

Abstract
Taphrina fungi are biotrophic plant pathogens that cause plant deformity diseases. We sequenced the genomes of four Taphrina species—Taphrina wiesneri, T. deformans, T. flavorubra, and T. populina—which parasitize Prunus, Cerasus, and Populus hosts with varying severity of disease symptoms. High levels of gene synteny within Taphrina species were observed, and our comparative analysis further revealed that these fungi may utilize multiple strategies in coping with the host environment that are also found in some specialized dimorphic species. These include species-specific aneuploidy and clusters of highly diverged secreted proteins located at subtelomeres. We also identified species differences in plant hormone biosynthesis pathways, which may contribute to varying degree of disease symptoms. The genomes provide a rich resource for investigation into Taphrina biology and evolutionary studies across the basal ascomycetes clade.
Keywords: Taphrina comparative genomics, divergence clusters, fungi aneuploidy
Introduction
Many different fungal pathogens induce deformities of host plants. One of the most well-known examples is the tumor-like plant structure caused by Taphrina species. The disease symptoms of deformity vary depending on the plant organ where infection occurs and which Taphrina species is the source of infection (Mix 1935). Infection symptoms include leaf spots, leaf curl, deformed fruits, and witches’ brooms. Taphrina deformans, which causes leaf curl of peach, is the most economically important species and has been extensively studied, and its genome sequence is now publicly available (Cissé et al. 2013). Another Taphrina species of interest is T. wiesneri, which causes serious damage to the culturally important “Sakura" (Cerasus spp.) in Japan. This fungus causes witches’ brooms on the twigs, loss of flowering, and early defoliation on Cerasus species. In particular, Cerasus × yedoensis cv. Somei-Yoshino, the most popular ornamental species in Japan, is highly susceptible to infection (Komatsu et al. 2010) and is endangered in some localities. The witches’ brooms is thought to be induced by plant hormones produced by systemically distributed perennial intercellular mycelium in twigs, hence the different disease symptoms induced by Taphrina species may be attributed to their ability to synthesize and modulate different plant hormones. In addition, T. flavorubra is the causal agent of deformed fruits or plum pocket on plums (Rodrigues and Fonseca 2003). Infected fruit increases abnormally in size, deforms, and becomes spongy. On the other hand, T. populina induces less deformity than others; this fungus infects Populus trees and causes only minor leaf spots (Rodrigues and Fonseca 2003).
The four Taphrina species are part of the order of Taphrinales, in the class Taphrinomycetes. Taphrinomycetes belong to the subphylum Taphrinomycotina, which includes the model fission yeast Schizosaccharomyces pombe and the human pathogen yeast-like fungus Pneumocystis jirovecii. Together they form a monophyletic group basal to the rest of ascomycetes (Liu et al. 2009). The genus Taphrina comprises about 28 species, which are distributed worldwide. Currently, all known Taphrina species are dimorphic: The mycelial (filamentous) state is strictly plant parasitic, whereas the yeast state is saprophytic and can be cultured on artificial media (Mix 1949; Rodrigues and Fonseca 2003). The yeast state of the life cycle begins with budding of ascospores discharged from infected leaf surfaces usually around late spring or early summer. These yeast-state cells make contact with host plants and switch to the parasitic mycelial state under favorable conditions. The parasitic phase is the predominant mode of the life cycle of these species, where the hyphae penetrate the cuticle and continue to invade the host tissue between the epidermal cells until they reach the parenchyma cells below (Mix 1935). Although all of the disease symptoms were thought to be attributed to the plant hormone-like substances such as indole-3-acetic acid (IAA) produced by Taphrina species, the exact mechanisms remain unclear. Two genes that are known to be required for IAA production for Ustilago maydis are also present in T. deformans (Cissé et al. 2013), suggesting that T. deformans and other Taphrina species might have a similar route of IAA biosynthesis.
Here, we report genome sequences of four major Taphrina species: T. wiesneri, T. flavorubra, T. populina, and a much-improved assembly of T. deformans. We have performed a comparative analysis with seven other fungal species and identified features unique to each Taphrina species that may be linked to their pathogenicity. This study provides valuable insights into the mechanisms underlying host specificity and different infection symptoms of Taphrina species.
Materials and Methods
Strain Collection and Condition
Taphrina deformans strain JCM 22205, T. wiesneri strain JCM 22204, T. populina strain JCM 22190, and T. flavorubra JCM 22207 were obtained from RIKEN BRC-JCM (Tsukuba, Japan). The isolation source of the strains used, host range, distribution, and disease symptom of the four Taphrina species are listed in supplementary table S1, Supplementary Material online.
DNA/RNA Preparation and Sequencing
Yeast-like fungi were cultivated in YPD medium (0.5% (w/v) yeast extract, 1% peptone, and 0.9% glucose) at 25 °C for 3 days. The cells were collected from the medium by centrifugation (for 5 min at 3,000 × g) and were suspended in cell-wall lysis buffer (0.1 M McILvaine buffer containing 270 U/ml Westase (Takara Bio) and 0.5 M sodium tartrate). After incubation at 30 °C for 1.5 h, genomic DNA was extracted using Genomic-tip 100/G (Qiagen) according to manufacturer’s instruction. One microgram of the DNA was used to construct standard 300 bp libraries using the TruSeq DNA Sample Preparation Kit with the standard protocol (Illumina). Libraries were sequenced on an Illumina HiSeq2000 following the manufacturer’s recommended protocol to produce 100-bp paired end reads (https://icom.illumina.com/, last accessed April 4, 2014). RNA was extracted from yeast cells grown on YPD agar medium using Sepasol-RNA I Super (Nacalai Tesque) after cell disruption by tungsten beads (φ0.1 mm). RNA libraries were prepared with an Illumina TruSeq RNA Sample Prep Kit and sequenced (100-bp paired end) on an Illumina HiSeq2000 sequencer using the standard protocol (Illumina).
