

Abstract
Many insects harbor inherited bacterial endosymbionts. Although some of them are not strictly essential and are considered facultative, they can be a key to host survival under specific environmental conditions, such as parasitoid attacks, climate changes, or insecticide pressures. The whitefly Bemisia tabaci is at the top of the list of organisms inflicting agricultural damage and outbreaks, and changes in its distribution may be associated to global warming. In this work, we have sequenced and analyzed the genome of Cardinium cBtQ1, a facultative bacterial endosymbiont of B. tabaci and propose that it belongs to a new taxonomic family, which also includes Candidatus Amoebophilus asiaticus and Cardinium cEper1, endosymbionts of amoeba and wasps, respectively. Reconstruction of their last common ancestors’ gene contents revealed an initial massive gene loss from the free-living ancestor. This was followed in Cardinium by smaller losses, associated with settlement in arthropods. Some of these losses, affecting cofactor and amino acid biosynthetic encoding genes, took place in Cardinium cBtQ1 after its divergence from the Cardinium cEper1 lineage and were related to its settlement in the whitefly and its endosymbionts. Furthermore, the Cardinium cBtQ1 genome displays a large proportion of transposable elements, which have recently inactivated genes and produced chromosomal rearrangements. The genome also contains a chromosomal duplication and a multicopy plasmid, which harbors several genes putatively associated with gliding motility, as well as two other genes encoding proteins with potential insecticidal activity. As gene amplification is very rare in endosymbionts, an important function of these genes cannot be ruled out.
Keywords: Amoebophilaceae, IS elements, gliding motility, Candidatus Cardinium hertigii, host–symbiont interaction
Introduction
The sweet potato whitefly Bemisia tabaci (Aleyrodidae) is an important polyphagous agricultural pest (Byrne and Bellows 1991). It feeds on plant phloem, causing many economic losses, affecting the plants directly and/or indirectly by transmitting several viruses. Bemisia tabaci was originally described as a complex species comprised by many biotypes (Brown et al. 1995), but recently it has been suggested that it is actually a complex of at least 24 morphologically indistinguishable species (De Barro et al. 2011), although classification in biotypes still persists. Among the most important biotypes are B (Middle East-Asia Minor 1 species), which arose in the Middle East but now has a cosmopolitan distribution, and Q (Mediterranean species), which originated in the Mediterranean basin and is also widely distributed at present. A phylogenetic analysis of mitochondrial biotype Q individuals worldwide has revealed the existence of three subclades (Q1–Q3) (Gueguen et al. 2010).
Similar to other phloem-feeding insects, which need essential amino acids and other nutrients due to their unbalanced diet, whiteflies have established mutualistic relationships with bacterial endosymbionts that provide essential compounds (Baumann 2005; Moran et al. 2008; Moya et al. 2008). All whiteflies species harbor the obligate, or primary endosymbiont, “Candidatus Portiera aleyrodidarum” (Gammaproteobacteria; hereafter Portiera) (Thao and Baumann 2004), which is restricted to specialized insect cells called bacteriocytes. In B. tabaci, several other bacterial species (secondary or facultative symbionts) may coexist with Portiera (Gottlieb et al. 2008). The most frequently observed are Arsenophonus sp. and “Candidatus Hamiltonella defensa” (hereafter Hamiltonella) (both Gammaproteobacteria), which share bacteriocytes with Portiera. Less frequently present are Rickettsia sp., Wolbachia sp. (both Alphaproteobacteria) and “Candidatus Cardinium sp.” (Bacteroidetes), detected in bacteriocytes but also in other insect tissues.
Symbiosis between whiteflies and Portiera has been shaped through a long-term relationship, mainly characterized by Portiera providing essential nutrients (amino acids and carotenoids) to the host, thus compensating for its deficient diet, as revealed by genome sequencing (Santos-Garcia et al. 2012; Sloan and Moran 2012). However, the effects of facultative endosymbionts need to be determined. So far, it has been noted that secondary symbionts in other insects can have different effects, such as reproductive manipulation, nutritional contribution, temperature tolerance, and defence against pathogens and parasitoids (Feldhaar 2011; White et al. 2011).
“Ca. Cardinium hertigii” (hereafter C. hertigii) was first characterized in Encarsia wasps, which are parasitoids of B. tabaci, and it was proposed as the species type (Zchori-Fein et al. 2004). However, in recent years, infections with bacteria belonging to the genus Cardinium have been detected not only in whiteflies but also in other insects (armored scale, sharpshooters, and Culicoides spp.) and other arthropods (mites, ticks, spiders, and copepods). Nowadays, the infection rate in arthropods has been estimated as close to 7% (Zchori-Fein and Perlman 2004; Gruwell et al. 2009; Nakamura et al. 2009). Based on molecular data (16S rRNA and gyrB genes) and the presence of microtubule-like complexes, a morphological feature shared by all known Cardinium, the genus has been divided into supergroups and strains, following a nomenclature similar to Wolbachia endosymbionts, with four described supergroups (A, B, C, and D) (Lo et al. 2002; Nakamura et al. 2009; Edlund et al. 2012). Similarly, in several arthropod taxa, C. hertigii has been described as a reproductive manipulator through diverse effects such as feminization, cytoplasmic incompatibility, and induction of parthenogenesis (White et al. 2011). However, these effects have not been found in other species (e.g., B. tabaci), suggesting that C. hertigii might also be a mutualistic endosymbiont. This aforesaid claim has been supported by the recently released genome of Cardinium cEper1 (endosymbiont of the wasp Encarsia pergandiella), which encodes a complete biotin biosynthetic pathway, suggesting a potential role in wasp nutrition (Penz et al. 2012). The 16S rRNA gene sequences from several Cardinium endosymbionts of B. tabaci reported to date are very similar (>99%) to those of the species type C. hertigii, suggesting that they are strains of the same species. Several analyses of secondary endosymbiont coinfections revealed that B. tabaci individuals infected only with C. hertigii are very unusual, especially for the C1 strain (supergroup A), the most widespread Cardinium among whiteflies (Gueguen et al. 2010). Generally, the coexistence with Hamiltonella is the most frequently observed, although combinations with other secondary symbionts, such as Wolbachia sp. or Rickettsia sp., are also possible (Gueguen et al. 2010; Skaljac et al. 2010; Park et al. 2012).
The laboratory strain B. tabaci QHC-VLC, belonging to the Q1 subclade, harbors Cardinium cBtQ1, which belongs to the C1 strain according to its 16S rRNA gene. This strain coexists within bacteriocytes harboring Portiera and Hamiltonella and can also be found scattered in different tissues of the whitefly (Gottlieb et al. 2008), similar to other hosts infected by C. hertigii (Bigliardi et al. 2006; Kitajima et al. 2007; Nakamura et al. 2009). In contrast, C. hertigii strains from different Encarsia species have only been detected in the ovaries and accessory cells (Zchori-Fein et al. 2004; Penz et al. 2012).
In this work, the genome of Cardinium cBtQ1 endosymbiont of B. tabaci QHC-VLC was sequenced and compared with that of the cEper1 strain and with several other Bacteroidetes, including the parasitic amoeba endosymbiont “Candidatus Amoebophilus asiaticus” (hereafter referred to as A. asiaticus) (Horn et al. 2001; Schmitz-Esser et al. 2010). Overall, all the analysis indicated that the Cardinium cBtQ1 genome has undergone changes to facilitate its recent establishment in B. tabaci.
