

Abstract
Mitochondria become targets for autophagic degradation after nutrient deprivation, a process also termed mitophagy. In this study, we used LysoTracker Red (LTR) and MitoTracker Green to characterize the kinetics of autophagosomal proliferation and mitophagy in cultured rat hepatocytes. Autophagy induced by nutrient deprivation plus glucagon increased LTR uptake assessed with a fluorescence plate reader and the number of LTR-labeled acidic organelles assessed with confocal microscopy in individual hepatocytes both by 4- to 6-fold. Serial imaging of hepatocytes coloaded with MitoTracker Green (MTG) revealed an average mitochondrial digestion time of 7.5 min after autophagic induction. In the presence of protease inhibitors, digestion time more than doubled, and the total number of LTR-labeled organelles increased about 40%, but the proportion of the LTR-labeled acidic organelles containing MTG fluorescence remained constant at about 75%. Autophagy inhibitors, 3-methyladenine, wortmannin and LY204002, suppressed the increase of LTR uptake after nutrient deprivation by up to 85%, confirming that increased LTR uptake reflected autophagy induction. Cyclosporin A and NIM811, specific inhibitors of the mitochondrial permeability transition (MPT), also decreased LTR uptake, whereas tacrolimus, an immunosuppressive reagent that does not inhibit the MPT, was without effect. In addition, the c-Jun N-terminal kinase (JNK) inhibitors, SCP25041 and SP600125, blocked LTR uptake by 47% and 61%, respectively, but ERK1, p38 and caspase inhibitors had no effect. The results show that mitochondria once selected for mitophagy are rapidly digested and support the concept that mitochondrial autophagy involves the MPT and signaling through PI3 kinase and possibly JNK.
Keywords: autophagy, fluorescence multiwell plate reader, Lyso Tracker Red, Mito Tracker Green, mitochondrial permeability transition, mitophagy
INTRODUCTION
Autophagy is a process that digests injured, worn out and surplus organelles, such as mitochondria, and recovers their amino acids and other nutrients for other uses. Fasting is a potent inducer of autophagy in the liver, an effect mediated in part by glucagon, a hormone also stimulating hepatic glycogen breakdown.1,2 During nutritional stress, autophagy is the major mechanism for protein degradation.3,4 By contrast after nutrient replenishment, insulin inhibits autophagy and promotes hepatic glycogen formation.5 Autophagy has two important functions in cells: (1) removal of excess and damaged organelles and (2) liberation of free amino acids and other nutrients at times of nutrient deprivation.6 When autophagy is impaired, as in Atg7-deficient mice, deformed mitochondria accumulate in the liver.7
As an organelle occupying 20% of the cytoplasmic volume of hepatocytes,8 mitochondria are a major target of autophagy. Even in well nourished young animals, mitochondria turn over every 15 to 25 days by autophagy.9,10 Mitophagy may also help clear mitochondria with mutations of mitochondrial DNA (mtDNA).11–13 With aging, mitochondrial degradation through autophagy appears to become compromised to promote accumulation of mtDNA mutations and damaged mitochondria. Lysosomes progressively accumulate lipofuscin to further impair mitochondrial degradation.12 Because mitochondria appear to be selectively targeted for autophagy in some settings,14,15 the term mitophagy has been introduced for the specific process of mitochondrial autophagy.13
Although mechanisms targeting mitochondria for mitophagy remain poorly understood, a possible component of the targeting process is a signal from the mitochondria that will undergo autophagy. Previously, we proposed that the mitochondrial permeability transition (MPT) represents one such signal for mitophagy.14 The MPT results from opening of high conductance permeability transition (PT) pores in the mitochondrial inner membrane. Swelling after the MPT causes rupture of the outer membrane, which releases cytochrome c and other pro-apoptotic factors into the cytosol. Cyclosporin A (CsA) is an immunosuppressive undecapeptide that blocks the MPT and prevents MPT-dependent necrotic and apoptotic cell killing to hepatocytes and other cell types.16–19 Previously using a confocal fluorescence resonance energy transfer (FRET) technique to identify depolarizing mitochondria, CsA was shown to block mitochondrial depolarization after autophagic stimulation and the autophagosomal proliferation that followed. These observations supported the conclusion that the MPT initiates mitochondrial depolarization in mitophagy and promotes sequestration of depolarized mitochondria into autophagosomes.14
Methods to assess autophagy and mitophagy rely on techniques such as quantitative electron microscopy and release of radioactivity after labeling cellular proteins with radioisotopes.10,20,21 More recently, markers of acidic organelles like monodansylcadaverine or LysoTracker Red (LTR) have been used to study autophagy by fluorescence microscopy.14,22 A drawback of microscopy is that relatively few cells can be studied at a time and the inability to perform high throughput screening. Here, we evaluated LTR and MitoTracker Green (MTG) as probes of mitochondrial autophagy using correlative total LTR fluorescence measurements and confocal microscopy. Our results show that total LTR uptake increases as the lysosomal/autophagosomal compartment expands after autophagic stimulation. This autophagy predominantly involves mitochondria, which undergo protease-dependent autophagic digestion within 10 min or less. 3-Methyladenine (3-MA), blockade of the MPT and inhibition of phosphatidylinositol-3 kinase (PI3K), which suppress autophagy, inhibited cellular LTR uptake. Inhibitors of c-Jun N-terminal kinase (JNK), but not inhibitors of other stress kinases or caspases, also block autophagy assessed by LTR uptake.
MATERIALS AND METHODS
Materials
LysoTracker Red and MitoTracker Green were obtained from Molecular Probes (Eugene, OR). CsA was obtained from Sigma Chemical (St. Louis, MO). SCP25041 was a gift of Celgene, Signal Research Division (San Diego, CA). SP600125 was obtained from A.G. Scientific (San Diego, CA). Wortmannin, LY294002, PD98059, SB203580, Z-VAD-fmk, DEVD-fmk, IETD-fmk, and LEHD-cho were purchased from Calbiochem-Novabiochem (La Jolla, CA). NIM-811 was the kind gift of Novartis (Basel, Switzerland). Tacrolimus was obtained from Fujisawa Healthcare (Deerfield, IL). All other reagents were of analytical grade from commercial sources.