Nuclear and Mitochondrial Genome Assembly
The initial assemblies of Taphrina genome sequences were produced using the MaSuRCA assembler (Zimin et al. 2013) from Illumina reads. These assemblies were further improved using the PAGIT approach (Swain et al. 2012). Gapfiller (Boetzer and Pirovano 2012) and IMAGE (Tsai et al. 2010) were used to close gaps and extend contig ends. The original Illumina reads were then remapped to the assemblies, and a separate assembly was produced with the unmapped Illumina reads using Velvet (Zerbino and Birney 2008) and further scaffolded into the main assemblies using SOPRA (Dayarian et al. 2010). Finally, iCORN v0.97 was run for three iterations to improve the consensus quality of the assemblies, and potential misassemblies were detected and broken using REAPR (Hunt et al. 2013). The CEGMA pipeline (Parra et al. 2007) was run to assess the genome completeness of these assemblies. The statistics of the final assemblies is shown in table 1.
Table 1.
Taphrina Genome Assemblies
| Species Name | Strain | Assembly Size (bp) | Number of Scaffolds | Average Scaff Size (kb) | Largest Scaff Size (kb) | N50 (kb) | N50 (Number) | N90 (kb) | N90 (Number) | Full Cegma (%) | GC Content (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Taphrina wiesneri | JCM 22204 | 13,070,656 | 225 | 58.1 | 584.8 | 304.1 | 17 | 71.6 | 47 | 98.79 | 47.8 |
| T. deformans | JCM 22205 (= NRRL Y-17787) | 13,778,166 | 529 | 26 | 398.7 | 182.6 | 28 | 25.9 | 97 | 98.79 | 48.9 |
| T. deformansa | PYCC 5710 (= CBS 356.35) | 13,364,053 | 394 | 33.9 | 244.1 | 71.9 | 61 | 17.5 | 203 | 98.7 | 49.5 |
| T. populina | JCM 22190 (= CBS 337.55) | 11,999,082 | 335 | 35.8 | 892.8 | 172.6 | 20 | 35.7 | 76 | 98.39 | 47.3 |
| T. flavorubra | JCM 22207 | 15,727,932 | 865 | 18.2 | 480.9 | 177.6 | 30 | 7.9 | 149 | 98.39 | 48.9 |
aPublished by Cissé et al. (2013).
Mitochondrial genome reads were assembled separately using MITObim (Hahn et al. 2013). Initial seeds were identified from the nuclear genome assemblies based on BLAST analyses using S. pombe mitochondrial genes as queries. Mitochondria assemblies were made by iterative mappings of the Illumina short reads to the initial seeds using kmers of 31 and 45. The MITOS web server (Bernt et al. 2013) was used for annotation of obtained assemblies.
Genome Feature Analysis
Transposable elements (TEs) in the assembly of the four Taphrina species were identified based on output from two repeat detection programs RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html, last accessed April 4, 2014) and TransposonPSI (http://transposonpsi.sourceforge.net/, last accessed April 4, 2014). UCLUST (Edgar 2010) was used to cluster these putative sequences (with 80% identity) to generate consensus sequences for a nonredundant library of repeat sequences. Annotations of these sequences were based on output of both programs and BLAST results from the National Center for Biotechnology Information (NCBI) nonredundant database. RepeatMasker (v3.2.8, http://www.repeatmasker.org, last accessed April 4, 2014) was used to calculate the distribution of each repeat and its abundance. Custom Perl scripts were used to choose the best match from overlapping matches in RepeatMasker output to avoid calculating the same region more than once. Illumina reads were remapped to the Taphrina genome assemblies using SMALT (v 0.7.5, available at http://www.sanger.ac.uk/resources/software/smalt/, last accessed April 4, 2014). The summary statistics of the read mappings is shown in supplementary table S9, Supplementary Material online). The GC content of 2-kb nonoverlapping windows in the assemblies was calculated using BEDtools (Quinlan and Hall 2010). Duplicates were called using GATK (McKenna et al. 2010), and median sequence coverage of 10-kb nonoverlapping window was calculated from the output of samtools (Li et al. 2009) and with a combination of custom perl and R scripts.
Gene Prediction and Functional Annotations
RNAseq reads were aligned to the respective Taphrina genome assemblies using Tophat (v2.0.4 using parameters –mate-std-dev 50 -a 6 -i 10 -I 10000 –min-segment-intron 10 –max-segment-intron 10000; summary of the mappings is shown in supplementary table S9, Supplementary Material online) (Kim et al. 2013), and the mappings were converted into intron hints. Additionally, cufflinks (v2.0.1) (Trapnell et al. 2012) was used to assemble transcript fragments, and this information was converted into exon hints. Two additional sources of hints were available: The published T. deformans PYCC5710 proteome (Cissé et al. 2013) was transferred using RATT (Otto et al. 2011), and 2,863 T. wiesneri expressed sequence tag (EST) sequences were aligned using BLAT (Kent 2002). Initial gene models were first predicted based on these hints using Augustus (initial parameters were generated using the etraining script; Stanke et al. 2006). About 500 genes were curated for each Taphrina species using Artemis (Carver et al. 2008), and these models were used to train species-specific parameters in Augustus. The final sets of gene models were predicted based on trained parameters and concatenated hints in Augustus. Using T. wiesneri as an example, exon prediction sensitivity was increased from 76.6% to 84.6%, but specificity was decreased from 85.2% to 83.2% when comparing the repredicted gene models against the manual curated models. Signal peptides and transmembrane domains were detected using SignalP 4.0 (Petersen et al. 2011) and TMHMM 2.0 (Krogh et al. 2001), respectively. Codon usage and GC content of the codons’ third position was calculated using codonW (http://sourceforge.net/projects/codonw/, last accessed April 4, 2014). Functional annotations were predicted as means of GO terms by argot2 (Falda et al. 2012) and by identifying protein domains against Pfam v27.0 (Punta et al. 2012) using pfam_scan.pl (ftp://ftp.sanger.ac.uk/pub/databases/Pfam/Tools, last accessed April 4, 2014).
The MEROPS server was used to identify Taphrina putative peptidases. The peptidase candidates were derived from MEROPS batch BLAST (Rawlings et al. 2006). The candidates were manually examined in terms of similarity (E-value cutoff 1 × 10−10) to MEROPS proteins and presence of all catalytic sites. CAZymes (carbohydrate active enzymes) were detected using the dbCAN HMMer-based classification system (Yin et al. 2012) with an E-value threshold of 1 × 10−5, alignment length cutoff of 80 amino acids and covered fraction threshold of 0.3.