Materials and Methods
Genome Assembly and Annotation
Enriched bacterial samples (Harrison et al. 1989) were collected from approximately 40,000 B. tabaci strain QHC-VLC adult whiteflies. DNA was extracted with the JetFlex Genomic DNA purification kit following the manufacturer’s instructions (Genomed). DNA was pyrosequenced using Roche 454 GS FLX Titanium single-end (shotgun) and paired-end (3 kb) libraries, and an Illumina HiSeq2000 mate-pair library (5 kb). The genome assembly was complex due to the high number of repetitive sequences, around 14% of the genome. This includes the presence of long duplications, almost 100% identical, and a large number of IS (Insertion Sequence elements). Duplicated regions were detected by abnormal coverage peaks, the differential genomic context (different presence of IS elements, genes, nonrepetitive intergenic regions), and paired-ends/mate-paired connections. The ends of most contigs were incomplete IS elements of different types. Because the contigs ending in the same IS type usually did not overlap, we expected a slight underestimation of the percentage of repetitive elements of Cardinium cBtQ1 genome. See supplementary materials and methods, Supplementary Material online (supplementary file S1, Supplementary Material online) for a detailed description of the assembling and annotation procedure, as well as the software used.
TBLASTX was used to compare the gene content between plasmids of Cardinium cBtQ1 and cEper1, and BLAST hits and gene order were plotted with the genoPlotR package (Guy et al. 2010) from R software (R Development Core Team 2011).
Phylogenetic and Phylogenomic Analyses
High-quality sequenced 16S rRNAs (including fragments of more than 900 bp) were downloaded from SILVA (Quast et al. 2013) and National Center for Biotechnology Information (NCBI) databases. ssu-aligner was employed for the alignment, because it takes into account the secondary structure (based on covariance models) of the 16S rRNA genes (Nawrocki 2009). Predefined masking was selected to ensure reproducibility in future alignments, and finally, the alignment was refined with Gblocks (Castresana 2000) allowing 50% in coverage gaps. General time reversible, with estimates of invariant sites and gamma-distribution among-site rate variation (GTR+I+G), was selected as the best model using jModeltest2 (Darriba et al. 2012). Additionally, amino acid sequences for the gyrB gene were downloaded from the NCBI database, aligned with MAFFT using the L-INS-i algorithm (Katoh et al. 2002) and refined with Gblocks. ProtTest3 (Darriba et al. 2011) gave the improved general amino-acid replacement matrix, with gamma distributed rates across sites (LG+G) as the best evolutionary model. All accession numbers are supplied in the supplementary table S1, Supplementary Material online.
For phylogenomic reconstructions, 37 orthologous single copy genes related to the translation/transduction machinery and protein folding functions that were present in 61 Bacteroidetes genomes (with the exception of Candidatus Sulcia mulleri CARI that lacked 9 of these genes) and a non-Bacteroidetes species, used as outgroup, were selected using the homology search tool from the Microbial Genome Database (Uchiyama 2003) (supplementary table S1, Supplementary Material online). Encoding proteins were downloaded and aligned with MAFFT v6.717b (L-INS-i) and concatenated. The selected best model was LG+G+F based on ProtTest3 results.
The first 15 BLASTP hits using the encoded proteins of Cardinium cBtQ1 cgl gene (CHV_c0068) and the Leishmania major genes cgl (cystathionine gamma-lyase, LmjF35.3230), cbl (cystathionine beta-lyase, LmjF32.2640), and cgs (cystathionine gamma-synthase, LmjF14.0460) were selected, and their amino acid sequences downloaded. In addition, the first 15 BLASTP hits against Bacteroidetes for the cystathionine beta-lyase protein (metC) from Escherichia coli str. K-12 (NP_417481.1) were also selected. Amino acid sequences were aligned with MAFFT v6.717b (L-INS-i), and ProtTest3 was used to select the appropriate model for the alignment, in this case, the LG+G model. For plasmid TraG protein, the first 50 BLASTP best hits were downloaded (including Cardinium cEper1 and A. asiaticus) and aligned with MAFFT v6.717b (L-INS-i). For TraG, the best model selected with ProtTest3 was LG+G+I.
RaxML (Stamatakis 2006) was used to calculate maximum likelihood phylogenetic trees for all the alignments, using optimizations for branch lengths and model parameters, and 1,000 rapid bootstrap replicates. Models were adjusted for each case. In addition, PhyloBayes3 (Lartillot et al. 2009) was used to infer Bayesian posterior distributions for each phylogenetic tree. In each case, evolutionary model was adjusted to the model selected (described above), and three independent chains were run for each alignment. Following Lartillot et al (2009) recommendations, all effective sizes were greater than 200 and maximum discrepancy between chains was less than 0.1. Finally, a majority rule consensus tree was calculated for each alignment.
Genomic Redundancy and Mobile Elements
NUCmer from MUMmer 3 (Kurtz et al. 2004) was used to plot repetitive regions of A. asiaticus (AmAs), Cardinium cBtQ1, and Cardinium cEper1, using each genome as query and subject. Results were filtered and only sequences with at least 95% identity and 500 bp length were used. To estimate the level of redundancy in the genomes, a BLASTN approach using each genome (chromosome plus plasmid concatenated) as query and subject was performed with e-value cutoff of 1e−20. The BLASTN results were transferred to a spreadsheet, where any alignment with an identity smaller than 95% was removed. Lines were sorted through several steps to identify the single copy segments of the genome. The repetitive fraction was estimated subtracting the summation of the single copy segments from the total genome length. IS elements were detected as described previously (Gil et al. 2008), refined using the web server ISsaga (Varani et al. 2011) and deposited in ISfinder database. Reference copies for each mobile element were used to search with BLASTX against the nonredundant NCBI database (1e−3 e-value cutoff). The 25 best hits for each mobile element were used as the input for MEGAN4 (Huson et al. 2011), and taxonomical assignments were done with the default LCA parameters. The genomes of A. asiaticus, Cardinium cEper1, and Cardinium cBtQ1 (with contigs concatenated in decreasing order, excluding the plasmid) were used to compute nucleotide synteny blocks with progressive Mauve aligner (Darling et al. 2010). Cardinium cBtQ1 was set as the reference for gene order and the alignment was plotted with the genoPlotR package.
Orthologous Gene Identification
After phylogenomic reconstruction, all genomes belonging to the order Cytophagales, including both Cardinium strains (cBtQ1 and cEper1) and A. asiaticus (AmAs), were used for orthologous gene identification. Flavobacterium johnsoniae (Bacteroidetes: Flavobacteriales), whose gliding motility has been broadly studied, was selected as the outgroup for the order Cytophagales. All proteins, including those from plasmids, were used as the input for OrthoMCL (Li et al. 2003). OrthoMCL and COG (Clusters of Orthologous Groups of proteins) (Tatusov et al. 2000) profile assignment pipelines were run as described previously (1.5 as inflation value, 70% of match cutoff, and an e-value cutoff of 1e−5) (Manzano-Marín et al. 2012). Gene clusters may contain zero, one, two, or more genes in each genome. Some gene clusters were manually refined because OrthoMCL failed to recognize some orthologous genes in endosymbionts.
