

Abstract
The revolution in sequencing techniques in the past decade has provided an extensive picture of the molecular mechanisms behind complex diseases such as cancer. The Cancer Cell Line Encyclopedia (CCLE) and The Cancer Genome Project (CGP) have provided an unprecedented opportunity to examine copy number, gene expression, and mutational information for over 1000 cell lines of multiple tumor types alongside IC50 values for over 150 different drugs and drug related compounds. We present a novel pipeline called DIRPP, Drug Intervention Response Predictions with PARADIGM7, which predicts a cell line’s response to a drug intervention from molecular data. PARADIGM (Pathway Recognition Algorithm using Data Integration on Genomic Models) is a probabilistic graphical model used to infer patient specific genetic activity by integrating copy number and gene expression data into a factor graph model of a cellular network. We evaluated the performance of DIRPP on endometrial, ovarian, and breast cancer related cell lines from the CCLE and CGP for nine drugs. The pipeline is sensitive enough to predict the response of a cell line with accuracy and precision across datasets as high as 80 and 88% respectively. We then classify drugs by the specific pathway mechanisms governing drug response. This classification allows us to compare drugs by cellular response mechanisms rather than simply by their specific gene targets. This pipeline represents a novel approach for predicting clinical drug response and generating novel candidates for drug repurposing and repositioning.
1. Introduction
The potential for bioinformatics techniques to bring about transformative results in personalized medicine is just beginning to be realized. Large scale studies such as The Cancer Genome Atlas (TCGA), the Cancer Cell Line Encyclopedia (CCLE) and the Cancer Genome Project (CGP) have provided bioinformaticians with a wealth of –omic and pharmacologic data to interrogate1–5. Novel algorithms have been developed to perform detailed signaling pathway analysis6, integrate diverse –omic data types7–11, and even predict markers of drug sensitivity and resistance12. Analytical efforts are also underway to identify candidates for drug repurposing or repositioning and to computationally predict new drug indications for disease13.
Despite this wealth of innovation, the complexity for interpretation and translation of results to cancer patients remains challenging. The diversity of computational approaches has made it difficult to identify which of these have the most potential to improve the treatment of patients and improve clinical outcomes14. Each algorithm relies on a different type of –omic or combination of –omic data making it difficult to integrate them in a single analytical pipeline12, 13.
An important goal of computational bioinformatics pipelines is to provide actionable results to help physicians make optimal therapeutic decisions for a patient. To this end, the patient’s likelihood to respond to a specific treatment regimen is of particular interest to clinicians. The typical clinical case includes investigators looking to discover alternative therapies for patients who demonstrate resistance to the primary treatment. Both drug repurposing, the recycling of shelved or failed drugs, and drug repositioning, the use of active therapies for new applications, represent opportunities for the development of second line therapies. In order to maximize the impact of such an analysis pipeline, it should be versatile enough to address a myriad of clinical and scientific questions and easily integrate with existing clinical pipelines to assist physicians.
To address these clinical and analytical challenges we propose an integrative pipeline called DIRPP, Drug Intervention Response Predictions with PARADIGM (Pathway Recognition Algorithm using Data Integration on Genomic Models)7. Our pipeline aims to classify a cell line as either sensitive or resistant to a given therapy and to define specific genetic backgrounds represented in the cell line, potentially applicable to specific patients, associated with drug response phenotypes. This classification is performed using an extension of an open source probabilistic graphical model called PARADIGM. Drawing on multiple data types, DIRPP proceeds to integrate the copy number and gene expression data for a cell line into a biological pathway activity score which includes the result of a simulated drug intervention. Once the cell line (which may be a surrogate for a patient of interest) has been classified as sensitive or resistant to a given therapy, downstream gene set enrichment analysis (GSEA) on the pathway activity scores illustrates the underlying biological pathway mechanisms at work driving the drug response phenotype. The method can be applied to assess the impact of a wide variety of therapies on one particular cancer, or multiple cancers at a time to develop precision medicine strategies.