Ortholog Clustering, Phylogeny, and Synteny Analysis
To construct a representative set of fungal gene families, nonredundant proteomes of Saccharomyces cerevisiae, Sclerotinia sclerotiorum, Fusarium graminearum, Neurospora crassa, U. maydis, and P. jirovecii were downloaded from Uniprot (version November 6, 2013), and S. pombe was downloaded from PomBase (Wood et al. 2012). Genes were clustered based on BLASTP (E value of 1 × 10−5) results and OrthoMCL (inflation parameter of 1.5, (Li et al. 2003). Amino acid alignment using MAFFT (Katoh and Standley 2013) of 75 single-copy orthologs with less than 10% alignment gaps were concatenated, and the distance matrix of the merged alignment was calculated using ProtTest (Abascal et al. 2005). The phylogenetic tree was constructed by maximum likelihood using RAxML (Stamatakis 2006) with 100 bootstrap replicates. From the OrthoMCL and Pfam output, we identified gene families that were lost and expanded in different positions along the global phylogeny leading to the Taphrina species. GO enrichments were carried out on these families using TopGO (version 2.10.0; Alexa and Rahnenfuhrer 2010).
The highly contiguated nature of the Taphrina spp. genome assemblies allowed for an analysis of scaffold synteny. To visualize synteny relationships between Taphrina species, dotplots of T. wiesneri versus the other three species were produced using the MUMmer 3.0 suite (Kurtz et al. 2004), and pairwise syntenic blocks were inferred using DAGchainer (Haas et al. 2004). To determine gene order rearrangements between T. wiesneri and S. pombe, we retrieved one-to-one orthologs between the two species from OrthoMCL output and visualized using circos (Krzywinski et al. 2009).
Results and Discussion
The Genomes and Gene Features of Taphrina Fungi
The nuclear genomes of T. wiesneri, T. deformans, T. populina, and T. flavorubra were selected for sequencing on the basis of the very different symptoms exhibited by their many Prunus, Cerasus, and Populus hosts worldwide (fig. 1A, supplementary table S1, Supplementary Material online). The Taphrina genomes were sequenced using Illumina technology with up to 450 × depth of coverage obtained. CEGMA analysis suggests that assemblies are 98.4–98.8% complete. The assemblies range from 12.0 to 15.7 Mb (table 1), similar to the 12.5-Mb genome of fission yeast S. pombe. Using transcriptomic evidence such as RNA Illumina sequenced reads and ESTs to guide de novo gene prediction, we identified 6,403–7,563 putative gene models per genome (table 2). Our T. deformans assembly is on average 99.7% identical with the published genome sequence of T. deformans PYCC5710 (Cissé et al. 2013) but is twice as contiguated. Similarly, the Taphrina mitochondrial genomes were assembled into single scaffolds ranging from 56 to 60 kb (supplementary fig. S1, Supplementary Material online), which is bigger than that reported for S. pombe (19.4 kb) (Bullerwell et al. 2003) but still smaller than Sa. cerevisiae (85.8 kb) (Foury et al. 1998). In the most contiguated case of T. wiesneri, the mitochondrial genome was assembled into a single sequence of 58,215 bp without any gaps, containing 39 genes (supplementary fig. S1A, Supplementary Material online), consisting of 14 proteins, 2 ribosomal RNAs, and 23 transfer RNAs. Our mitochondrial assembly of T. deformans is 56,006 bp in size, which is 20 kb bigger and identified 16 more tRNA genes than previously reported (Cissé et al. 2013). The gene orders of the four mitochondrial genomes are in almost perfect synteny to each other, with only minor differences in the order of tRNAs (supplementary fig. S1B, Supplementary Material online). Together these assemblies allow us to build upon what is already known about Taphrina species using a comparative genomics approach.
Fig. 1.—

Taphrina infection symptoms and phylogeny. (A) Leaf curl (a), witches’ broom (b), leaf spots (c), and deformed fruits (d) that were caused by four different Taphrina species indicated by arrows. (a) Image no. 5435623: Yuan-Min Shen, Taichung District Agricultural Research and Extension Station, Bugwood.org; (c) image no. 3046084: Theodor D. Leininger, USDA Forest Service, Bugwood.org. (B) A maximum-likelihood phylogeny of four Taphrina species and seven other fungi from 75 single copy orthologs. Blue dots indicate 100% bootstrap support.
Table 2.
Taphrina Gene Models Statistics
| Species Name | Number of Genes | Gene Density (Genes/Mb) | Mean Exon per Gene | Mean Exon Length (bp) | Mean Intron Length (bp) |
|---|---|---|---|---|---|
| Schizosaccharomyces pombe | 5,123 | 554 | 2.0 | 1,015 | 81 |
| Pneumocystis jirovecii | 3,898 | 481 | 3.7 | 211 | 61 |
| Taphrina wiesneri | 6,403 | 490 | 2.0 | 691 | 48 |
| T. deformans | 6,433 | 467 | 2.0 | 689 | 48 |
| T. populina | 6,481 | 540 | 2.0 | 656 | 51 |
| T. flavorubra | 7,563 | 481 | 1.9 | 704 | 50 |
The Taphrina genomes contain low numbers of TEs, with the largest class being LTR retrotransposons, which occupy about 1% of the genomes (supplementary table S2, Supplementary Material online). This observation partly explains that the highly contiguated nature of Taphrina assemblies, where 50% of the genomes are present in scaffolds at least 172 kb in length in spite of the fact that only short-insert Illumina libraries were used. A loss of TEs has occurred in T. wiesneri when compared with the others, where only 0.4% of the genome has signatures to any types of TEs.