Orthologous genes for A. asiaticus and both Cardinium strains were classified as core genome (genes shared by the three genomes), genes shared by two of the three organisms and strain-specific genes (supplementary table S2, Supplementary Material online). Euler diagrams were obtained with gplots package (Warnes et al. 2013) from R.
Last Common Ancestor Reconstruction
All genomes used in the OrthoMCL clustering method were used to reconstruct the putative last common ancestor (LCA) gene contents for each node in the phylogeny (fig. 1 and supplementary table S3, Supplementary Material online). The MPR (most parsimonious reconstruction) function in ape package (Paradis et al. 2004) from R was used to infer the ancestral state for each character (gene clusters) in each node (LCA). Pseudogenes for all genomes were manually revised. For orthologous cluster assignments of the pseudogenes, TBLASTX was used (e value of 1e−5 and an overlap of 80% query subject) against the proteins present in the orthologous clusters. Pseudogenes that did not modify the LCA reconstruction (strain-specific genes) were not considered. Pseudogenes that were mobile elements were also excluded. Parsimony reconstruction for orthologous groups that included the previously selected pseudogenes were checked using parsimony reconstruction of discrete characters in Mesquite (Maddison WP and Maddison DR 2011).
Fig. 1.—

Phylogenomic maximum likelihood tree of 61 Bacteroidetes. Phylogenomic reconstruction was done under the LG+G+F model on a concatenated alignment of 37 proteins. Cardinium genomes fall in the Cytophagales clade, with Marivirga tractuosa and Cy. marinum as the closest free-living relatives. Cardinium cBtQ1 is displayed in bold. Family names are displayed on the right delimited by a horizontal red line. The genomes used for the LCA reconstruction are shown in blue. Numbers inside gray dots show the LCAs reconstructed in each node. Only maximum likelihood bootstrap values below 95% are displayed. Bayesian posterior probabilities for each node were above 0.95 and are also not displayed. Chlorobaculum tepidum was used as outgroup.
For each reconstructed LCA and genome, COG categories were assigned for each orthologous cluster based on the initial OrthoMCL results and the COG assignment described above (Manzano-Marín et al. 2012). For each orthologous cluster, COG categories with less than a 10% of a cluster, as well as the unassigned category, were removed. The LCA, indeterminations (the presence/absence of the gene in the LCA node could not be determined) were counted as half (0.5), instead of presence (1) or absence (0). Relative percentages of each COG type using LCA4 or LCA2 as reference were plotted using the gplots heatmap2 function without dendograms and reordering functions. Euler diagram was plotted using gplots. COG profiles, stated as the absolute number of COG categories divided by the total number of COG for each genome or LCA, were plotted as a heatmap with gplots allowing hierarchical clustering (dendograms are grouping the most similar rows or columns together). Cyclobacteraceae family habitats were obtained from GOLD database (supplementary table S3, Supplementary Material online).
Analyses of B. tabaci and Encarsia spp. Samples
See supplementary materials and methods and tables S4–S6, Supplementary Material online (supplementary file S1, Supplementary Material online).
Fluorescent In Situ Hybridization
See supplementary materials and methods, Supplementary Material online (supplementary file S1, Supplementary Material online).
Results
General Features of Cardinium cBtQ1 Genome
Cardinium cBtQ1 has a relatively small genome (1.065 Mb) composed of a chromosome (1.013 Mb) and a circular plasmid (52 kb) named pcBtQ1 (table 1). The chromosomal sequence is composed of 11 contigs (612, 80.4, 77.6, 73.8, 66.9, 41.4, 30.3, 13.5, 7.4, 5.1, and 4.1 kb, respectively) with average coverages of 90× and 547× for 454 and Illumina platforms, respectively, whereas the plasmid is in a single contig with coverages of 595× (454) and 4,046× (Illumina). Paired-end and mate-pair information confirmed that the plasmid was a single closed circular contig. Also, the higher coverage of the plasmid compared with the chromosomal contigs is indicative of a multicopy plasmid, probably between six and seven copies relative to the chromosome.
Table 1.
General Genomic Features of Cardinium Strains and Amoebophilus asiaticus
| Bacterial Genome | Cardinium cBtQ1a | Cardinium cEper1b | A. asiaticus 5a2 | ||
|---|---|---|---|---|---|
| Host | Bemisia tabaci | Encarsia pergandiella | Acanthamoeba spp. | ||
| Chromosome | Plasmid | Chromosome | Plasmid | Chromosome | |
| Contigs | 11 | 1 | 1 | 1 | 1 |
| Size (kb) | 1,013 | 52 | 887 | 58 | 1,884 |
| GC (%) | 35 | 32 | 36 | 31 | 35 |
| CDS | 709 | 30 | 841 | 65 | 1,557 |
| Average CDS length (bp) | 1,033 | 1,389 | 911 | 733 | 990 |
| Coding density (%) | 79.7 | 80.1 | 85.5 | 82.1 | 81.8 |
| rRNAs | 3 | — | 3 | — | 3 |
| tRNAs | 35 | — | 37 | — | 35 |
| Other RNA genes | 2 | — | — | — | — |
| Pseudogenes (total) | 156 | 4 | 3 | — | 222 |
| Pseudogenes (transposase) | 132 | 3 | 3 | — | — |
| Pseudogene (other CDS) | 24 | 1 | — | — | — |
| Reference | This study | Penz et al. (2012) | Schmitz-Esser et al. (2010) | ||
aThe chromosome of Cardinium cBtQ1 is not closed, but it is considered a high-quality draft genome.
bThe chromosome of Cardinium cEper1 contains a single gap not closed due to repetitive elements.
We found 709 and 30 coding genes on the chromosome and plasmid, respectively. Many of them were annotated as encoding conserved or hypothetical proteins. We also annotated 156 pseudogenes in the chromosome, 132 derived from transposases and 24 from nontransposase genes (supplementary table S7, Supplementary Material online), and 4 in the plasmid (3 transposases and 1 resolvase). The genome contains one set of rRNA genes distributed in two segments, one including the 16S rRNA and the other the 23S and the 5S rRNA genes. It also contains a set of 35 tRNA genes, which are able to completely decode the mRNA sequences, and two other noncoding RNA genes (rnpB and tmRNA) (table 1).
Taxonomic Status of Cardinium cBtQ1 Endosymbiont of B. tabaci Biotype Q
We performed a phylogenomic reconstruction of a Bacteroidetes phylogeny (see supplementary table S1, Supplementary Material online, for locus tags in each genome), which showed that both Cardinium and A. asiaticus formed a well-differentiated clade, related to the families Cyclobacteriaceae and Flammeovirgaceae, with the family Cytophagaceae slightly more distant (fig. 1). This reconstruction was used to select the genomes for subsequent analyses.
Because this phylogenomic reconstruction is consistent with other reported studies (Gupta and Lorenzini 2007; Karlsson et al. 2011), and due to the high bootstrap values obtained, we propose that the Cardinium/Amoebophilus clade should be assigned to the order Cytophagales, instead of remaining in the nonclassified Bacteroidetes. Furthermore, the analysis suggests that within the Cytophagales, Cardinium forms a new family phylogenetically allied with the Cyclobacteriaceae and Flammeovirgaceae, to which we propose the name Amoebophilaceae (fig. 1).