Hepatocyte isolation and culture
Primary rat hepatocytes were isolated from overnight fasted male Sprague-Dawley rats (200–250 g) by collagenase perfusion, as described previously.23 Cell viability routinely exceeded 90%, as assessed by trypan blue exclusion. Hepatocytes were plated on Type 1 collagen-coated 48-well microtiter plates (Falcon, Lincoln Park, NJ) at a density of 75,000 cells per well and cultured overnight in Waymouth's MB-742/1 growth medium containing 27 mM NaHCO3, 2 mM L-glutamine, 10% fetal calf serum, 100 nM insulin and 10 nM dexamethasone, pH 7.4 at 37°C in 5% CO2/air. To induce autophagy, hepatocyte cultures were switched from serum-containing complete growth medium to serum-free Krebs-Ringer-HEPES buffer (KRH, in mM: 25 HEPES, 115 NaCl, 5 KCl, 1 KH2PO4, 1.2 MgSO4, and 2 CaCl2, pH 7.4 at 37°C in air) containing 1 μM glucagon. In some experiments, 3-MA (10 mM), CsA (5 μM), NIM811 (5 μM), tacrolimus (5 μM), wortmannin (0.5 μM), 2-(4-morpholinyl)-8-phenylchromone (LY-294003, 10 μM), PD98059 (100 μM), SB203580 (100 μM), SCP25041 (100 μM), SP600125 (20 μM), Z-VAD-fmk (100 μM), DEVD-fmk (100 μM), IETD-fmk (100 μM), and LEHD-cho (100 μM) were added 30 min before and then during autophagic induction.
Loading of LysoTracker Red
After 70 min of nutrient deprivation with glucagon, LTR (25 to 500 nM) was added. After 20 min, each well was washed two times with fresh KRH and fixed with 2% paraformaldehyde in phosphate-buffered saline for 10 min at 4°C. The red fluorescence of LTR (>590 nm) was measured immediately using a 544-nm (15-nm band pass) excitation filter and a 590-nm long pass emission filter with a FLUOstar multi-well fluorescence plate reader (BMG LabTechnologies, Offenburg, Germany). LTR fluorescence after various treatments was expressed as the percentage of LTR fluorescence of hepatocytes incubated in complete growth medium.
Laser scanning confocal microscopy of LTR-labeled hepatocytes
Some hepatocytes were cultured overnight on Type I collagen-coated 40-mm round number 1.5 glass coverslips inside 60-mm Petri dishes at a density of 600,000 cells per dish.24 Until induction of autophagy, cells were kept in complete serum-containing Waymouth's MB-742/1 growth medium. On the microscope stage, complete growth medium was switched to KRH plus 1 μM glucagon. After 70 min, the hepatocytes were loaded with red-fluorescing LTR (200 nM) for 20 min and through-focus confocal images of LTR-loaded hepatocytes were taken at 2 μm intervals with a Zeiss LSM 410 inverted laser scanning confocal microscope (Carl Zeiss, Oberkochen, Germany) using a 63× oil/1.4 N.A. planapochromat objective lens. Excitation at 568 nm was provided by an argon/krypton laser, and fluorescence emission was measured through a 590-nm long pass barrier filter. Laser excitation energy was attenuated 100- to 1000-fold to minimize photobleaching and photodamage.
Laser scanning confocal microscopy of hepatocytes coloaded with LTR and MTG
In other experiments, hepatocytes were cultured overnight on Type I collagen-coated 7-mm round number 1.5 coverslips fixed to the bottom of 35-mm Petri dishes (Corning, Ashland, MA) at a density of 300,000 cells per dish. Until induction of autophagy, cells were kept in complete Waymouth's MB-742/1 growth medium. Hepatocytes were loaded with green-fluorescing MitoTracker Green (MTG, 0.5 μM) for 60 min in humidified air at 37°C in Waymouth's MB-742/1 growth medium containing 25 mM Na-HEPES. Afterwards, the MTG-loaded hepatocytes were washed 2 times with fresh complete growth medium and incubated with red-fluorescing LysoTracker Red (LTR, 0.5 μM) for 20 min under identical conditions. After MTG and LTR had been loaded, one third of the initial concentration of LTR was kept in the medium for the duration of the experiment to maintain steady state loading. On the microscope stage, complete growth medium was switched to KRH plus 1 μM glucagon in the presence or absence of proteases inhibitors (7.5 μM pepstatin A or 10 μM leupeptin) at 37°C. Time series of confocal images were collected every 1 to 2 min for up to 90 min after autophagic induction with a Zeiss LSM 510 inverted laser scanning confocal microscope (Carl Zeiss, Oberkochen, Germany) using a 63× oil 1.4 N.A. planapochromat objective lens. Excitation of LTR at 543 nm was provided by a helium/neon laser, and fluorescence emission was measured through a 560-nm long pass barrier filter. Excitation of MTG at 488 nm was provided by an argon laser, and fluorescence emission was measured through a 500–550-nm band pass barrier filter. Laser excitation energy was attenuated 100- to 1000-fold to minimize photobleaching and photodamage, which was negligible in control experiments in complete growth medium.
Measurement of the cellular content of lysosomes
The number of LTR-labeled organelles in confocal images was quantified from serial confocal images using the computer program Image Processing Kit 4.0.25 Briefly, a superimposing image was generated from two single slices through a difference of Gaussians. In each superimposing image, the threshold of each structure was adjusted and processed to get outlined structures. Each outlined structures were quantified using a mathematical function in Adobe Photoshop 7.0.
Statistical analysis
Data are presented as means ± S.E.M. Differences between means were analyzed by the student's t test using p < 0.05 as the criterion of significance.
RESULTS
Increase of LTR uptake by cultured hepatocytes after nutrient deprivation plus glucagon
LTR accumulates inside endosomes, autophagosomes, lysosomes and autolysosomes (autophagosomes fused with lysosomes) by virtue of their low internal pH. To determine the appropriate concentration of LTR for the microplate reader assay, hepatocytes were incubated for 70 min in KRH medium or complete growth medium followed by loading with various concentrations of LTR from 25 nM to 500 nM (Fig. 1A). Fluorescence values were normalized to baseline values of LTR fluorescence in complete growth medium, which represented 100%. The greatest relative increase in LTR fluorescence after incubation in KRH plus glucagon compared to complete growth medium occurred at 50 nM, and LTR uptake increased from 100 ± 39% in complete growth medium to 580 ± 70% in KRH plus glucagon (n = 12, p < 0.001). In comparison, lower relative increases of LTR fluorescence were observed after incubation with 25, 200 and 500 nM LTR. These data indicated that 50 nM LTR provided the maximal fluorescence response to nutrient deprivation. To determine if 70 min represented an adequate time for autophagy to develop, hepatocytes were exposed to nutrient deprivation plus glucagon for 15, 30, 70 and 90 min and then loaded with 50 nM LTR. An increase of LTR fluorescence was observed within 15 min of exposure to nutrient deprivation plus glucagons. A maximal fluorescence signal was reached at 70 min (Fig. 1B). Accordingly, 50 nM LTR and 70 min of incubation were used in subsequent experiments.