Phylogeny, Synteny, and Aneuploidy
To identify gene gain and loss in Taphrina species, orthologous relationships were established using OrthoMCL (Li et al. 2003) against seven other fungi (fig. 1B). A phylogeny based on 75 single-copy orthologs placed the Taphrina species into a monophyletic clade relative to other Taphrinomycotina members, such as S. pombe and the human pathogen P. jirovecii. Together they form a clade of early diverging ascomycetes (fig. 1B). Unlike the fission yeasts (Rhind et al. 2011), the Taphrina genomes exhibit a high degree of synteny (supplementary fig. S2, Supplementary Material online). For the most closely related species of T. wiesneri and T. deformans, single-copy orthologs have on average 86.2% amino acid identity and show a strikingly high gene colinearity of 89%. Gene order and chromosomal synteny have been lost when comparing with S. pombe (fig. 2), presumably because of the highly reorganized chromosome structure to retain centromere function in fission yeasts (Rhind et al. 2011). Interestingly, we observed disomy of the second largest scaffold (385 kb) in T. deformans and not the corresponding syntenic scaffolds in other Taphrina species (fig. 3). Although it remains to be clear whether this is proper aneuploidy or duplications of a large part of a scaffolds,.aneuploidy is common in fungi and plays a role in increased virulence and better survival when cells undergo severe natural or induced environmental conditions (Kwon-Chung and Chang 2012). For the genes that have functional annotations, GO term enrichment revealed that the genes are mostly enriched in organic acid metabolic process (supplementary table S3, Supplementary Material online) and very interestingly also “leaf development” on this scaffold. Additionally, Taphrina spp. encode a single copy of a phosphoinositide-interacting protein (Td_0030400.1) and PFS2 (Td_0031400.1) on this scaffold of which the homologs in Arabidopsis thaliana are involved in leaf senescence and control of flowering time (Henderson et al. 2005; Xing et al. 2008), respectively. The exact mechanism by which aneuploidies confer an advantage to T. deformans is unclear, as we found no allelic differences in the additional scaffold copy and the median expression level of the scaffold is similar to the rest of scaffolds (supplementary fig. S3, Supplementary Material online). It remains to be determined whether aneuploidy is also apparent in isolates of different Taphrina species and whether the genes contained in these scaffolds have a role in interactions of the fungi with their hosts.
Fig. 2.—
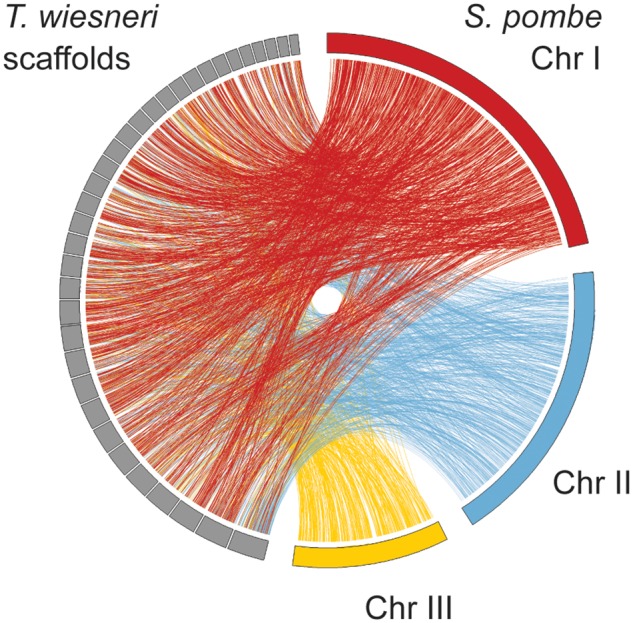
Lack of synteny between Taphrina wiesneri and Schizosaccharomyces pombe. One-to-one orthologs inferred from OrthoMCL (indicated by lines) connecting three S. pombe chromosomes against T. wiesneri scaffolds.
Fig. 3.—
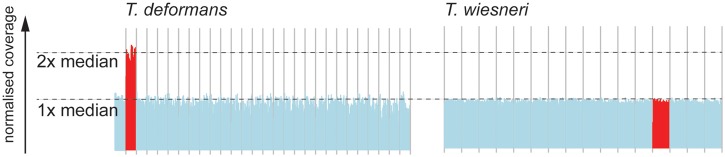
Distribution of normalized genome coverage on Taphrina wiesneri and T. deformans. Each vertical line indicates the median coverage of 100-kb windows normalized against the median coverage of either species. Even coverage was observed across the scaffolds of T. wiesneri, but in the case of T. deformans, the coverage was doubled across the second largest scaffold. Red color lines represent the coverage of such scaffold and the corresponding syntenic scaffold in T. deformans.
The average GC contents of the Taphrina genomes range from 47.3% to 48.9%, which is higher than that of compared with other Taphrinomycotina members. Density plots of GC content in 2-kb windows of Taphrina genomes (fig. 4A) show a single peak suggesting that the GC content is evenly distributed along the scaffolds. Neither extreme GC nor AT-rich isochores were detected as observed in other yeasts. Interestingly, GC content profiles of T. wiesneri and T. populina are more similar and lower compared with other two species, which are in different positions in the phylogeny (fig. 4A). Consistent with our findings, T. deformans PYCC5710 also shows a higher average GC content. One of the major determinants of GC content in fungi is that more highly expressed genes preferentially use a set of codons that have particular GC biases (Bennetzen and Hall 1982). We first assessed codon usage by calculating the effective number of codons (ENC) and GC content at the third nucleotide positions (GC3) of codons across the four Taphrina proteomes. A gene that has a low ENC values indicates greater biases in codon usage, and we found the Taphrina genes show ENCs that were negatively correlated with higher expression values (fig. 4B, P value < 0.001). Additionally, we found that the GC3 was positively correlated with gene expression levels in all four Taphrina species (P value < 0.001). The interplay between codon usage and GC content is apparent in Taphrina species and is consistent with findings in other fungi (Hershberg and Petrov 2009). The two sets of GC profiles different to the Taphrina phylogenetic relationship may suggest similar gene expression profiles due to overlap in host environment during speciation of Taphrina genomes.
Fig. 4.—

(A) Distribution of GC content in Taphrina species and (B) scatterplot showing relationship between codon usage (ENC) and expression count (FPKM) in Taphrina wiesneri.