2. Materials and Methods
2.1. Datasets, Pathway Sources, and Pharmacologic Profile Data
Copy number, gene expression, and drug sensitivity data for 202 cancer cell lines from two recently published preclinical studies, the cancer genome project (CGP)4 and the cancer cell line encyclopedia (CCLE)5 were used for analysis. The distribution of cell lines by cancer type was: 20 ovarian, 39 breast, and 6 endometrial cancer cell lines from the CGP for testing of the algorithm and 51 ovarian, 59 breast, and 27 endometrial cancer cell lines from the CCLE for an independent dataset to validate the algorithm. Of the 16 drugs in common between the two studies, 9 inhibitory drugs were selected for analysis based on their clinical potential for treatment of ovarian cancer and repurposing/repositioning in breast and endometrial cancers (Table 1). Genetically similar sub-types of these cancers represented in this array of cell lines have been the subject of numerous genomic and drug repositioning studies and provide a robust sample set for analysis.
Table 1.
Nine (9) anticancer inhibitory drugs analyzed in both the CGP and CCLE with primary clinical relevance to ovarian cancer and secondary clinical relevance to breast, and endometrial cancer.
| Drug Name | Target(s) | Class |
|---|---|---|
| Erlotinib | EGFR | Kinase Inhibitor |
| Irinotecan | Topoisomerase | Cytotoxic |
| AZD0530 | Src, ABL/BCR-ABL, EGFR | Kinase Inhibitor |
| AZD6244 | MEK, ERK, MAPK | Kinase Inhibitor |
| PD0325901 | MEK, RAF, MAPK | Kinase Inhibitor |
| Lapatinib | EGFR, HER2 | Kinase Inhibitor |
| 17-AAG | HSP90 | Other |
| Sorafenib | KIT, PDGFRB, FLT3, FLT4, KDR, RAF1, BRAF | Kinase Inhibitor |
| Paclitaxel | Microtubules | Cytotoxic |
All cell line drug sensitivity values were reported as IC50 values, the concentration at which a drug inhibits 50% of cellular growth4, 5. Gene expression probes were normalized by centering on the gene’s median expression across all cell lines and then taking the base 2 log of that value7. SuperPathway, a merged biological pathway of 1,441 curated signal transduction, transcriptional, and metabolic pathways, was used to analyze the comprehensive cellular network of activity in the cell lines. This framework captures the global interactions of any perturbation in a cell while removing redundant pathway elements15. For each drug of interest, detailed pharmacological information about gene targets and mechanism of action was obtained from the drugbank and selleckchem databases16–18.
2.2. Analysis Pipeline
The DIRPP7 pipeline was implemented and tested using the overall scheme and specific steps laid out in Figure 1. Two runs of the PARADIGM algorithm are completed, one with –omic data, the two factor analysis, the other with –omic data and a simulated drug intervention, a three factor analysis. PARADIGM represents each entity in a biological pathway as a node whose value depends upon a defined internal set of “evidence nodes” whose connectivity mirrors the central dogma of molecular biology (Figure 2). These “evidence nodes” enable the integration of patient data into the biological pathway network. After assessing the signaling pathway activity of the cell lines with an initial run of the PARADIGM algorithm, where a DNA node interacts with a mRNA node to propagate biological information to the cellular network7, a second run of PARADIGM is performed while including a drug induced re-wiring of the cellular network (Figure 2). The resulting IPLs were then compared on a per-patient-per-gene basis to assess the impact of the drug intervention on perturbing the signaling network of a cancer cell line by computing a paired t-test p-value using the IPLs of the two PARADIGM runs for each cell line. The least perturbed cell lines were deemed the most resistant (least sensitive). All cell lines were then ranked in order of increasing sensitivity. Biological pathways involved in drug sensitivity and resistance were then identified using Gene Set Enrichment Analysis (GSEA) 6.
Figure 1.

Experimental design of DIRPP. For each cell line dataset, gene expression and copy number data were analyzed in 2-factor PARADIGM analysis. These inferred pathway levels IPL’s were compared to those from 3-factor PARADIGM analysis with a simulated drug intervention to generate a ranking by drug sensitivity. This ranking was then validated on the CGP and CCLE data. Response mechanisms were classified with GSEA.
Figure 2.