Figure 1.

Increased LTR fluorescence in cultured hepatocytes after autophagic stimulation. In (A), cultured hepatocytes were loaded with 25, 50, 200 and 500 nM LTR after 70 min incubation in complete growth medium (GM) or KRH plus 1 μM glucagon (KRH+G). After another 20 min, LTR fluorescence was measured using a fluorescence plate reader, as described in Materials and Methods. In (B), hepatocytes were loaded with 50 nM LTR, as described in (A). LTR fluorescence was then measured after various times of incubation. *, p < 0.001 vs. complete media (n = 12 except for 25 nM LTR where n = 4).
To examine the intracellular distribution of LTR fluorescence, hepatocytes cultured on coverslips were incubated 70 min in complete growth medium or KRH plus glucagon. Afterwards, the hepatocytes were loaded with 200 nM LTR for 20 min. For these experiments, a higher concentration of LTR was employed because preliminary experiments showed that more LTR was needed to produce bright, well resolved confocal fluorescence images. Serial confocal images were then collected through the entire thickness of individual cells, and the optical sections were superimposed to represent the entire cellular complement of organelles taking up LTR. LTR uptake occurred in discrete subcellular organelles (Fig. 2). In hepatocytes incubated in KRH plus glucagon, the number of LTR-labeled organelles was markedly increased compared to hepatocytes incubated in complete growth medium, but diffuse background fluorescence of the cytoplasm was unchanged (Fig. 2, compare upper and lower left panels). The increased number of LTR-labeled organelles in hepatocytes incubated in KRH plus glucagon was indicative of autolysosome and autophagosome formation. Counts of the number of LTR-labeled organelles per hepatocyte correlated well with total LTR fluorescence measured in the multi-well fluorescence plate reader. Specifically, the number of LTR-labeled organelles per hepatocyte increased from 35 ± 9 acidic organelles/cell (n = 5 cells) after 70 min of incubation in complete growth medium to 182 ± 22 acidic organelles/cell after incubation in KRH plus glucagon (p < 0.001, n = 6 cells). This 520% change compared closely with the 580% change of LTR fluorescence measured by the plate reader (Fig. 1A). By contrast, bright field microscopy revealed little obvious change to the cells beyond that accounted for by cell to cell heterogeneity (Fig. 2, right panels). Thus, an increase in the number of LTR-labeled organelles appeared to account for increased LTR uptake measured by fluorescence plate reading after nutrient deprivation.
Figure 2.
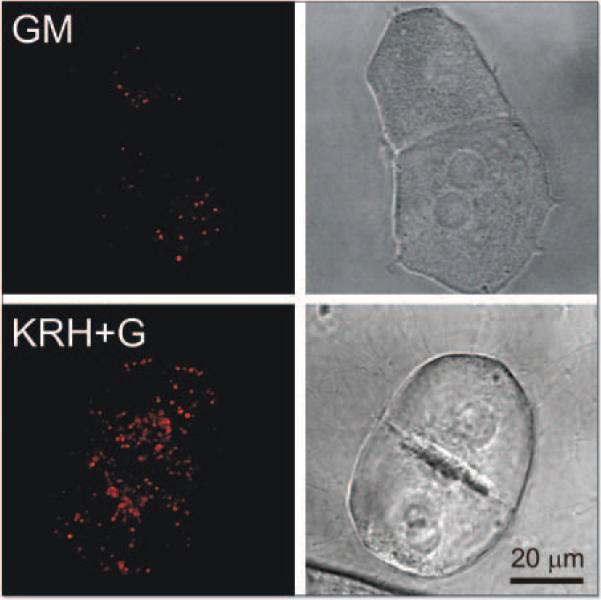
Confocal and wide field microscopy of LysoTracker Red (LTR) uptake in hepatocytes after autophagic induction. Cultured hepatocytes were loaded with LTR (200 nM, 20 min) and incubated for 70 min in complete growth medium (GM, upper panels) or in KRH plus 1 μM glucagon (KRH+G, lower panels). The left panels show representative superimposed through-focus confocal images of red LTR fluorescence of hepatocytes in growth medium (upper left panel) or KRH plus glucagon (lower left panel). The right panels are corresponding bright images.
Mitochondrial digestion by acidic organelles after autophagy induction
Time series were also collected of confocal images of hepatocytes coloaded with green-fluorescing MTG to visualize mitochondria and red-fluorescing LTR to visualize acidic organelles. MTG is taken up electrophoretically by mitochondria in response to the negative mitochondrial membrane potential. After uptake, MTG becomes covalently bound to mitochondrial proteins and remains in mitochondria even if a mitochondrion subsequently depolarizes.14,26 By colabeling with MTG and LTR, we could visualize mitochondrial entrapment inside acidic autolysosomes. In the experiment shown in Figure 3, no colabeling with LTR was evident after 76.5 min in KRH plus glucagon in the field of view shown. After another 1.5 min, the mitochondrion at the arrow began to label with LTR and turn yellow. This labeling became successively stronger over the next minutes. These images showed directly the transformation of an individual mitochondrion into a mitochondrion-containing autophagosome/autolysosome. After about 9 min, the LTR-labeled structure disappeared. Disappearance of autolysosomes was due either to completion of autophagic digestion or to movement of the structures out of the confocal image plane. Disappearance by movement out of the confocal plane was minimized by opening the pinhole of the confocal microscope for an optical slice thickness of 2 μm. Colabeling did not represent random overlap of organelles within the plane of confocal sections, since entrapped MTG-labeled mitochondria were consistently totally within LTR-labeled autophagosomes/autolysosomes. MTG fluorescence in autophagosomal/autolysosomal structures progressively diminished compared to normal mitochondria due to hydrolytic digestion (Figs. 3 and 4A).
Figure 3.