Taphrina Proteome Specializations
We sought to identify specialized and expanded gene families in Taphrina species by first assigning Pfam domains to identify any genes with new combination of protein domains. Pairwise comparisons of domain numbers between the four Taphrina species show no significant differences, suggesting that the repertoire of annotated genes is similar across Taphrina species. Next, we sought to identify clusters of genes that are highly diverged within Taphrina spp. within syntenic regions, which have been shown to associate with virulence strength in other pathogenic fungi (Schirawski et al. 2010). Our analyses revealed a total of 58 regions containing 3–10 highly diverged genes adjacent to each other in all possible pairwise comparisons of four Taphrina species. Interestingly, the largest divergence cluster is enriched with genes encoding secreted products indicating their potential to be effectors, which are all located at scaffold ends with the T. wiesneri scaffold containing the telomeric repeat TTAGGG (fig. 5). This cluster is present in three out of four Taphrina species with no similar proteins found in T. populina assembly. Rapid expansion and diversification of gene families at subtelomeres in yeasts are not uncommon (Brown et al. 2010). It remains to be elucidated whether Taphrina utilize this region as the hotbeds for genomic innovation against plant immune response and the rapid diversification of secreted proteins as effectors that deals with specific hosts.
Fig. 5.—

Divergent cluster in Taphrina species. Genes with signal peptide predicted are marked in red. Shaded bars connect homolog pairs as suggested by DAGchainer and are marked in yellow if divergence is less than 5% quantile of all possible pairwise comparisons. Average similarities between all homologs of pairwise species comparisons and putative telomeres are denoted.
Predicted proteins from Taphrina genomes were clustered into a total of 10,356 gene families using OrthoMCL. Of those, 4,574 families contain at least one gene from Taphrina species and one from other fungi. A total of 1,324 families contain only genes from Taphrina members. For those that have functional annotation, topGO analysis suggests that the Taphrina species are particularly enriched in categories such as “regulation of transcription from RNA polymerase II promoter” (supplementary table S4, Supplementary Material online), which suggests that the mechanism for transcription regulation in Taphrina species may be diversified compared with other fungi. In each of the four Taphrina species, we also identified a single copy of perilipin, a protein responsible for lipid-based homeostasis in mammals, which was previously thought to be present only in pezizomycotinal ascomycetes (Bickel et al. 2009). Inhibition of perilipin like protein MPL1 expression in the insect pathogen Metarhizium anisopliae shows reduced ability to breach insect cuticles (Wang and St Leger 2007). This suggests certain specialized genes that were involved in lipid metabolism have been found in Taphrina genomes and may be part of infection strategy. In the remainder of the Results section, we focus on topics that are of particular interest to Taphrina biology.
Phosphorelay Signaling System through Taphrina Histidine Kinases
The two-component phosphorelay signaling system is one of the most extensively studied systems in fungi where they comprise histidine kinases (HKs), response regulators, and phosphotransfer proteins. Together these play important roles in osmosensing, oxidative stress, virulence, and even dimorphic switch between nonpathogenic to pathogenic states (Schaller et al. 2011). The model yeast Sa. cerevisiae and S. pombe encode one (SLN1) and three copies (MAK1/MAK2/MAK3) of HKs, respectively, and filamentous fungi in general possess an expanded repertoire of HKs allowing for different input into the phosphorelay. We identified 16–19 copies of HKs in the Taphrina genomes, a similar complement to the numbers found in filamentous fungi and more than any other fungal species in our selected proteomes (table 3; typically 1–19 copies in fungi as reviewed in Schaller et al. 2011). Taphrina species contain HKs with a range of domain combinations including both SLN1/MAK1 and MAK2/MAK3, suggesting that the two distinct osmo- and oxidative stress response pathways are present in Taphrina species, similar to fission yeast S. pombe (Quinn et al. 2011). Additionally, all Taphrina species possess a single copy of an HK that contains an MHYT domain (Galperin et al. 2001), which is present in many bacterial HKs but not in our nine selected fungal proteomes. Searching through NCBI nr database reveals very few copies present in only the Pezizomycotina yeasts. Together they suggest the two component phosphorelay system in Taphrina are adapted to sense a diverse set of environment responses with a repertoire of HKs similar to many filamentous fungi some of which are also plant pathogens (Schaller et al. 2011).
Table 3.
Summary of HKs Present in Taphrina and Seven Other Fungi Species Used in This Study
| HK Family | Fusarium graminearum | Sclerotinia sclerotiorum | Neurospora crassa | Saccharomyces cerevisiae | Schizosaccharomyces pombe | Ustilago maydis | Taphrina populina | T. flavorubra | T. deformans | T. wiesneri |
|---|---|---|---|---|---|---|---|---|---|---|
| RIM15 | 1 | 1 | 1 | 1 | 1 a | 1 | 1 | 1 | 1 | 1 |
| SSK1 | 1 | 2 | 2 | 1 | 1 | 4 | 1 | 1 | 1 | 1 |
| SKN7 | 1 | 0 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 |
| SLN1/MAK1 like | 14 | 18 | 12 | 1 | 1 | 6 | 14 | 15 | 15 | 15 |
| MAK2/MAK3 like | 1 | 1 | 1 | 0 | 2 | 0 | 2 | 4 | 4 | 4 |
| Total | 15 | 19 | 13 | 1 | 3 | 6 | 16 | 19 | 19 | 19 |
Note.—Pneumocystis jirovecii contains 0 copies in all.
aThrough literature search.
Host Infection Signaling by Mating and Dimorphism-Related Genes
Changing from yeast to mycelium phases is critical for Taphrina fungi to establish an infection in the host (Mix 1935). The triggers and molecular mechanisms of dimorphism in Taphrina remain unclear, but ploidy change by mating and/or host signals is likely to be important as described in U. maydis (Nadal et al. 2008). We identified homologs of most of the S. pombe sex-related genes in the four Taphrina species (supplementary table S5, Supplementary Material online). Those include genes involved in mating type switching (for instance, swi1, swi5, swi6, and swi10) and genes in the pheromone-activated MAPK signaling pathway (e.g., bry1 and bry2). Interestingly, Rik1-associated factor 1 (raf-1) and meiosis protein mei2 are absent only in T. populina. Mei2 is a RNA-binding protein that is important for induction of meiosis in S. pombe (Sherwood and Bennett 2009), suggesting that a Mei2-indipendent pathway may exist for activation of meiosis in T. populina.