To establish the relationship of Cardinium cBtQ1 to other Cardinium endosymbionts based on 16S rRNA sequences, a covariance model aligner was employed. The 16S rRNA alignment was used to infer a phylogeny, showing that almost all Cardinium endosymbionts of B. tabaci (including cBtQ1) were present in a clade with other Cardinium endosymbionts of several Encarsia species (99.14% identity with the whole 16SrRNA of Cardinium from E. pergandiella) (fig. 2, left). A phylogeny with gyrB was also performed, which corroborated the close phylogenetic relation with Cardinium from Encarsia spp. and also showed that Cardinium cBtQ1 was embedded in the Cardinium–Encarsia clade (fig. 2, right). Because 16S rRNA sequences of Cardinium cBtQ1 and the species type C. hertigii (symbiont of E. hispida) show only 1.2% of differences, we propose that Cardinium cBtQ1 is a strain of the latter, in agreement with previous reports (Zchori-Fein and Perlman 2004). The Bemisia/Encarsia clade belongs to the Cardinium group A, which is well differentiated from the other two groups included in the analysis: Group C, which is specific to the genus Culicoides (Nakamura et al. 2009) and group D, which is present in some Copepoda spp. (Edlund et al. 2012) (fig. 2).
Fig. 2.—

Bayesian phylogenetic tree for Cardinium from different arthropods species. Phylogenetic relationships among different Cardinium supergroups are shown. The Cardinium group A strains (green vertical arrow) seem the most widespread group, infecting different arthropod orders including the subclass Neoptera (which includes whiteflies). Groups C (blue arrow) and D (red arrow) showed a more restricted pattern of hosts. Group C is restricted to the order Diptera, whereas Group D is detected on subclass Copepoda. On the 16S rRNA phylogeny, Cardinium endosymbionts of Bemisia tabaci are grouped in two different strains, the C1 (red dot) that belongs to the Mediterranean species (Q biotype) and the CI1 (blue dot) that belongs to the Asia II species. The GTR+I+G model was selected for 16S rRNA and the LG+G model for gyrB. Cardinium strain cBtQ1, endosymbiont of B. tabaci QHC-VLC, is displayed in bold type. In both cases, Amoebophilus asiaticus was used as the outgroup but was excluded from the figure for plotting reasons. Triangles represent collapsed branches of the same species or genus with the number of collapsed sequences between square brackets. Arthropoda taxonomic names and their respective colors are shown in the upper box. Accession numbers for noncollapsed branches are displayed. Only Bayesian posterior values above 0.5 are displayed (blue), and branches under this score were condensed. Bootstrap values from maximum likelihood phylogenetic reconstruction above 50% are also displayed (black).
Genome Comparison of Cardinium Strains and A. asiaticus
Compared with A. asiaticus (Schmitz-Esser et al. 2010), the genomes of Cardinium cEper1 (Penz et al. 2012) and Cardinium cBtQ1 (this work) contain a smaller number of coding genes (table 1). However, Cardinium cEper1 has almost no annotated pseudogenes, a difference that might reflect gene annotation criteria because open reading frames that belong to transposase fragments are annotated as coding sequences (CDSs). The average gene identity between Cardinium cEper1 and cBtQ1 was 92.9%, whereas the average protein identity was 91.8%. The genome fraction assigned to coding genes (labeled as coding density in table 1) was approximately 6% smaller in Cardinium cBtQ1 than in Cardinium cEper1.
Cardinium cEper1 also contains a plasmid (pCher) of similar size to pcBtQ1. However, the gene content of both plasmids is different, with only a few shared genes in a syntenic segment showing a high level of nucleotide identity (fig. 3). For example, CHV_p004 (virE, virulence-associated E family protein), CHV_p006 (pre, plasmid recombination enzyme), and CHV_p011 (traG, putative conjugal transfer protein TraG) all show nucleotide identities that range from 76% to 83% between both plasmids. Phylogenetic reconstructions for each of these genes support the close relation between both plasmids (see TraG phylogeny in supplementary file S1: supplementary fig. S1, Supplementary Material online). Nucleotide, gene order conservation, and phylogeny suggest that both plasmids derive from a common ancestral plasmid present before the split of both Cardinium lineages. A putative traG gene (Aasi_0886), closely related to Cardinium traG phylogenetically (supplementary fig. S1, Supplementary Material online), is harbored in the A. asiaticus chromosome, suggesting that the origin of the plasmid can be traced to the family Amoebophilaceae, with its subsequent chromosomal insertion in A. asiaticus.
Fig. 3.—
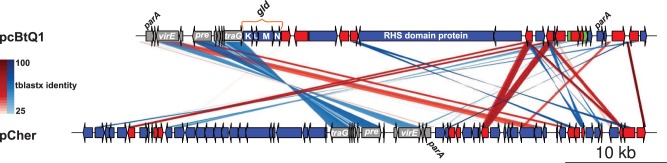
Comparison of Cardinium plasmids. TBLASTX comparison of the plasmids from Cardinium cBtQ1 (pcBtQ1) and cEper1 (pCher). Gray arrows are genes included in the syntenic block, blue arrows nontransposase genes, red arrows transposase genes, and the green arrow is a resolvase pseudogene. Red lines show genes in the same orientation and blue lines in reverse orientation. Some gene names are shown in the plot.
All three genomes share a core of 468 gene clusters (supplementary table S2, Supplementary Material online, and fig. 4), of which six encode putative host interacting proteins (Penz et al. 2012). There are 140 unique gene clusters that were present in both Cardinium but not in A. asiaticus, 46 of which encode hypothetical proteins, 15 are membrane transport related proteins, and 13 are putative host interacting proteins. Among the remaining Cardinium shared genes, there are six coding for transposases, two for phage-derived proteins (or afp-like proteins), and five for vitamin biosynthetic proteins (supplementary table S2, Supplementary Material online).
Fig. 4.—
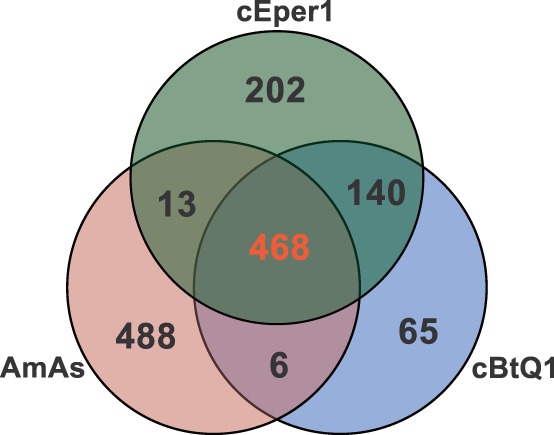
Euler diagram of orthologous clusters. Euler diagram representing the core genome, the strain-specific orthologous clusters, and the orthologous clusters shared by only two organisms. Numbers inside each subspace represent the number of orthologous gene clusters assigned to the subspace. Core genome set is displayed in orange. cBtQ1, Cardinium cBtQ1; cEper1, Cardinium cEper1; AmAs, Amoebophilus asiaticus.
Cardinium cEper1 has 202 strain-specific gene clusters, which include, among others, those encoding hypothetical proteins (145), transposases (30), host-interacting proteins (6), and biotin (2) and pyridoxal (1) biosynthetic enzymes. Cardinium cEper1 and A. asiaticus share 13 gene clusters with most of them defined as hypothetical proteins (6), mobile elements (3), a cell-wall-related protein, a membrane protein, and a host-manipulation protein (supplementary table S2, Supplementary Material online).