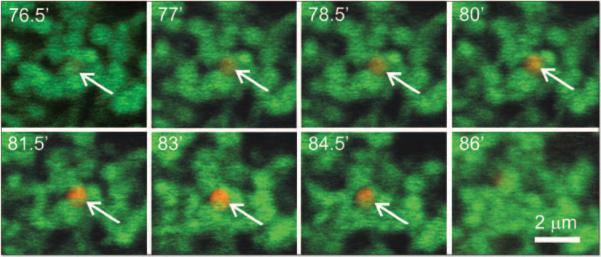
Mitochondrial digestion by during autophagy. Cultured hepatocytes were coloaded with MTG (0.5 μM) for 60 min and LTR (0.5 μM) for 20 min at 37°C in complete growth medium. On the microscope stage, complete growth medium was replaced KRH plus 1 μM glucagon, and a time series of confocal images of green MTG fluorescence and red LTR was collected. Time points were selected to illustrate the onset and completion of mitochondrial digestion by autophagy. One experiment representative of 10.
Figure 4.

Prolongation of autophagic digestion of mitochondria by proteases inhibitors. In (A), cultured hepatocytes were coloaded with MTG and LTR and placed in KRH plus glucagon, as described in Figure 3, in the absence (left panels) and presence of 7.5 μM pepstatin A (right panels). After 70 min, single confocal images of green MTG and red LTR fluorescence were collected. Upward-pointing white arrows represent the LTR-labeled acidic organelles that colocalized with MTG-labeled mitochondria. Downward-pointing yellow arrows represent LTR-labeled lysosomes that did not colocalize with MTG fluorescence. In (B), the number of LTR-labeled acidic organelles containing MTG fluorescence (black bars) and the total number of LTR-labeled organelles per hepatocyte containing MTG fluorescence in single confocal optical sections (~2 μm thickness) (black bars) and the total number of LTR-labeled organelles per hepatocyte optical section (white bars) are plotted in the absence (None) and presence of pepstatin A (Pep A, 7.5 μM) or leupeptin (Leu, 10 μM). In (C), the duration of colocalization of MTG and LTR in individual autophagosomes was determined from time series of confocal images after autophagic stimulation by KRH plus glucagon in the presence and absence of protease inhibitors, as illustrated in (A). *, p < 0.001 vs. None (n = 6).
Suppression of mitochondrial digestion by protease inhibitors
Confocal images of hepatocytes coloaded with MTG and LTR were also collected in the presence and absence of protease inhibitors. After 70 min incubation with KRH plus glucagon, the total number of LTR labeled organelles was greater in the presence of protease inhibitors like pepstatin A (7.5 μM) than in their absence (Fig. 4A, compare lower and upper panels). However, on average the proportion of LTR-labeled organelles containing MTG fluorescence (upward-pointing white arrows in Fig. 4A) to those without MTG fluorescence (downward-pointing yellow arrows) did not change (Fig. 4B). Pepstatin A (7.5 μM) and leupeptin (10 μM) promoted increases of 46 ± 2% and 54 ± 2%, respectively, in the number of organelles coloaded with both MTG and LTR (Fig. 4B). Figure 4B shows that about 75% of total LTR-labeled acidic organelles contained MTG-labeled mitochondria both in the presence and absence of proteases inhibitors. However, the total number of acidic organelles quantified in the first minute after replacing complete growth with KRH plus glucagon was similar in the presence and absence of protease inhibitors (8 ± 1.5, 7.5 ± 2 and 6.3 ± 0.5 LTR-labeled organelles/hepatocyte after no addition, leupeptin, and pepstatin A, respectively; n = 6 cells). Thus, formation of LTR-labeled structures increased over time in both the presence and absence of proteases inhibitors.
From temporal series of confocal images (see Fig. 3), the duration of colocalization of MTG and LTR in individual autolysosomes was determined after autophagic stimulation by KRH plus glucagon. The time of colocalization (coincidence of MTG and LTR fluorescence) averaged 7.5 min and increased 2.5-fold in the presence of either leupeptin or pepstatin A (Fig. 4C). Thus, protease inhibitors increased the time required for autophagic digestion of individual mitochondria within autolysosomes.
Suppression of autophagosomal/autolysosomal proliferation by 3-methyladenine, cyclosporin A and NIM811 after autophagic stimulation
3-MA is a well characterized inhibitor of the early stages of autophagy.27 Accordingly, we tested if 3-MA (10 mM) inhibits the increase of LTR fluorescence after autophagic stimulation. Hepatocytes were preincubated with 3-MA in complete growth medium for 30 min before and then during induction of autophagy in KRH plus glucagon. 3-MA suppressed the increase of LTR fluorescence induced by nutrient deprivation by 85% (Fig. 5A). To assess the effect of inhibition of the MPT on autophagy assessed by the LTR multi-well assay, CsA (5 μM) was added before and then during the induction of autophagy by nutrient deprivation. CsA treatment suppressed the increase of LTR fluorescence by 63% (Fig. 5A).
Figure 5.

LTR fluorescence plate reader screening assay: inhibition of autophagy by agents inhibiting the MPT, PI3 kinase and JNK but not by agents inhibiting caspases, ERK1 and p38. Cultured hepatocytes in growth medium were preincubated 30 min in complete growth medium followed by 70 min incubation in growth medium (GM) or KRH plus 1 μM glucagon (KRH+G). LTR (50 nM) was added, and LTR fluorescence was measured as described in Materials and Methods. As indicated, various inhibitors were added from the beginning of the 30 min preincubation. Agents used were: (A), 10 mM 3-MA, 5 μM tacrolimus (calcineurin inhibitor), 5 μM NIM811 (MPT inhibitor), 5 μM CsA (calcineurin and MPT inhibitor); (B), 0.5 μM wortmannin, 10 μM LY294002 (PI3 kinase inhibitors); C, 100 μM DEVD-fmk (caspase 3 inhibitor), 100 μM IETD-fmk (caspase 8 inhibitor), 100 μM LEHD-cho (caspase 9 inhibitor), 100 μM Z-VAD-fmk (pan caspase inhibitor); D, 10 mM 3-MA, 100 μM PD98059 (ERK1 inhibitor), 100 μM SB203580 (p38 inhibitor), 100 μM SCP25041 (JNK inhibitor), 20 μM SP600125 (JNK inhibitor). *, p < 0.001 compared to KRH+G (n = 12–16 per group except SP600125 where n = 4).
Although CsA specifically inhibits the MPT at the concentration used, CsA also inhibits the Ca2+-dependent protein phosphatase, calcineurin, an effect underlying the immunosuppressive properties of CsA.28 Therefore, hepatocytes were incubated with N-methyl-4-isoleucine cyclosporin (NIM811, 5 μM), a nonimmunosuppressive CsA analog that blocks the MPT but does not inhibit calcineurin.29 NIM811 blocked the increase of LTR fluorescence after nutrient deprivation to nearly same extent as CsA (Fig. 5A). By comparison, tacrolimus (5 μM), an immunosuppressive calcineurin inhibitor that does not block the MPT, did not inhibit autophagy (Fig. 5A).