Invasion through Cell Wall Degradation
The plant cell wall represents the primary barrier to most plant pathogens and CAZymes (carbohydrate active enzymes) that are able to break down plant cell wall are thus important for plant pathogens in establishing infection or in accessing nutrients. CAZymes are categorized into different classes and families in the CAZy database (http://www.cazy.org, last accessed April 5, 2014) including glycoside hydrolases (GH), glycosyl transferases, polysaccharide lyases, carbohydrate esterases, and carbohydrate-binding modules (Cantarel et al. 2009). The high diversity of CAZymes in Taphrina species suggest that they target a wide aspect of plant cell wall components including cellulose, hemicellulose, and pectin (supplementary table S6, Supplementary Material online). The genomes of the four Taphrina species contain 226–237 copies of putative of CAZymes, which is greater than other members in Taphrinomycotina but smaller than other plant parasitic fungi including the necrotrophic Sc. sclerotiorum and hemibiotrophic F. graminearum (supplementary table S6, Supplementary Material online). This observation is consistent with a previous study that suggests biotrophic fungi tend to have fewer CAZymes than necrotrophic or hemibiotrophic fungi (Zhao et al. 2013). In addition, Taphrina species also have GH enzymes that are active to fungal cell wall components including β-1,3 glucan and chitin. The four Taphrina species share similar repertories of CAZymes to each other but some species specific differences are apparent especially in T. populina. For instance, most of the GH families such as GH38, GH53, and GH79 are shared among all four species, but T. populina lacks some GH families that are present in all other three species (supplementary table S7, Supplementary Material online), which may relate to differences in host specificity. For instance, GH30, GH32, and GH10 genes are not found in T. populina but present in other two or three Taphrina species with signal peptides and may secret into host tissues to establish infections. In particular, GH10 proteins have ability to degrade xylan, a main component of secondary plant cell wall, which may relate to T. populina’s relatively weak symptom and localization on leaves.
Imbalance of Plant Hormones in Infected Tissues Caused by Pathogens
The distinctive characteristics of the plant symptoms caused by Taphrina species are deformation of plant tissues. These deformations are probably due to induced imbalances of plant hormones in the host tissues. In particular, production of auxin and IAA by fungi is thought to be causally related to the symptoms. An IAA biosynthesis pathway via indole-3-acetaldehyde (IAAld pathway: Tryptophan–Indole-3-pyrvic acid–IAAld–IAA) has been identified in the smut fungus U. maydis as the IAA main biosynthesis pathway. Each of the four Taphrina species contains orthologs of genes involved in this fungal IAA biosynthetic pathway; two orthologs of the tryptophan aminotranferases (encoded by Tam genes) and two orthologs of indole-3-acetaldehyde dehydrogenates (encoded by iad genes) (supplementary table S8, Supplementary Material online). This suggests that Taphrina spp. have a similar fungal IAA biosynthesis pathway to U. maydis (Reineke et al. 2008). Interestingly, putative orthologs of a plant YUC gene (Mashiguchi et al. 2011), which encodes flavin monooxygenase and is involved in plant IAA biosynthesis, were found in all four Taphrina species (one orthologs in T. populina and two orthologs in the three other species) (supplementary table S8, Supplementary Material online). Although Arabidopsis tryptophan aminotransferase gene (TAA1) ortholog is not present, tryptophan aminotransferase genes (Tam) are present in Taphrina species as stated above, suggesting the Taphrina species may have another IAA biosynthesis pathway (Tryptophan–Indole-3-pyrvic acid–IAA) resembling that used by the plants in addition to the fungal IAAld pathway (Mashiguchi et al. 2011). No orthologs of tryptophan monooxygenase genes or indole acetamide hydrolase genes were found in Taphrina species, suggesting the fungi did not have the indole-acetamide pathway that is present in bacteria (e.g., Agrobacterium). In addition, no orthologs of IAA conjugate-related genes (e.g., ILr1, IAR3, ILL1, ILL2, GH3, and UGT84B1) were present in Taphrina species, suggesting that auxin conjugates were not associated with IAA imbalance in host plants (Woodward and Bartel 2005).
Cytokinin also may be responsible for the deformation symptoms, and cytokinins production by Taphrina species has been reported (Johnston and Trione 1974; Sziraki et al. 1975). Each Taphrina species harbored putative orthologs of principal cytokinin biosynthesis genes, that is, tRNA-isopentenyltransferase (encoded by tRNA-IPT), cytokinin hydroxylases (encoded by CYP735A gene), and phosphoribohydrolases (encoded by LOG gene) (supplementary table S8, Supplementary Material online), confirming the ability of Taphrina species to produce cytokinin. We also identified putative orthologs of GA conversion-related genes (GA2ox, GA3oc, and GA20ox) and G4-desaturase (Hedden et al. 2001) in three of the four species (supplementary table S8, Supplementary Material online), suggesting that these genes may have a role in converting GAs in plant tissues during the colonization by Taphrina species. However, interestingly, no orthologs of those gibberellin-related genes were identified in T. populina, which may be related to the less severe symptom by the fungus than other three species.
Putative genes involved in abscisic acid (ABA) biosynthesis have been identified in T. deformans (Cissé et al. 2013), although the ABA production capability of the Taphrina species remains to be confirmed. In this study, we also found that each Taphrina species contain orthologs of the Botrytis cinerea ABA biosynthesis genes, for example, P450 monooxygenase gene (BcABA1, BcABA2) and short chain dehydrogenase/reductase gene (BcABA4) (Siewers et al. 2006) (supplementary table S8, Supplementary Material online). However, BcABA3 was not found in Taphrina species. The role of BcABA3 in ABA biosynthesis pathway is still unknown. In addition, each Taphrina species genome harbored orthologous copies of plant ABA biosynthesis genes, for example, xanthoxin dehydrogenase gene, 9-cis-epoxycarotenoid dioxygenase gene, and abscisic-aldehyde oxidase gene but lacked a neoxanthin synthase gene (supplementary table S8, Supplementary Material online). Together they suggest all Taphrina species studied can synthesize ABA but are unable to synthesize neoxanthin.