Cardinium cBtQ1 contains 71 gene clusters, 65 strain–specific, and 6 shared with A. asiaticus, which are not present in Cardinium cEper1. These gene clusters include ankyrin-domain-containing proteins (14), hypothetical proteins (35), transposases, and other mobile elements (4). A set of very interesting genes is located in the multicopy plasmid of Cardinium cBtQ1. They include four gliding genes (gldK, gldL, gldM, and gldN, see fig. 3) that are related to motility in members of the phylum Bacteroidetes (shared with A. asiaticus). The fact that the chromosome of Cardinium cBtQ1 contains four duplicated genes rtxBDE (A. asiaticus contained a single copy of the paralogous genes rtxBE) and tolC related to type I secretion system (T1SS) is also remarkable because only a few sequenced Bacteroidetes harbor secretion system types I, III, IV, or VI (McBride and Zhu 2013). The rtxBDE genes, related to the hemolysin secretion proteins (Hly), seem to be an event of horizontal gene transfer (HGT), with RTX toxin transport system of Vibrio as best BLAST hits. The chromosomal segment involving these genes is duplicated in Cardinium cBtQ1 (fig. 5A).
Fig. 5.—

Redundancy and synteny between Cardinium and Amoebophilus asiaticus genomes. (A) Putative linear representation of the ancestral genomic region before duplication (on top) and the present state of the two duplications, which are distributed in five contigs (on bottom). Note that lpxH, mreB, tolC, rtxBDE, and yitW conserve two intact copies, whereas the other duplicated genes only maintain one intact copy. Red arrows are mobile elements, blue arrows genes in the duplicated region, green arrows pseudogenized genes, and gray arrows adjacent genes outside the duplication. Orange bars connect the two duplicated copies of each gene. Contig names are plotted at the beginning or the end of the contig (CHV_), and only regions that contain the duplications are shown. The right ends of contigs CHV_g and CHV_e are connected through paired-end information with the right ends of either contig CHV_j or CHV_i. In both cases, a complete ISCca1 copy, whose fragments are detected at the end of the contigs, is required for joining. (B) Mummer plot showing direct (red) and inverted (blue) genomic repeats with at least 500 bp lengths and 95% similarity. For A. asiaticus (AmAs) and Cardinium cEper1 (cEper1), inner plot lines denote the division of the chromosome in base pairs sections. Black arrows point contig ends for the largest contigs in Cardinium cBtQ1. These contigs were placed in order of decreasing length. Because plots are not scaled to genome size due to limitations of the software, it is noteworthy that the A. asiaticus genome is less repetitive than Cardinium cBtQ1 although the more compact plot in the former may alter that impression. (C) Common pairwise syntenic blocks of more than 1 kb for A. asiaticus (AmAs), Cardinium cBtQ1 (cBtQ1), and cEper1 (cEper1). The chromosome of cBtQ1 was taken as reference. Contigs in cBtQ1 are ordered in order of decreasing length and denoted by double backslashes. For plotting reasons, only the seven longest cBtQ1contigs are shown. Red and blue lines show blocks in direct and inverted orientation. The stronger the line, the more nucleotide identity between synteny blocks.
Other interesting Cardinium cBtQ1-specific genes, also located in the plasmid, are the putative toxin-related genes CHV_p018 and CHV_p021. The latter encodes a long (4,603 amino acids) RHS-repeat-associated core-domain protein with C-terminus ankyrin repeats. Large proteins with RHS domains have been related with bacterial insecticidal toxins and intercellular signaling proteins (TIGR03696). Although no clear signal peptide was detected in CHV_p021, the presence of ankyrins in the C-terminus domain in combination with a signal peptide has been attributed to protein secretion by T1SS (Kaur et al. 2012). Because the best BLASTX hits, with 63% query coverage and 36% identity (on average), belong to Daphnia, Wolbachia (HGT event), and mosquitoes, it suggests that the target of this protein can be a conserved protein in arthropods.
The level of redundancy in the genome of Cardinium cBtQ1 (∼14%) was twice as high as the level found in Cardinium cEper1 and A. asiaticus (∼7% in both cases), most of which is associated with mobile elements (fig. 5B). The mobile elements of Cardinium cBtQ1, and their inactive derivatives, account for approximately 166 kb of the chromosome (196 copies) and 12.5 kb of the plasmid (12 copies) (supplementary table S8, Supplementary Material online). From this number of mobile element copies, only 48 contained a functional transposase gene, whereas 135 were transposase pseudogenes. These transposase proteins were classified in 20 different IS families, with only eight being complete IS elements (containing intact transposase genes and inverted repeats at their ends) and could be named according to the ISfinder recommendations and deposited under the names ISCca1-8 (supplementary table S8, Supplementary Material online). Only three mobile element types were specific of the Cardinium cBtQ1 (ISCca6, nv_IS3, and the Retron type one), whereas the rest of transposases were shared with A. asiaticus, Cardinium cEper1 or both.
It would appear that at least some IS in Cardinium are still active, in contrast with A. asiaticus (Schmitz-Esser et al. 2011), given we can observe very recent gene duplication events (based on > 99.9% nucleotide identity), with one of the copies interrupted by the insertion of an IS (e.g., pseudogenes recG or ftsK) (fig. 5A). Another important feature is the presence of a repetitive element composed of a copy of ISCca4 and a copy of nv_IS2, resulting in an IS that is apparently active. The inactivation of ftsK was produced by the insertion of this chimeric IS. Recombination associated to IS may be the cause of the high number of rearrangements in the Cardinium cBtQ1, with only some microsyntenic blocks maintained (fig. 5C).
Finally, Cardinium cBtQ1 contains a recent chromosome segmental duplication (almost 100% identical) involving at least 11 genes and around 17 kb, distributed in five contigs (fig. 5A). The duplicated region contains the genes cpsA (a protease), recG (recombination and DNA repair), glyS (aminoacyl-tRNA-ligase), lpxH (lipid A biosynthesis), mreB (actin-like bacterial cytoskeleton), tolC (T1SS transmembrane transporter), rtxB, rtxD, and rtxE (T1SS ABC transporters HlyB and HlyD), ftsK (cell cycle and chromosome partitioning), and yitW (putative chromosome partitioning related function). Although one of the two copies of cpsA, recG, glyS, and ftsK is pseudogenized by IS insertions, the rest of the genes conserve the two functional copies (fig. 5A), indicating that their retention could be advantageous for the organism fitness. However, the fact that the two copies are still active due to their recent duplication cannot be ruled out.
Evolution of Gene Repertoires in Lineages of Amoebophilus and Cardinium
Gene clusters obtained with OrthoMCL were classified according to COG categories and used to reconstruct by maximum parsimony the gene cluster content in the nine LCA corresponding to each node denoted with gray circles (fig. 1). Several analyses were performed to compare the gene content of the present and reconstructed ancestral genomes.