Inhibition of autophagy by phosphatidylinositol-3 kinase and c-Jun N-terminal kinase inhibitors but not by ERK1, p38 and caspase inhibitors
Autophagy is frequently prominent in cells undergoing apoptosis, especially in cells containing many membranous organelles.30 In hepatocytes and hepatoma cells, the MPT is a critical step in the progression of apoptosis induced by TNFα, Fas, TGFβ, TRAIL and other inducers (reviewed in ref. 17). Upstream to the MPT during apoptosis is caspase 8 activation, whereas downstream is caspase 9 and 3 activation. By contrast, phosphatidylinositol-3 kinase (PI3 kinase) is antiapoptotic in hepatocytes and other cells.31,32 Since both PI3 kinase and the MPT are implicated in the regulation of autophagy,14,33,34 we examined the effects of PI3 kinase and caspases inhibitors on autophagy assessed by the LTR fluorescence plate reader assay. To inhibit PI3 kinase, hepatocytes were incubated with wortmannin (0.5 μM) and 2-(4-morpholinyl)-8-phenylchromone (LY294002, 10 μM) 30 min before and then during the induction of autophagy for 70 min. As measured by LTR fluorescence, wortmannin and LY294002 inhibited autophagy by 54 ± 4% and 42 ± 7%, respectively (Fig. 5B). These findings are consistent with previous reports showing inhibition of autophagy by PI3 kinase inhibitors.33,34 To inhibit caspase 3, caspase 8, caspase 9 and pan-caspase activity, hepatocytes were incubated with 100 μM of DEVD-fmk, IETD-fmk, LEHD-cho and Z-VAD-fmk, respectively. However, none of the caspase inhibitors altered LTR uptake (Fig. 5C).
Mitogen-activated protein kinases (MAPKs) act in signaling cascades that control cell differentiation, proliferation and death.35,36 Stress activates MAPKs, and autophagy often implies nutrient deficiency stress for cells. To screen for a possible role of MAPKs in autophagy, hepatocytes were subjected to autophagic stimulation in the presence and absence of MAPK inhibitors, including PD98059 (100 μM) for ERK1, SB203580 (100 μM) for p38 and SCP25041 (100 μM) and SP600125 (20 μM) for JNK. Both JNK inhibitors blocked LTR uptake to nearly the same extent as 3-MA, whereas the ERK1 and p38 inhibitors were without effect (Fig. 5D).
DISCUSSION
Autophagy is responsible for degradation and turnover of cell organelles, including mitochondria (reviewed in refs. 37 and 38). As organelles occupying 20% of the volume of hepatocyte cytoplasm,8 mitochondria are a frequent target of autophagic digestion after fasting in vivo and nutrient deprivation in vitro. Here, we used LTR and MTG to characterize the kinetics of autophagy and specifically of mitochondrial autophagy (mitophagy) during nutrient deprivation to rat hepatocytes. We also developed a high throughput multi-well plate assay to measure autophagy in cultured hepatocytes based on uptake of the acidic organelle marker, LTR. LTR uptake, as measured by its red fluorescence in a plate reader, increased 4- to 6-fold after nutrient deprivation and was blocked by 3-MA, the classical inhibitor of autophagy (Figs. 1, 5A and D). Increased LTR uptake measured by fluorescence plate reading correlated with the proliferation of large acidic structures visualized by confocal microscopy (Fig. 2, right panel), and the majority of large acidic organelles after nutrient deprivation contained MTG-labeled mitochondrial remnants, which positively identified the structures as autophagosomes/autolysosomes (Fig. 3).
Proliferation of autophagosomes and autolysosomes is a basic characteristic of autophagic progression. Acidification of newly sequestered autophagosomes occurs almost immediately and is required for subsequent fusion with lysosomal vesicles.39–41 Here using confocal microscopy we show the time course of mitochondrial autophagy, or mitophagy, that had previously been inferred only by electron microscopy (Fig. 3).10,39,42 By temporal imaging of hepatocytes coloaded with LTR, a marker of autophagosomes and other acidic organelles, and MTG, a marker of mitochondria, we directly visualized the progression of mitophagy and observed that autophagic digestion of mitochondria progressed to completion in about 7.5 min on average. Thus, MTG in approximately half the autophagosomes disappeared within 7.5 min of autophagic sequestration. MTG binds covalently to mitochondrial proteins after accumulating electrophoretically into polarized mitochondria.14,26 Loss of MTG from autolysosomes thus represents release due to lysosomal digestion of mitochondrial peptide fragments or breakage of covalent bonds between MTG and mitochondrial proteins. Our observed half-time of digestion agrees well with earlier estimates by electron microscopy that the half time of disappearance of autophagosomes and autolysosomes is about 9 min in rat livers after in vivo treatment with insulin to block formation of new autophagosomes.10 Additionally, we showed that this digestion was markedly slowed by the proteases inhibitors, leupeptin and pepstatin A, as expected for the autophagic process (Fig. 4). In the cultured hepatocytes studied here, autophagic stimulation produced predominantly mitochondrial autophagy, since after coloading with LTR and MTG 75% of LTR-labeled acidic organelles contained MTG-labeled mitochondria (Fig. 4A and B). The remaining 25% may represent nonmitochondrial autophagy or represent lysosomal remnants after the completion of mitochondrial digestion.
Using the LTR fluorescence assay, we showed that the MPT pore inhibitors, CsA and NIM811, inhibited autophagy and that the calcineurin inhibitor, tacrolimus, did not (Fig. 5A). Additionally, the PI3 kinase inhibitors, wortmannin and LY294002, suppressed autophagy induced by nutrient deprivation (Fig. 5B). The last finding is consistent with previous reports that PI3 kinase inhibitors suppress autophagy, that 3-MA is itself a PI3 kinase inhibitor, and that Class III PI3 kinase is an important regulator of intracellular membrane trafficking.33,34,43 The findings are also consistent with a previous report from our laboratory that CsA inhibits mitochondrial autophagy.14 Taken together, these findings provide strong validation of the LTR multi-well plate assay for autophagy. We also examined the effects of inhibition of various caspases on autophagy, including pan-caspase inhibition (z-VAD-fmk), inhibition of the initiator caspase-8 (IETD-fmk) and inhibition of the executioner caspases 9 and 3 (DEVD-fmk and LEHD-cho, respectively) (Fig. 5C). However, none of the caspase inhibitors tested produced any statistically significant change of autophagy.