Conclusion
In addition to their status as important plant pathogens, the Taphrina yeasts are also of particular evolutionary interest as they form part of an ancient clade within the Ascomycete fungi. Unlike its related fission yeast genus Schizosaccharomyces, the Taphrina fungi display a high degree of gene colinearity. Our comparative analysis of the Taphrina genome structures has revealed biology that shows these organisms use various strategies similar to those used by yeasts to adapt to the host environment. For example, aneuploidy results in additional copies of genes that may have host-modulating roles, whereas secreted proteins that may be putative effectors are highly diverged between Taphrina species are located in subtelomeres. We have also highlighted a few potential areas that could follow. First, the most closely related Taphrina perilipin and MHYT HK orthologs in ascomycetes were found only in the Pezizomycotina subphylum but neither Taphrinomycotina nor Saccharomycotina, suggesting that the origin of these genes in Taphrina may be obtained via horizontal gene transfer. More genome sequences in the order of Taphrinales are needed to confirm this observation. Second, although inoculation and recovering of some Taphrina fungi in hosts still remain technically difficult, infection assays with divergent gene clusters deletion lines would demonstrate whether these genes attribute to virulence in hosts. Finally, the most interesting topic may be the copy number differences in repertoires of genes that may modulate plant hormone production. It remains to be elucidated how these genes are regulated between Taphrina species to cause differences in degrees of plant deformity. As these results demonstrate, comparative analyses make these Taphrina genome sequences a valuable resource for application of further omics technologies and the evolutionary studies on early evolution of yeasts.
Supplementary Material
Supplementary figures S1–S3 and tables S1–S9 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank John Jones, Daryi Wang, Tzi-Yuan Wang, Yoshitaka Takano, and Anna Avrova for reading and commenting the manuscript, Masabumi Komatsu and Yu Ichihara for providing fungal strains and useful suggestions, and Yuan-Min Shen, Taichung District Agricultural Research and Extension Station, and Theodor D. Leininger, USDA Forest Service for providing photographs of Taphrina deformans and T. populina. They also thank Dr Sayaka Ban for her support for verifying the isolation source of Taphrina strains used. This study was partly supported by JSPS KAKENHI (grants numbers 20353659 and 23248024), by JSPS Postdoctoral Fellowship Program for Foreign Researchers to I.J.T., and by JSPS Postdoctoral Fellowship Program (PD) to R.T.
Literature Cited
- Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- Alexa A, Rahnenfuhrer J. topGO: topGO: enrichment analysis for gene ontology. 2010 R package version 2.10.0. [cited 2014 Apr 4]. Available from: http://www.bioconductor.org/packages/2.12/bioc/html/topGO.html. [Google Scholar]
- Bennetzen JL, Hall BD. Codon selection in yeast. J Biol Chem. 1982;257:3026–3031. [PubMed] [Google Scholar]
- Bernt M, et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013;69:313–319. doi: 10.1016/j.ympev.2012.08.023. [DOI] [PubMed] [Google Scholar]
- Bickel PE, Tansey JT, Welte MA. PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim Biophys Acta. 2009;1791:419–440. doi: 10.1016/j.bbalip.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M, Pirovano W. Toward almost closed genomes with GapFiller. Genome Biol. 2012;13:R56. doi: 10.1186/gb-2012-13-6-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CA, Murray AW, Verstrepen KJ. Rapid expansion and functional divergence of subtelomeric gene families in yeasts. Curr Biol. 2010;20:895–903. doi: 10.1016/j.cub.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullerwell CE, Leigh J, Forget L, Lang BF. A comparison of three fission yeast mitochondrial genomes. Nucleic Acids Res. 2003;31:759–768. doi: 10.1093/nar/gkg134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver T, et al. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics. 2008;24:2672–2676. doi: 10.1093/bioinformatics/btn529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cissé O, Almeida J, Fonseca Á. Genome sequencing of the plant pathogen Taphrina deformans, the causal agent of peach leaf curl. MBio. 2013;4:e00055–13. doi: 10.1128/mBio.00055-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayarian A, Michael TP, Sengupta AM. SOPRA: scaffolding algorithm for paired reads via statistical optimization. BMC Bioinformatics. 2010;11:345. doi: 10.1186/1471-2105-11-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Falda M, et al. Argot2: a large scale function prediction tool relying on semantic similarity of weighted gene ontology terms. BMC Bioinformatics. 2012;13(Suppl 4):S14. doi: 10.1186/1471-2105-13-S4-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foury F, Roganti T, Lecrenier N, Purnelle B. The complete sequence of the mitochondrial genome of Saccharomyces cerevisiae. FEBS Lett. 1998;440:325–331. doi: 10.1016/s0014-5793(98)01467-7. [DOI] [PubMed] [Google Scholar]
- Galperin MY, Gaidenko TA, Mulkidjanian AY, Nakano M, Price CW. MHYT, a new integral membrane sensor domain. FEMS Microbiol Lett. 2001;205:17–23. doi: 10.1111/j.1574-6968.2001.tb10919.x. [DOI] [PubMed] [Google Scholar]
- Haas BJ, Delcher AL, Wortman JR, Salzberg SL. DAGchainer: a tool for mining segmental genome duplications and synteny. Bioinformatics. 2004;20:3643–3646. doi: 10.1093/bioinformatics/bth397. [DOI] [PubMed] [Google Scholar]
- Hahn C, Bachmann L, Chevreux B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 2013;41:e129. doi: 10.1093/nar/gkt371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedden P, Phillips AL, Rojas MC, Carrera E, Tudzynski B. Gibberellin biosynthesis in plants and fungi: a case of convergent evolution? J Plant Growth Regul. 2001;20:319–331. doi: 10.1007/s003440010037. [DOI] [PubMed] [Google Scholar]