First, hierarchical clustering based on the relative abundance (percentage) of each COG category in each genome was performed (fig. 6). Three main clusters were observed: One that contained the endosymbiotic genomes and the LCA1 and 2; a second that grouped Marivirgia tractuosa and Cyclobacterium marinum with the LCA3, 4, 8, and 9; and a third that contained the rest of the genomes and LCAs. The second cluster (fig. 6, blue) showed a clear reduction in some COG groups as G and K but an enrichment in the H and J groups when it was compared with the third cluster (fig. 6, yellow). Also, hierarchical clustering grouped both Cardinium strains with A. asiaticus and LCA1 and LCA2. They showed a stronger conservation of genes in some COG categories such as J and O, when they were compared with the free-living Bacteroidetes, a signal also observed in other symbiotic reduced genomes. LCA4, the ancestor of the Cardinium/A. asiaticus lineage and family Cyclobacteriaceae, was close to the free-living Cyclobacteriaceae (see habitats in supplementary table S3, Supplementary Material online), suggesting a similar free-living style. Parsimony reconstruction assigned 1,301 gene clusters to LCA4 and the equally parsimonious presence/absence of other 684 gene clusters.
Fig. 6.—

Relative abundance of gene clusters in Bacteroidetes and LCAs. Hierarchical clustering heatmap representing the relative abundance (percentage) of each COG category in relation to the total number of gene clusters in each genome. Three main COG clusters (left) are observed: Highly retained categories (J, L, and R), medium retained categories (I, H, G, T, O, E, P, C, S, K, and M), and low retained categories (V, F, Q, U, D, N, and Z). Three main species/LCA cluster (up) are cEper1, AmAs, cBtQ1, LCA1, and LCA2 (only symbionts, left cluster); MaTr, CyHu, LCA3, LCA4, LCA8, and LCA9 (middle cluster), and FlJh, SpLi, DyFe, LeBy, CyMa, RuSl, LCA5, LCA6, and LCA7. Species clustering together by COG categories could have similar metabolic features and consequently, a similar ecological niche. Cardinium cEper1 (cEper1), Ca. Amoebophilus (AmAs), Cardinium cBtQ1 (cBtQ1), Marivirga tractuosa (MaTr), Cytophaga hutchinsonii (CyHu), Flavobacterium johnsoniae (FlJh), Spirosoma linguale (SpLi), Dyadobacter fermentans (DyFe), Leadbetterella byssophila (LeBy), Cyclobacterium marinum (CyMa), and Runella slithyformis (RuSl).
Second, the transition from LCA4 to LCA2 was examined comparing the percentages of gene clusters in each COG category regarding LCA4 (100%) (fig. 7A). It had a strong impact in the number of gene clusters with more than half of them being lost (LCA2, 655 gene clusters plus 36 present/absent) (supplementary table S3, Supplementary Material online). Relatively to LCA4 (100%), there was a high reduction in all categories except for some housekeeping categories such as J, L, and D with more than 80% of gene clusters conserved. Biosynthetic capabilities of LCA2 were clearly reduced (e.g., COG categories C, E, F, and H).
Fig. 7.—

Last free-living common ancestor comparison. (A) Heatmap showing the percentage of genes in each COG category, compared with the number of the same category in LC4 (100%). In left, reduced phylogenomic reconstruction with the name of each LCA reconstructed. (B) The same heatmap type comparing to LCA2 (100%). L category in Cardinium cEper1 is an artifact produced by an incorrect annotation of inactivated transposases as CDS instead of pseudogenes. COG definitions are below. cBtQ1, Cardinium cBtQ1; cEper1, Cardinium cEper1; AmAs, Amoebophilus asiaticus.
Third, the transition from LCA2 to LCA1 and to both Cardinium and A. asiaticus was examined comparing the percentages of gene clusters in each COG category regarding LCA2 (100%) (fig. 7B). The transition to LCA1 (649 gene clusters) produced the loss of 160 gene clusters, although 118 new gene clusters were acquired. Comparing the number of gene clusters of LCA2 to LCA1, and to both Cardinium and A. asiaticus, we observed several differences among COG categories (fig. 7B, supplementary table S3, Supplementary Material online). First, A. asiaticus showed 331 strain-specific gene clusters, distributed in several categories, not present in LCA2. Second, the reductive evolution of the Cardinium lineage was more clearly observed in several COG categories, such as E, G, H, S, T, and V. The absence of gene clusters in Cardinium for the N category was probably due to the fact that some genes related with motility have not been yet annotated in the COG database, especially those involved in gliding motility (see later) that are, in fact, present in Cardinium cBtQ1, A. asiaticus, LCA1, LCA2, and LCA4.
Biosynthetic Capabilities in Cardinium cBtQ1
Cardinium cBtQ1, according to KEGG classification pathways, presents low biosynthetic capabilities, similar to those observed in Cardinium cEper1 and A. asiaticus (Karlsson et al. 2011; Penz et al. 2012).The main differences between the biosynthetic capabilities of both Cardinium strains are in the biosynthesis of vitamins and cofactors. Both bacteria are able to produce lipoate, a key cofactor for intermediate metabolism and an important antioxidant molecule (Spalding and Prigge 2010). Cardinium cEper1 has the genes pdxS and pdxT and can synthesize pyridoxal 5-phosphate (precursor of vitamin B6). However, pdxT was pseudogenized by an IS transposition in cBtQ1. This event seems to have happened recently, because the pdxT pseudogene is 93.5% identical to the cEper1 gene, a percentage higher than the average gene identity between these two strains.
In addition, it is noteworthy that in Cardinium cEper1, the presence of a complete biotin operon, a coenzyme belonging to vitamin B class, is an event of HGT from Alphaproteobacteria to the genus Cardinium (Penz et al. 2012). The loss of the ability to synthesize biotin in Cardinium cBtQ1 seems to have taken place by the combined effect of the insertion of an IS and a later deletion event, removing the complete bioB gene and almost the complete sequence of the adjacent bioF gene (92.5% identical to cEper1 in the remnant segment). Another recent signal of the loss of a nutritional contribution is the pyridoxal-dependent enzyme cystathionine gamma-lyase (involved in the synthesis of cysteine) whose CDS contains an internal stop codon mutation that produces the pseudogene CHV_c0068 in cBtQ1 (94.9% identical to cEper1 gene). A phylogenetic analysis showed that the functional gene, present in this state in cEper1, was acquired by an ancestor through HGT from a eukaryote, perhaps an amoeba (supplementary file S1: supplementary fig. S2, Supplementary Material online). The phylogenetic analysis, including the three in silico identified cystathionine metabolizing enzymes of L. major (Williams et al. 2009), showed its closer relation with L. major cystathionine gamma-lyase rather than with L. major cystathionine beta-lyase, as previously annotated in Cardinium cEper1 (Penz et al. 2012).
In Cardinium cBtQ1, the inability to synthesize pyridoxal and biotin may be complemented by other facultative endosymbionts of the host. In the case of B. tabaci strain QHC-VLC, such enzyme synthesis could be achieved by Hamiltonella, which is located inside the bacteriocytes (supplementary fig. S3, Supplementary Material online). The complete sets of genes required for the synthesis of both cofactors was described for the Hamiltonella secondary endosymbiont of the aphid Acyrthosiphon pisum (Degnan et al. 2009), and all of them were also found by BLAST similarity in the draft genome of Hamiltonella from B. tabaci strain QHC-VLC (unpublished data).