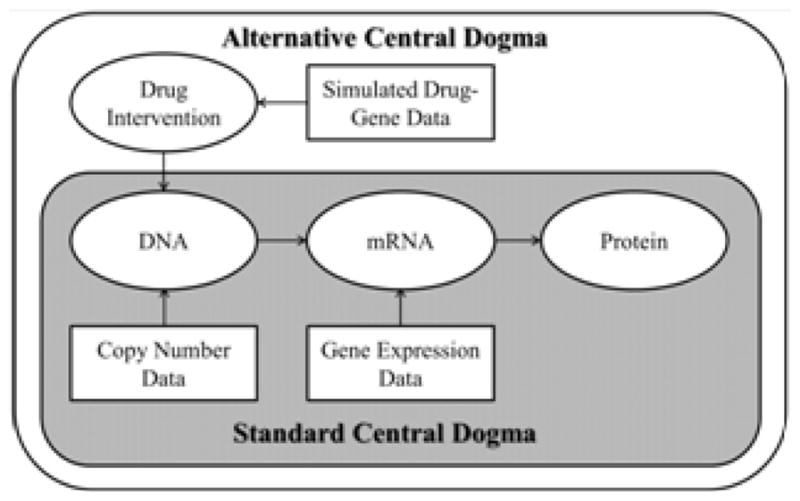
Comparison of the PARADIGM standard central dogma with an alternative dogma which represents a drug induced re-wiring of the network. The drug intervention propagates through the network based on an inferred interaction at the DNA node as a surrogate for its actual influence on protein activity.
2.3. PARADIGM Model
Briefly, PARADIGM is a factor-graph-based approach which quantifies the activity of a gene given a pathway diagram and dataset of observations8. For the model proposed here, SuperPathway was used to define this pathway diagram where each gene, protein, or process is connected by a series of factors which encode the probabilistic constraints between variables7, 15. Each entity in the pathway infers its activity from a set of nodes which define an internal set of rules for how these data types interact to assign a value to the pathway entity. Nodes for DNA and mRNA connect to the active protein node which then passes information through the entire pathway diagram via the dependencies encoded in the factors. The DNA and RNA nodes of each gene in the pathway are assigned values as a function of the copy number and gene expression data respectively to include biological information from the cell lines. For each gene, PARADIGM is capable of integrating these diverse –omic data types to compute an inferred pathway level (IPL) for each gene in the pathway. These IPL scores were computed using a belief-propagation algorithm on the factor graph diagram of the pathway. Each score represents a log-posterior odds (LPO) ratio of the state of a pathway entity given the observed data. Positive IPLs correspond to an entity being active in a tumor relative to normal tissue and negative to inactivity7, 8.
2.4. Drug Intervention Simulation
DIRPP exploits a versatile feature of PARADIGM which allows the user to define a drug induced re-wiring for a gene in a pathway. As designed, PARADIGM is capable of integrating DNA methylation data by including an extra node in a gene’s normal wiring connected at the DNA node7. The current algorithm utilized the DNA methylation feature to encode the action of a drug on that particular gene’s regulatory structure (Figure 2). A drug’s mechanism of action was retrieved from drugbank and selleckchem databases, which provide a list of genes (proteins) the drug targets16–18. A matrix of genes that correspond to a drug intervention was then defined. The edge connecting the intervention node to the DNA node encoded a factor which signaled a downregulation to the gene (similar to the standard use of methylation). Only genes listed in this intervention matrix had the extra node added to their wiring diagrams. In principle, the edge connecting the intervention node to the DNA node could be changed to act in an amplifying manner for an agonist.
To assess the significance of a drug intervention, two runs of the PARADIGM algorithm were completed: one with copy number and gene expression data, the other with the addition of a third data type, the simulated drug intervention, with each run generating a matrix of IPL scores. The two resulting matrices of IPL scores were then compared on a per-cell line-per-gene basis using a paired t-test to calculate a p-value for that cell line. The cell lines were ranked in order of largest to smallest p-value corresponding to a ranking of least to most sensitive cell lines for a given drug.
2.5. Validation
To validate our approach, analysis of the CGP and CCLE data were independently performed by calculating the accuracy and precision statistics for each ranking. Accuracy assesses the algorithm’s overall performance for distinguishing between sensitive and resistant cancer cell lines while precision is used to assess the positive predictive value of the algorithm at identifying drug resistant cancer cell lines.