- Henderson IR, Liu F, Drea S, Simpson GG, Dean C. An allelic series reveals essential roles for FY in plant development in addition to flowering-time control. Development. 2005;132:3597–3607. doi: 10.1242/dev.01924. [DOI] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. General rules for optimal codon choice. PLoS Genet. 2009;5:e1000556. doi: 10.1371/journal.pgen.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt M, et al. REAPR: a universal tool for genome assembly evaluation. Genome Biol. 2013;14:R47. doi: 10.1186/gb-2013-14-5-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JC, Trione EJ. Cytokinin production by the fungi Taphrina cerasi and T. deformans. Can J Bot. 1974;52:1583–1589. [Google Scholar]
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Taniguchi M, Matsushita N, Takahashi Y, Hogetsu T. Overwintering of Taphrina wiesneri within cherry shoots monitored with species-specific PCR. J Gen Plant Pathol. 2010;76:363–369. [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Krzywinski M, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon-Chung KJ, Chang YC. Aneuploidy and drug resistance in pathogenic fungi. PLoS Pathog. 2012;8:e1003022. doi: 10.1371/journal.ppat.1003022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, et al. Phylogenomic analyses support the monophyly of Taphrinomycotina, including Schizosaccharomyces fission yeasts. Mol Biol Evol. 2009;26:27–34. doi: 10.1093/molbev/msn221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashiguchi K, et al. The main auxin biosynthesis pathway in Arabidopsis. Proc Natl Acad Sci U S A. 2011;108:18512–18517. doi: 10.1073/pnas.1108434108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mix AJ. The life history of Taphrina deformans. Phytopathology. 1935;25:41–46. [Google Scholar]
- Mix AJ. A monograph of the genus Taphrina. 1949 Univ Kansas Sci Bull. 33:3–167. [Google Scholar]
- Nadal M, García-Pedrajas MD, Gold SE. Dimorphism in fungal plant pathogens. FEMS Microbiol Lett. 2008;284(2):127–134. doi: 10.1111/j.1574-6968.2008.01173.x. [DOI] [PubMed] [Google Scholar]
- Otto TD, Dillon GP, Degrave WS, Berriman M. RATT: rapid annotation transfer tool. Nucleic Acids Res. 2011;39:e57. doi: 10.1093/nar/gkq1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Punta M, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn J, et al. Two-component mediated peroxide sensing and signal transduction in fission yeast. Antioxidants Redox Signal. 2011;15:153–165. doi: 10.1089/ars.2010.3345. [DOI] [PubMed] [Google Scholar]
- Rawlings ND, Morton FR, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res. 2006;34:D270–D272. doi: 10.1093/nar/gkj089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reineke G, et al. Indole-3-acetic acid (IAA) biosynthesis in the smut fungus Ustilago maydis and its relevance for increased IAA levels in infected tissue and host tumour formation. Mol Plant Pathol. 2008;9:339–355. doi: 10.1111/j.1364-3703.2008.00470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, et al. Comparative functional genomics of the fission yeasts. Science. 2011;332:930–936. doi: 10.1126/science.1203357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues MG, Fonseca A. Molecular systematics of the dimorphic ascomycete genus Taphrina. Int J Syst Evol Microbiol. 2003;53:607–616. doi: 10.1099/ijs.0.02437-0. [DOI] [PubMed] [Google Scholar]
- Schaller GE, Shiu S-H, Armitage JP. Two-component systems and their co-option for eukaryotic signal transduction. Curr Biol. 2011;21:R320–R330. doi: 10.1016/j.cub.2011.02.045. [DOI] [PubMed] [Google Scholar]
- Schirawski J, et al. Pathogenicity determinants in smut fungi revealed by genome comparison. Science. 2010;330:1546–1548. doi: 10.1126/science.1195330. [DOI] [PubMed] [Google Scholar]
- Sherwood RK, Bennett RJ. Fungal meiosis and parasexual reproduction—lessons from pathogenic yeast. Curr Opin Microbiol. 2009;12:599–607. doi: 10.1016/j.mib.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siewers V, Kokkelink L, Smedsgaard J, Tudzynski P. Identification of an abscisic acid gene cluster in the grey mold Botrytis cinerea. Appl Environ Microbiol. 2006;72:4619–4626. doi: 10.1128/AEM.02919-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Stanke M, Tzvetkova A, Morgenstern B. AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol. 2006;7(Suppl 1) doi: 10.1186/gb-2006-7-s1-s11. :S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain MT, et al. A post-assembly genome-improvement toolkit (PAGIT) to obtain annotated genomes from contigs. Nat Protoc. 2012;7:1260–1284. doi: 10.1038/nprot.2012.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sziraki I, Balazs E, Kiraly Z. Increased levels of cytokinin and indoleacetic acid in peach leaves infected with Taphrina deformans. Physiol Plant Pathol. 1975;5:45–50. [Google Scholar]
- Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai IJ, Otto TD, Berriman M. Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol. 2010;11:R41. doi: 10.1186/gb-2010-11-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, St Leger RJ. The Metarhizium anisopliae perilipin homolog MPL1 regulates lipid metabolism, appressorial turgor pressure, and virulence. J Biol Chem. 2007;282:21110–21115. doi: 10.1074/jbc.M609592200. [DOI] [PubMed] [Google Scholar]
- Wood V, et al. PomBase: a comprehensive online resource for fission yeast. Nucleic Acids Res. 2012;40:D695–D699. doi: 10.1093/nar/gkr853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward AW, Bartel B. Auxin: regulation, action, and interaction. Ann Bot. 2005;95:707–735. doi: 10.1093/aob/mci083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing D, Zhao H, Xu R, Li QQ. Arabidopsis PCFS4, a homologue of yeast polyadenylation factor Pcf11p, regulates FCA alternative processing and promotes flowering time. Plant J Cell Mol Biol. 2008;54:899–910. doi: 10.1111/j.1365-313X.2008.03455.x. [DOI] [PubMed] [Google Scholar]
- Yin Y, et al. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40:W445–W451. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Liu H, Wang C, Xu J-R. Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics. 2013;14:274. doi: 10.1186/1471-2164-14-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin A, et al. The MaSuRCA genome assembler. 2013 doi: 10.1093/bioinformatics/btt476. Bioinformatics 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.