Gliding Genes in Cardinium
As stated earlier, the genome of Cardinium cBtQ1 harbors four gliding genes organized in an operon (gldK, gldL, gldM, and gldN) in the pcBtQ1 plasmid (fig. 3). To state that the differential tissue locations of Cardinium endosymbionts of B. tabaci (scattered) and Encarsia spp. (restricted to ovaries) were correlated with the presence of the four gliding genes, two populations of E. pergandiella (USA and Brazil), one of E. hispida (Italy) and one from E. inaron (USA) were screened for their presence. None of these species gave a positive result for any of the gliding genes, whereas the presence of Cardinium was stated in all of them through the amplification of the control gene gyrB (supplementary file S1: supplementary materials and methods and tables S4 and S6, Supplementary Material online).
For B. tabaci, adult whiteflies were sampled in 12 points from four localities of the province of Valencia (Spain) (supplementary file S1: supplementary materials and methods and tables S5 and S6, Supplementary Material online). Polymerase chain reaction (PCR) amplification revealed the presence of the four gliding genes (as well as of the large plasmid gene CHV_p021) in all the samples (except one that failed in all the PCR amplifications). To determine insect biotypes, a mitochondrial cytochrome oxidase I (COI) fragment was sequenced in four females per sampling point. Nine of the samples belonged to the biotype Q (Mediterranean species), whereas two were from the biotype S (Sub-Saharan Africa species), an uncommon biotype in Spain (Moya et al. 2001) (supplementary file S1: supplementary fig. S4, Supplementary Material online). All the samples, including those of biotype S, harbored Cardinium, which was determined by PCR amplification of a fragment of the 16S rRNA gene. PCR products from biotype S (sample F) and biotype Q (sample B) (1,100 bp) were sequenced and resulted 100% identical to Cardinium cBtQ1.
The presence of additional secondary endosymbionts was also checked finding that biotype Q individuals also contained Hamiltonella, whereas biotype S contained Arsenophonus and Wolbachia (supplementary file S1: supplementary materials and methods and tables S5 and S6, Supplementary Material online).
Discussion
Although the cost of maintaining an obligate mutualistic endosymbiont may be compensated in many cases by its supplementation of the host diet, the maintenance of stable associations with facultative symbionts may require either reproductive manipulation or compensatory benefits from the endosymbiont (Oliver et al. 2010; White et al. 2011; Wernegreen 2012). Both facultative and obligate endosymbionts are transmitted vertically, but facultative symbionts may retain the ability to be horizontally transmitted as revealed by the incongruence of host and symbiont phylogenetic reconstructions (Russell et al. 2003).
Symbionts belonging to the genus Cardinium are present in many types of arthropods including arachnids (Nakamura et al. 2009), crustaceans (Edlund et al. 2012), and insects (Zchori-Fein et al. 2004; Zchori-Fein and Perlman 2004; Gruwell et al. 2009; Nakamura et al. 2009), indicating that members of this genus can colonize new niches. Phylogenies and nucleotide conservation of two genes (16S rRNA and gyrB) show that Cardinium from B. tabaci QHC-VLC was closely related to Cardinium from armored scale insects and parasitoid wasps, including the species type C. hertigii from the wasp E. hispida (Zchori-Fein et al. 2004). Researchers have also proposed that Cardinium cBtQ1 was a strain of C. hertigii, closely related to Cardinium cEper1 (symbiont of E. pergandiella) whose genome has recently been reported (Penz et al. 2012). In this work, we have sequenced the genome of Cardinium cBtQ1, and all the analyses carried out confirm that this strain is closely related to Cardinium cEper1, despite being endosymbionts of whiteflies and parasitoids, respectively. There are, however, some differences that could be related to the massive presence of IS in Cardinium cBtQ1, as well as to the adaptation to the specific hosts of each strain (see later).
The high number of available Bacteroidetes genomes provided a robust phylogeny (fig. 1), which showed that both Cardinium (cEper1and cBtQ1) and A. asiaticus formed a well-defined clade, distant from other family members of Cyclobacteriaceae, Cytophagaceae, and Flammeovirgaceae in the order Cytophagales. Thus, we propose that they form a new family, to be named Amoebophilaceae. This name is proposed because A. asiaticus has a larger genome than Cardinium spp. and shares more genes with LCA2 than either Cardinium strain. Also on the basis of this phylogeny, we were able to infer the coding gene contents (clusters of genes) of the most recent common ancestor in the Cardinium/Amoebophilus clade (LCA4, LCA2, and LCA1) and to analyze the evolution of the gene repertoires (figs. 6 and 7).
Despite it is not the objective of this work, we can extract general ideas from the cluster analysis (fig. 6). The distribution of genes in COG categories is a well predictor of the way of life of the organisms. Hierarchical clustering indicates that LCA3 to 9 were, similar to free-living Bacteroidetes, able to occupy different niches (fig. 6). For example, the differences between the abundance of G category in the middle and right clusters could be related to a more restricted source of carbohydrates (niche specialization). It also seems that the increase of the coenzyme metabolism (H) in the middle cluster could be advantageous for the establishment of symbiotic relationships (Cyclobacterium was found in the celomic fluid of a sand dollar, supplementary table S3, Supplementary Material online).
The number of gene clusters estimated in LCA4 was between 1,301 and 1,985, showing a COG profile similar to those of M. tractuosa (Flammeovirgaceae) and Cy. marinum (Cyclobacteriaceae) (fig. 6), and differing from the COG profiles of LCA2 and LCA1, which clustered with those of the symbiotic bacteria A. asiaticus and Cardinium (fig. 6). Because species of the family Cyclobacteriaceae are mostly marine free-living bacteria, and associations with animal hosts have been described (supplementary table S3, Supplementary Material online), we propose that LCA4 was probably a marine free-living bacterium, with a wide range of functional capabilities and the ability to establish symbiotic relationships. Also, it is likely that LCA4 was able to glide because it contained the whole set of gliding genes essential for gliding, including the sprATE genes (McBride and Zhu 2013).
The transition from LCA4 to LCA2 was clearly a reductive process that affected almost all COG categories (fig. 7A) producing an ancestral endosymbiont with few biosynthetic capabilities. Considering that the species derived from LCA2 were endosymbionts of amoebas (Horn et al. 2001; Schmitz-Esser et al. 2010) or insects (Zchori-Fein and Perlman 2004; Penz et al. 2012), the most probable reason for this reduction was the transition from a free living to intracellular life style, to start a symbiotic (either mutualistic or parasitic) relationship with a eukaryotic host. During this transition, the number of gene clusters and associated functions was reduced, although LCA2 maintained the ability to acquire new genes by HGT. The higher number of gene clusters in A. asiaticus versus LCA2 (298) could be due to this fact, although other reasons, such as the possibility of a biased sample of genomes, or different annotation problems, would also explain its high number of specific gene clusters.
Genome reduction was an ongoing process in the LCA1/Cardinium clade, and it was notorious for some categories such as E and G (fig. 7B). Moreover, the comparative analysis of the gene contents of the two Cardinium strains revealed that, despite being very similar at nucleotide level (92.9% nucleotide identity), revealing a recent evolutionary divergence, there are some relevant differences between the genomes of both strains, indicating differences in the evolution of endosymbiosis in Encarsia spp. and B. tabaci.
First, both Cardinium contain a plasmid of 50–60 kb with many differences in gene content (fig. 3). However, both plasmids contain a short syntenic block of genes, whose nucleotide content and gene order conservation, as well as the gene phylogenies carried out, suggests that both derived from an ancestral plasmid present in their LCA (LCA1), and the differences must have been accumulated after the split of both lineages. These differences are due to the insertion of mobile elements, sometimes carrying accessory genes, and to the transfer of genes from the chromosome.