The MAPK superfamily of protein kinases is used by mammalian cells to transduce extracellular signals into cellular responses, and MAPKs play an important role regulating apoptotic signaling.35,36 Here, we showed that ERK1 inhibition with PD98059 and p38 inhibition with SB203580 did not decrease autophagy induced by nutrient deprivation plus glucagon. By contrast, JNK inhibition with SCP25041 and SP600125 decreased autophagy assessed by LTR uptake, suggesting a role of this kinase in the induction of autophagy (Fig. 5D). JNK inhibition also inhibited autophagosomal proliferation as assessed by confocal microscopy of LTR-labeled hepatocytes (data not shown). Recent studies show that JNK inhibition blocks a form of programmed cell death called autophagic cell death.37,44,45 However, under our conditions of autophagic stimulation by nutrient deprivation, cell killing did not occur, as assessed by nuclear propidium iodide staining and morphological changes (Fig. 2 and data not shown). Thus, effects of JNK on induction of autophagy may be direct rather than secondary to the induction of the cell death process. Nonetheless, confirmation of this pharmacological evidence and characterization of the participation of JNK in autophagy will require future study.
LTR is an acidotropic fluorescent probe used to label and track acidic organelles in living cells (see ref. 46). LTR labels all acidic organelles, including lysosomes, autophagosomes, late endosomes and, to a lesser extent, early endosomes that are not as acidic as other acid organelles. LTR fluorescence measured by a fluorescence plate reader represents the relative overall mass of acidic organelles, and the increase of LTR fluorescence we observe after nutrient deprivation plus glucagon might represent proliferation of any of the acidic organelles. However, in the context of our assay, increased LTR fluorescence most likely represents an increase of autophagosomes and autolysosomes for several reasons. First, nutrient deprivation and glucagon, both strong stimuli of autophagy, induced the increase of LTR fluorescence (Figs. 1 and 5A–D). Second, agents that block autophagy, including 3-MA, MPT blockers and PI3 kinase inhibitors, inhibited the increase of LTR fluorescence after nutrient deprivation plus glucagon (Fig. 5A and B). Third, imaging of hepatocytes by confocal microscopy revealed proliferation of relatively large LTR-stained structures whose size and heterogeneous structure were consistent with autophagosomes and autolysosomes rather than with endosomes and primary lysosomes (Figs. 2–4). Fourth, most of these structures contained MTG-labeled mitochondria, which positively identified them as autophagosomes/autolysosomes (Fig. 3).
Endocytosis is also upregulated by nutrient deprivation, which is inhibited by 3-MA.47,48 Although individual pinosomes/endosomes are below the resolution of light microscopy, an increase of LTR fluorescence due to increased endocytosis would be evident as an increase of diffuse cellular LTR fluorescence, which was not observed (see Fig. 2). This observation combined with the small size of endosomes and their weaker acidification than lysosomes implies that changes of endocytosis are likely not contributing to observed changes of LTR fluorescence after autophagic stimulation.
The extent of LTR uptake by cells depends not only on the size of the lysosomal/autophagosomal compartment but also on the degree of acidity of these compartments. Thus, LTR uptake will be suppressed by agents like bafilomycin and monensin that collapse ΔpH across lysosomal/autophagosomal membranes. Accordingly, “hits” using the high through-put fluorescence plate reader screening assay should be followed up by more specific techniques, such as confocal microscopy, to determine the basis for changes of LTR accumulation. Decreased ΔpH should be suspected if microscopy shows weak LTR uptake by lysosomes and autophagosomes without a decrease of the number of labeled vesicles. Similarly, increased intraluminal acidity can be expected to increase LTR uptake.
Previously, our laboratory showed that monodansylcadaverine labels these proliferating acidic organelles after exposure of rat hepatocytes to nutrient deprivation plus glucagon,14 in agreement with earlier reports that monodansylcadaverine is an autophagosomal marker.22 As a weak amine, monodansylcadaverine accumulates into acidic organelles much like LTR.46 Inside such organelles, the fluorescence of monodansylcadaverine is enhanced by a hydrophobic environment, i.e., when the acidic compartment contains lipid as occurs after autophagy of membranous organelles.49 However, we found that monodansylcadaverine was unsuitable for use in a multi-well fluorescence assay because of high levels of background fluorescence after monodansylcadaverine loading (data not shown). Confocal microscopy revealed that this background fluorescence was diffuse throughout cells and may represent insertion of the amphipathic monodansylcadaverine into various endomembranes. LTR produced much less background fluorescence.
In the fluorescence plate reader assay, the greatest relative increase of fluorescence after nutrient deprivation occurred after 50 nM LTR, presumably because nonspecific background fluorescence was minimal at this low concentration. For confocal imaging, however, a higher concentration of LTR was necessary for adequate sensitivity and spatial signal to noise ratio. Confocal imaging easily distinguished specific LTR-labeled organelles from diffuse nonspecific background fluorescence. In general, the lowest possible concentration of LTR should be used to avoid LTR-induced disturbances of intraorganellar acidity.
CsA has a dual inhibitory effect on mammalian cells.28,50 One effect is to bind to cyclophilin D in the mitochondrial matrix space to inhibit the MPT pore, and the other is to bind cyclophilin A in the cytosol to inhibit calcineurin. Calcineurin is a protein phosphatase, and inhibition of calcineurin leads to increased phosphorylation of many proteins, possibly including those regulating autophagy. A new finding here was that NIM811 blocked autophagic stimulation during nutrient deprivation to the same degree as CsA (Fig. 5A). NIM811, an analog of CsA, blocks the MPT but does not inhibit calcineurin.29 This observation and the fact that another calcineurin inhibitor, tacrolimus, did not block autophagy support the conclusion that the MPT is an important event initiating mitochondrial autophagy.
Although long assumed to be a random process, increasing evidence suggests that autophagy of mitochondria, peroxisomes and possibly other organelles can be selective.13–15,17,51–53 Accordingly, the term mitophagy is appropriate to describe autophagy of mitochondria in analogy to the term pexophagy for peroxisomal autophagy.13 The involvement of the MPT in autophagy implies that factors which promote the MPT, such as mitochondrial Ca2+ uptake, increased Pi, reactive oxygen and nitrogen species, certain sphingolipids, and alterations of cellular redox state, may also modulate the rate and extent of autophagy. An elucidation of these factors in autophagy is now ongoing.