| (1) |
| (2) |
A ranking of cell lines by p-value was first constructed using the results of the t-test. This ranking by p-value was compared to the actual ranking by IC50 value measuring drug sensitivity. The accuracy and precision statistics were calculated by defining a cutoff in the ranking where the cell lines change from primarily resistant (IC50>1) to primarily sensitive (IC50<0.1), where intermediately sensitive lines (0.1< IC50<1) were treated as resistant. There were generally more drug-resistant cancer cell lines than sensitive ones and for some drugs; no sensitive cell lines were available for comparison. For validation of these difficult drugs we defined our cutoff for drug resistance detection at an IC50 value of 8μM, where we considered values greater than 8μM to correspond to highly drug resistant cell lines and everything below to moderately drug resistant cell lines. The CGP did not have any ovarian, breast, or endometrial cancer cell lines with IC50 less than 8μM for Erlotinib. We then calculated DIRPP’s accuracy (1) and precision (2) for each dataset for each of the three cancers studied individually and together as a whole. Previous studies have indicated 78% accuracy as being a very high level, others have used a concordance index and set the cutoff at 0.6 to measure correlations12,13. We chose to use accuracy and precision cutoffs at 0.67 to define an “acceptable” level of validation between these two cutoffs.
3. Results
3.1. Drug Simulations
We simulated drug interventions for each of the drugs in Table 1 by defining mechanism-specific drug intervention files. The drug’s mechanism of action, the genes it targets, was propagated through the cancer cell line’s network via a drug intervention node coded in the PARADIGM algorithm’s rewiring for each effected gene. Four interventions were simulated for each drug in each dataset, one which included all breast, ovarian, and endometrial cancer cell lines as one cohort, and three other simulations for each cancer-type individually.
The ranking of cell lines by p-value was compared to the ranking of cell lines by IC50 for each drug and the accuracy and precision of that ranking was assessed using the cutoffs for resistance and sensitivity either by IC50 value, or by the highly-moderately resistant cutoff previously described. Certain ovarian cell lines have been shown to be hypermutated or were potentially mislabeled as they are more similar to other tumor types19. These cell lines were excluded to ensure the consistency of this analysis for only breast, endometrial, and ovarian cancer. Except for AZD0530, the overall response of all drugs across both datasets was predicted within 67% average accuracy or greater, with most being predicted with over 75% accuracy (Table 2). DIRPP predicted the resistance of cell lines with precision of 0.67 or greater for all drugs except for Paclitaxel. Some drugs such as Irinotecan performed distinctly different between datasets (Table 2). DIRPP was able to detect resistant cell lines with an overall precision of 0.81 across all datasets (Table 3). Across all cancers studied combined DIRPP performed with a precision of 0.78 and accuracy of 0.73. Ovarian cancer drug response was predicted better than the other cancers with an overall precision of 0.81 and accuracy of 0.79 (Table 3).
Table 2.
Precision and accuracy statistics for each drug across all cancer types combined by dataset and overall.
| CGP Data | CCLE Data | Overall | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Resistance Precision | Accuracy | Resistance Precision | Accuracy | Average Precision | Average Accuracy | |
| 17AAG | 0.88 | 0.77 | 0.72 | 0.59 | 0.80 | 0.68 |
| AZD0530 | 0.73 | 0.57 | 0.62 | 0.58 | 0.67 | 0.58 |
| AZD6244 | 0.84 | 0.76 | 0.90 | 0.81 | 0.87 | 0.79 |
| Erlotinib | - | - | 0.88 | 0.80 | 0.88 | 0.80 |
| Irinotecan | 0.44 | 0.63 | 0.91 | 0.88 | 0.68 | 0.75 |
| Lapatinib | 0.90 | 0.75 | 0.72 | 0.58 | 0.81 | 0.67 |
| Paclitaxel | 0.80 | 0.83 | 0.43 | 0.60 | 0.61 | 0.72 |
| PD0325901 | 0.79 | 0.69 | 0.93 | 0.86 | 0.86 | 0.78 |
| Sorafenib | 1.0 | 1.0 | 0.71 | 0.59 | 0.86 | 0.80 |
Table 3.
