Acad. Sci. U.S.A. 95, 10914–10919 [DOI] [PMC free article] [PubMed] [Google Scholar]