In conclusion, we characterize here LTR as a fluorescent probe of autophagy by cultured rat hepatocytes. LTR fluorescence measured with a fluorescence plate reader provides a rapid and sensitive means to monitor autophagy stimulated by nutrient deprivation and glucagon that is amenable to high throughput screening. Correlative confocal microscopy shows that this autophagy predominantly involves mitochondria and that protease-dependent autophagic digestion of individual mitochondria occurs within 10 min or less. We validate the fluorescence reader assay by showing that MPT and PI3 kinase inhibitors suppress LTR uptake, and we provide new evidence that JNK activity may also be involved in autophagy. However, despite the involvement of JNK, the MPT and PI3 kinase in pro- and anti-apoptotic regulation, caspases did not appear to have a role in signaling the autophagic response. Total fluorescence measurement and fluorescence imaging of LTR should be useful in screening and characterizing other agents acting on autophagic pathways.
ACKNOWLEDGEMENTS
We thank Dr. Lihua He and Dr. Jae-Sung Kim for helpful discussions and Ms. Sherry Grissom for expert technical assistance. This work was supported, in part, by Grants 1 P01 DK59340 and 5-R01 AG07218 from the National Institutes of Health. Dr. Rodriguez-Enriquez was supported by a fellowship from CONACyT-Mexico. Imaging facilities were supported, in part, by center grants 5-P30-DK34987 and 1-P50-AA11605 from the National Institutes of Health.
ABBREVIATIONS
- 3-MA
3-methyladenine
- CsA
cyclosporin A
- JNK
c-Jun N-terminal kinase
- LTR
LysoTracker Red
- MTG
MitoTracker Green
- MAPK
mitogen-activated protein kinase
- MPT
mitochondrial permeability transition
- PI3K
phosphatidylinositol-3 kinase
References
- 1.Arstila AU, Shelburne DJ, Trump BF. Studies on cellular autophagocytosis. A histochemical study on sequential alterations of mitochondria in the glucagon-induced autophagy vacuoles of rat liver. Lab Invest. 1972;27:317–23. [PubMed] [Google Scholar]
- 2.Lardeux BR, Mortimore GE. Amino acid and hormonal control of macromolecular turnover in perfused rat liver. Evidence for selective autophagy. J Biol Chem. 1987;262:14514–9. [PubMed] [Google Scholar]
- 3.Shworer CM, Shiffer KA, Mortimer GE. Quantitative relationship between autophagy and the proteolysis during graded amino acid deprivation in perfused rat liver. J Biol Chem. 1981;256:7652–8. [PubMed] [Google Scholar]
- 4.Mortimore GE, Poso AR. Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu Rev Nutr. 1987;5:49–70. doi: 10.1146/annurev.nu.07.070187.002543. [DOI] [PubMed] [Google Scholar]
- 5.Yu QC, Marzella L. Response of autophagic protein degradation to physiologic and pathologic stimuli in rat hepatocyte monolayer cultures. Lab Inves. 1988;58:643–52. [PubMed] [Google Scholar]
- 6.Codogno P, Ogier-Denis E, Houri JJ. Signal transduction pathways in macroautophagy. Cell Signal. 1997;9:125–30. doi: 10.1016/s0898-6568(96)00130-1. [DOI] [PubMed] [Google Scholar]
- 7.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loud AV. A quantitative stereological description of the ultrastructure of normal rat liver parenchymal cells. J Cell Biol. 1968;37:27–46. doi: 10.1083/jcb.37.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menzies RA, Gold PH. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J Biol Chem. 1971;246:2425–9. [PubMed] [Google Scholar]
- 10.Pfeifer U. Inhibition by insulin of the formation of autophagic vacuoles in rat liver. A morphometric approach to the kinetics of intracellular degradation by autophagy. J Cell Biol. 1978;78:142–67. doi: 10.1083/jcb.78.1.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Grey AD. A proposed refinement of the mitochondrial free radical theory of aging. BioEssays. 1997;19:161–6. doi: 10.1002/bies.950190211. [DOI] [PubMed] [Google Scholar]
- 12.Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging: Accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem. 2002;269:1996–2002. doi: 10.1046/j.1432-1033.2002.02869.x. [DOI] [PubMed] [Google Scholar]
- 13.Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction and aging. Rejuvenation Res. 2005;8:3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- 14.Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15:2286–7. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- 15.Kissova I, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem. 2004;279:39068–74. doi: 10.1074/jbc.M406960200. [DOI] [PubMed] [Google Scholar]
- 16.Zamzami N, Marchetti P, Castedo M, Hirsch T, Susin SA, Masse B, Kroemer G. Inhibitors of permeability transition interfere with the disruption of the mitochondrial transmembrane potential during apoptosis. FEBS Lett. 1996;384:53–7. doi: 10.1016/0014-5793(96)00280-3. [DOI] [PubMed] [Google Scholar]
- 17.Kim J-S, He L, Lemasters JJ. Mitochondrial permeability transition: A common pathway to necrosis and apoptosis. Biochem Biophys Res Commun. 2003;304:463–70. doi: 10.1016/s0006-291x(03)00618-1. [DOI] [PubMed] [Google Scholar]
- 18.Kim J-S, Qian T, Lemasters JJ. ATP supply and the mitochondrial permeability transition in the switch from necrotic to apoptotic cell death after ischemia/reperfusion to cultured rat hepatocytes. Gastroenterology. 2003;124:494–503. doi: 10.1053/gast.2003.50059. [DOI] [PubMed] [Google Scholar]
- 19.Assuncao-Guimaraes C, Linden R. Programmed cell deaths. Apoptosis and alternative deathstyles. Eur J Biochem. 2004;271:1638–50. doi: 10.1111/j.1432-1033.2004.04084.x. [DOI] [PubMed] [Google Scholar]
- 20.Seglen PO, Gordon PB, Holen I, Hoyvik H. Radiolabelled sugars as probes of hepatocytic autophagy. Biomed Biochim Acta. 1986;50:373–81. [PubMed] [Google Scholar]
- 21.Tuskada M, Ohsumi Y. Isolation of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333:169–74. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- 22.Munafó DB, Colombo MI. A novel assay to study autophagy: Regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114:3619–28. doi: 10.1242/jcs.114.20.3619. [DOI] [PubMed] [Google Scholar]
- 23.Gores GJ, Nieminen AL, Fleishman KE, Dawson TL, Herman B, Lemasters JJ. Extracellular acidosis delays onset of cell death in ATP-depleted hepatocytes. Am J Physiol. 1988;255:C315–22. doi: 10.1152/ajpcell.1988.255.3.C315. [DOI] [PubMed] [Google Scholar]