Precision and accuracy statistics by dataset for all cancer types combined and by cancer type individually
| CGP Data | CCLE Data | Overall | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Resistance Precision | Accuracy | Resistance Precision | Accuracy | Precision | Accuracy | |
| All Cancers | 0.81 | 0.78 | 0.76 | 0.70 | 0.78 | 0.73 |
| Breast | 0.83 | 0.80 | 0.73 | 0.67 | 0.78 | 0.73 |
| Ovarian | 0.75 | 0.81 | 0.84 | 0.78 | 0.81 | 0.79 |
| Endometrial | 0.83 | 0.83 | 0.74 | 0.65 | 0.76 | 0.70 |
3.2. Mechanisms of Drug Resistance
Once the cell lines were classified as either sensitive or resistant to a drug we performed gene set enrichment analysis (GSEA) by drug response phenotype to uncover the biological pathway mechanisms driving drug resistance. For this analysis we required cell lines with IC50 values greater than 1 or less than 0.1 to define, resistant and sensitive, respectively. Only 17AAG, Irinotecan, Paclitaxel, and PD0325901 had sufficiently diverse drug sensitivity profiles to classify cell lines using the above definition in order to perform GSEA. Each of these drugs has a distinct mechanism of action and no overlapping molecular targets. Despite this, we were able to identify several signaling pathway mechanisms that these cell lines shared related to drug resistance.
We ran GSEA using the IPL values generated by PARADIGM using the simulation that combined copy number and gene expression data. Permutation analysis of the phenotypes (sensitive or resistant) was used to judge significance. Pathways which had nominal p-values less than 0.05 were selected for further comparison across drugs. There was a common activation of PDGF signaling associated with resistance to PD0325901, Paclitaxel, and Irinotecan in the resistant endometrial, breast, and ovarian cancer cell lines. This confirms previous work which associates PDGF upregulation with Paclitaxel resistance in breast and ovarian cancer20, 21 and suggests that the genetically similar endometrial cancer1 may also share this mechanism of drug resistance. Irinotecan and Paclitaxel shared 9 mechanisms of resistance with each cancer. Paclitaxel also shared 8 mechanisms of drug resistance with 17AAG when comparing all; however none of these pathways overlapped between 17AAG and Irinotecan. Our results suggest that resistance to Paclitaxel is closely tied to that of Irinotecan and 17AAG.
We were able to identify common pathways which confer drug sensitivity in all three cancers to multiple drugs. 17AAG shared 7 sensitivity based biological pathways with Irinotecan and 4 with Paclitaxel. This is contrasted by the single biological pathway Paclitaxel and Irinotecan share associated with drug sensitivity. We can then begin to compare drugs on the basis of which biological pathways play a role in conferring drug sensitivity or resistance. Hierarchical schemas of drug similarity are illustrated in Figures 3 and 4.
Figure 3.

Number of common pathways implicated in drug resistance between 17AAG, Irinotecan, Paclitaxel, and PD0325901 (1,441 total pathways tested).
Figure 4.

Number of common pathways implicated in drug sensitivity between 17AAG, Irinotecan, Paclitaxel, and PD0325901 (1,441 total pathways tested).
Our results suggest that cancer cell resistant to Paclitaxel is likely to also resist 17AAG and Irinotecan. As Irinotecan and 17AAG appear to have quite distinct biological pathway mechanisms of action for drug resistance, it is less likely that a cancer cell line resistant to one will be resistant to another (Figure 3). On the other hand, as sensitivity to Irinotecan has some pathway similarities to sensitivity to 17AAG it is more likely that a cell line that is sensitive to one is sensitive to another (Figure 4). These results may suggest that a good starting point for the repurposing and repositioning of drugs is to classify them by their impact on the biological network of a cancer cell rather than by their distinct mechanism of action.
4. Discussion
Though there are some success stories, many clinical biomarkers have had limited impact7, and a shift is needed to more global explanations of disease and drug response phenotypes. Since a single gene is often involved in multiple pathways, it is difficult to assess the significance of a given genetic aberration without considering the broader context in which the dysregulation occurs7. In addition, many cancer patients have multiple genetic aberrations and multiple signaling pathways may be dysregulated and associated with drug resistance. However, the current analysis suggests that these signaling pathways related to drug resistance are shared by four drugs with completely different mechanisms of action. These results suggest that grouping drugs for treatment, repurposing, and repositioning by shared mechanisms which govern resistance and sensitivity may be more accurate than grouping them by the specific genes they appear to target. Such classification allows for simplification of the drug repurposing and repositioning process by making it a simple matter of counting and comparing biological pathway mechanisms.