- 24.Nieminen AL, Saylor AK, Herman B, Lemasters JJ. ATP depletion rather than mitochondrial depolarization mediates hepatocyte killing after metabolic inhibition. Am J Physiol. 1994;267:C67–74. doi: 10.1152/ajpcell.1994.267.1.C67. [DOI] [PubMed] [Google Scholar]
- 25.Russ JC. The image processing handbook. CRC Press; Boca Raton, Florida: 1999. [Google Scholar]
- 26.Elmore SP, Nishimura Y, Qian T, Herman B, Lemasters JJ. Discrimination of depolarized from polarized mitochondria by confocal fluorescence resonance energy transfer. Arch Biochem Biophys. 2004;422:145–52. doi: 10.1016/j.abb.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 27.Seglen PO, Gordon PB. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA. 1982;79:1889–92. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perrino BA, Wilson AJ, Ellison P, Clap LH. Substrate selectivity to inhibition by FK506 and cyclosporin A of calcineurin heterodimers composed of the alpha or beta catalytic subunit. Eur J Biochem. 2002;262:3540–8. doi: 10.1046/j.1432-1033.2002.03040.x. [DOI] [PubMed] [Google Scholar]
- 29.Waldmeier PC, Feldtrauer JJ, Qian T, Lemasters JJ. Inhibition of the mitochondrial permeability transition by the nonimmunosuppresive cyclosporin derivative NIM811. Mol Pharmacol. 2002;62:22–9. doi: 10.1124/mol.62.1.22. [DOI] [PubMed] [Google Scholar]
- 30.Schulte-Hermann R, Bursch W, Grasl-Kraupp B, Marian B, Torok L, Kahl-Rainer P, Ellinger A. Concepts of cell death and application to carcinogenesis. Toxicol Pathol. 1997;25:89–93. doi: 10.1177/019262339702500117. [DOI] [PubMed] [Google Scholar]
- 31.Kulik G, Klippel A, Weber MJ. Antiapoptotic signaling by the insulin-like growth factor 1 receptor, phosphatidylinositol 3-kinase and Akt. Mol Cell Biol. 1997;17:1595–606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen RH, Su YH, Chuang RL, Chang TY. Suppression of transforming growth factor-beta induced apoptosis through a phosphatidylinositol 3-kinase/Akt-dependent pathway. Oncogene. 1998;17:1959–68. doi: 10.1038/sj.onc.1202111. [DOI] [PubMed] [Google Scholar]
- 33.Blommaart EF, Krause U, Schellens JPM, Vreeling-Sindelarova H, Meijer AJ. The phosphatidilynositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240–6. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- 34.Petriot E, Ogier-Denis EF, Blommaart AJ, Meijer P, Codogno P. Distinct classes of phosphatidylinositol 3'-kinases are involved in the signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–8. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 35.Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderblit C, Cobb MH. MAP kinases. Chem Rev. 2001;101:2449–76. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- 36.Arbabi S, Maier RV. Mitogen-activated protein kinase. Crit Care Med. 2002;30:S74–S79. [PubMed] [Google Scholar]
- 37.Levine B, Klionsky DJ. Development by self-digestion: Molecular mechanism and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 38.Yoshimori T. Autophagy: A regulated bulk degradation process inside cells. Biochem Biophys Res Commun. 2004;313:453–8. doi: 10.1016/j.bbrc.2003.07.023. [DOI] [PubMed] [Google Scholar]
- 39.Dunn WA. Studies on the mechanisms of autophagy: Formation of the autophagic vacuole. J Cell Biol. 1990;110:1923–33. doi: 10.1083/jcb.110.6.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- 41.Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27:421–9. doi: 10.1247/csf.27.421. [DOI] [PubMed] [Google Scholar]
- 42.Shelburne JD, Arstila AU, Trump BF. Studies on cellular autophagocytosis. Cyclic AMP-and dibutyryl cyclic AMP-stimulated autophagy in rat liver. Am J Pathol. 1973;72:521–40. [PMC free article] [PubMed] [Google Scholar]
- 43.Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol. 2004;36:2445–62. doi: 10.1016/j.biocel.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 44.Borsello T, Croquelois K, Hornung JP, Clarke PG. N-methyl-d-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur J Neurosci. 2003;18:473–85. doi: 10.1046/j.1460-9568.2003.02757.x. [DOI] [PubMed] [Google Scholar]
- 45.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–2. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 46.Bampton ETW, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AV. The dynamics of autophagy visualised in live cells: From autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36. doi: 10.4161/auto.1.1.1495. [DOI] [PubMed] [Google Scholar]
- 47.Besterman M, Airhart JA, Low RB, Rannels DE. Pinocytosis and intracellular degradation of exogenous protein: Modulation by amino acids. J Cell Biol. 1983;96:1586–91. doi: 10.1083/jcb.96.6.1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Punnonen EL, Marjomaki VS, Reunanen H. 3-Methyladenine inhibits transport from late endosomes to lysosomes in cultured rat and mouse fibroblasts. Eur J Cell Biol. 1994;65:14–25. [PubMed] [Google Scholar]
- 49.Swanson SJ, Bethke PC, Jones RL, Barley F. Aleurone cells contain two types of vacuoles: Characterization of lytic organelles by use of fluorescence probes. Plant Cell. 1998;10:685–98. doi: 10.1105/tpc.10.5.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waldmeier PC, Zimmermann K, Qian T, Tintelnot-Blomley M, Lemasters JJ. Cyclophilin D as a drug target. Curr Med Chem. 2003;10:1485–506. doi: 10.2174/0929867033457160. [DOI] [PubMed] [Google Scholar]
- 51.Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA, Brenner DA, Herman B. The mitochondrial permeability transition in cell death: A common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta. 1998;1366:177–96. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- 52.Tolkovsky AM, Xue L, Fletcher GC, Borutaite V. Mitochondrial disappearance from cells: A clue to the role of autophagy in programmed cell death and disease? Biochimie. 2002;84:233–40. doi: 10.1016/s0300-9084(02)01371-8. [DOI] [PubMed] [Google Scholar]
- 53.Kiel JA, Komduur JA, Van der Klei IJ, Veenhuis M. Macropexophagy in Hansenula polymorpha: Facts and views. FEBS Lett. 2003;549:1–6. doi: 10.1016/s0014-5793(03)00794-4. [DOI] [PubMed] [Google Scholar]