DIRPP is a novel pipeline for classifying cell lines by drug sensitivity and for elucidating biological pathway mechanisms that drive drug response. PARADIGM forms the foundation of DIRPP and thus its scalability and comparability to other pathway based ones will be similar to that of PARADIGM. PARADIGM has been utilized in the hallmark TCGA papers and is an integral part of their pipeline easily scaling up to over 400 patient samples1–3. When compared to other pathway based methods, PARADIGM was shown to perform better compared to other methods7,9. Though PARADIGM has been used to compare separate groups of patients known to respond better to a selected therapies than others, it has never been used in a discovery manner as presented here. The DIRPP pipeline thus represents a novel extension of PARADIGM’s capabilities. Though we chose to connect the drug-intervention node to the DNA entity in PARADIGM’s central dogma, many drug targets are proteins. This could be reflected in future refinements of the method by modifying the connecting point for the drug-intervention node.
DIRPP performs comparably well on two independent datasets and is generalizable to other datasets with gene expression, copy number data, or both. The high predictive power of DIRPP across multiple drugs and cancers makes it a versatile tool to aid pre-clinical research. Further work to assess the utility of DIRPP is required. The CCLE and CGP datasets contain cell lines for ovarian cancer which were not screened for drug response and do not have IC50 values. Once a robust ranking of cell lines with known drug response is built and the accuracy is validated, DIRPP can be used to classify the unknown cell line(s) as either sensitive or resistant to a particular drug. Further analysis will utilize the –omic data for tumor samples from TCGA and other publically available datasets to predict drug response phenotypes by applying the knowledge learned and methods developed from the current analysis.
The complexity of cancer presents many challenges to predicting therapeutic effectiveness if using individual biomarkers alone. Pathway level approaches such as DIRPP bring us one step closer to the goal of personalized medicine by utilizing complex –omic data and knowledge on biological pathways in order to robustly identify drug sensitivity.
Acknowledgments
We would like to thank the investigators of the cancer genome project and cancer cell line encyclopedia for their comprehensive data which enabled this analysis. We especially thank Dr. Steve Benz for his assistance with running PARADIGM and the entire team at Five3genomics for their support. This work was supported in part by the Case Comprehensive Cancer Center Support Grant NCI 5P30 CA043703.
References
- 1.TCGA. Nat. 2013;497:67–73. [Google Scholar]
- 2.TCGA. Nat. 2011;474:609–615. [Google Scholar]
- 3.TCGA. Nat. 2012;490:61–70. [Google Scholar]
- 4.Garnett MJ, et al. Nat. 2012;483(7391):570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barretina J, et al. Nat. 2012;483(7391):603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Subramaian A, et al. PNAS. 2005;102:15545–15550. [Google Scholar]
- 7.Vaske CJ, et al. Bioinf. 2010;26:i237–i247. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng S, et al. Bioinf. 2012;28:i640–i646. doi: 10.1093/bioinformatics/bts402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varadan V, et al. IEEE Sig Proc. 2011:35–49. [Google Scholar]
- 10.Nibbe RK, et al. PLOS Comp Bio. 2009;6(1) [Google Scholar]
- 11.Nibbe RK, et al. Journal of Comp Bio. 2010;18(3):263–281. doi: 10.1089/cmb.2010.0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papillon-Cavanagh S, et al. J Am Med Inform Assoc. 2013;20:597–602. doi: 10.1136/amiajnl-2012-001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Napolitano F, et al. Journal of Chemiinformatics. 2013;5:30. [Google Scholar]
- 14.McCarthy JJ, et al. Science Translational Med. 2013;5(189):1–17. doi: 10.1126/scitranslmed.3005785. [DOI] [PubMed] [Google Scholar]
- 15.Heiser LM, et al. PNAS. 2011;109:2724–2729. [Google Scholar]
- 16.Knox C, et al. Nucleic Acids Res. 2011;39(Database issue):D1035–41. doi: 10.1093/nar/gkq1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wishart DS, et al. Nucleic Acids Res. 2008;36(Database issue):D901–6. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wishart DS, et al. Nucleic Acids Res. 2006;34(Database issue):D668–72. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Domcke S, et al. Nat Commun. 2013;4:2126. doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Isonishi S, et al. Oncology Reports. 2007;18(1):195–201. [PubMed] [Google Scholar]
- 21.Weigel MT, et al. Annals of Oncology. 2013;24(1):126–133. doi: 10.1093/annonc/mds240. [DOI] [PubMed] [Google Scholar]