Second, there are also differences in the chromosome of both Cardinium strains. The Cardinium cBtQ1 chromosome is 126 kb larger (probably a bit larger because, as a draft genome, gaps are not taken into account for this calculation) than that of Cardinium cEper1; nevertheless, this difference is not associated with a greater number of genes but to the presence of a large number of pseudogenes, most of them due to defective transposase encoding genes (table 1).The number of repeated sequences in the genome of Cardinium cBtQ1 is twice that of Cardinium cEper1, and most of these differences are due to a large number of IS in the former genome (fig. 5B). Moreover, some IS types seem to have their origins in Alphaproteobacteria, probably related to the genera Rickettsia or Wolbachia, supporting the idea of HGT events between secondary endosymbionts harbored by the same host (Toft and Andersson 2010; Schmitz-Esser et al. 2011; Duron 2013). A large number of mobile elements is a typical feature of endosymbionts that have established a recent relationship with their hosts, such as Candidatus Sodalis pierantonius str. SOPE (Gil et al. 2008, Oakeson et al. 2014) or Sodalis glossinidius (Belda et al. 2010). Also, enrichment in mobile elements seems to be linked to genome plasticity in some facultative symbionts (Gillespie et al. 2012). Several lines of evidence indicate that IS elements (supplementary table S8, Supplementary Material online) are already active in Cardinium cBtQ1. This activity combined with both the high number of copies throughout the genome, and a complete replication and repair machinery that can produce recombination, probably underlies the massive number of rearrangements in the genome of Cardinium cBtQ1, compared with Cardinium cEper1 and A. asiaticus (fig. 5C).
Finally, among the genes with an annotated function and differential presence in the two Cardinium strains, we consider some may give clues about the relationship between Cardinium cBtQ1 and B. tabaci. First, the presence of pseudogenes for pdxT (pyridoxal biosynthesis) and cgl (cystathionine gamma-lyase) and the loss of two genes of the biotin operon indicate that these genes were present in LCA1. This also indicates that the loss of these functions in Cardinium cBtQ1 is associated with settlement in a new environment composed by B. tabaci and its facultative symbiont Hamiltonella defensa. This symbiont seems to have become established in the populations of B. tabaci Q1 from Western Mediterranean based on the detection of frequencies of 100% (or almost 100%) in populations from North Africa and south-western Europe (Gueguen et al. 2010). Second, among 71 gene clusters of Cardinium cBtQ1, which are absent in Cardinium cEper1, there are up to 14 specific genes encoding ankyrin-domain proteins, which can interact with the host's machinery, but further studies are needed to understand their functions. Finally, the most interesting genes are those whose expression has been amplified by either gene duplication (mreB, lpxH, yitW, and rtxBDE/tolC) or by their presence in a multicopy plasmid (e.g., CHV_p018, a putative toxin-related gene, CHV_p021, a RHS-domain protein, and the gliding genes gldKLMN) (supplementary table S2, Supplementary Material online).
On the basis of the ancestor reconstruction analysis, we can predict that the four gliding genes were present in LCA4, LCA2, and LCA1 and were lost in Cardinium cEper1. Although LCA4 conserved full gliding machinery, sprATE was lost in LCA2 possibly due to its accommodation to an intracellular environment. Because LCA1 conserved the gldKLMN operon, this suggests that gldKLMN was lost in the Cardinium cEper1 lineage. Also, as in the closest Bacteroidetes genomes, such as A. asiaticus, M. tractuosa, or Cy. marinum, these genes are located in the chromosome, and we can postulate that in Cardinium cBtQ1, they have been translocated to a multicopy plasmid conserving the operon order. This supports the importance of these genes for Cardinium cBtQ1 and suggests that they may explain why the strain is not confined to a single tissue in B. tabaci (Gottlieb et al. 2008) (supplementary fig. S3, Supplementary Material online), in opposition to Cardinium cEper1 that is restricted to the ovaries of Encarsia (Zchori-Fein et al. 2004; Penz et al. 2012). Moreover, the gene amplification in Cardinium cBtQ1 not only of the four gliding genes but also of mreB and of the cluster rtxBDE/tolC suggests that they may play an important role in this organism, as genome reduction is an ongoing process in this strain. There are two possible hypotheses: 1) those genes are involved in gliding as in other genomes; 2) they are involved in the novel type 9 secretion system (PorSS), which is also associated with the secretion of proteins involved in motility and toxins (Sato et al. 2010; McBride and Zhu 2013).
Different Bacteroidetes possess the ability to move by a gliding mechanism, which is related to the ability to degrade some components present in the environment such as chitin and cellulose (Spormann 1999; McBride 2004; Braun et al. 2005). Several examples of gliding have been reported in species of the class Cytophagia where C. hertigii was included (Xie et al. 2007; McBride and Zhu 2013), and the scattered pattern detected for the tissue distribution of Cardinium cBtQ1 in B. tabaci might be caused by a similar mechanism.
The motor model (or focal adhesion) proposed for gliding in myxobacteria (Spormann 1999; Mignot et al. 2007; Jarrell and McBride 2008; Nakane et al. 2013; Nan et al. 2013) could be extended to Bacteroidetes, including Cardinium cBTQ1, although in a more simplified manner. This model considers molecular motors that are associated with cytoskeletal filaments and use proton motive force to transmit force through the cell wall to attached dynamic focal adhesion complexes (adhesins) to the substrate, causing the cell to move forward (Mignot et al. 2007; Sun et al. 2011; Nan et al. 2013). MreB, the homolog of the eukaryotic actin, has been proposed as the cytoskeletal part of the gliding machinery (Kearns 2007; Mauriello et al. 2010). Also, there may be an association with FtsZ, a protein that is part of the bacterium cytoskeleton and can produce force by itself (Erickson et al. 2010). Linkage between the cytoskeleton and gliding is supported by experimental data with the use of compound A22, which is able to affect the MreB structure and inhibits the gliding motility in Myxobacteria (Nan et al. 2011). Eleven genes, detected in most Bacteroidetes, have been defined as the core of the gliding machinery. Four of these genes (gldB, gldD, gldH, and gldJ) have unknown function, whereas the remaining seven genes (gldK, gldL, gldM, gldN, sprA, sprE, and sprT) also encode the PorSS system (McBride and Zhu 2013).
Cardinium cBtQ1 does not show the complete gliding machinery as it only contains four gliding core genes (gldKLMN). Neither homologous nor potential analogous genes of gldBDHJ have been detected. The sprAET genes are also absent, but their function would potentially be substituted by the cluster rtxBDE/tolC, which is also duplicated. RTX secretion system belongs to the T1SS and is able to transport proteins from the cytosol to the extracellular space in a SecYEG independent manner. Also, T1SS is able to secrete many different RTX family proteins and proteins without the C-terminal RTX nonapeptide (Linhartová et al. 2010; Kaur et al. 2012). The RTX system would secrete the adhesins (or other proteins that could interact with the host) across the bacterial membrane. However, we did not detect any orthologs to known adhesin proteins, but proteins with eukaryotic domains, such as ankyrins, TPR or WH2 (found in this strain), may function as adhesins in a multicellular eukaryotic organism. Moreover, Cardinium may be able to manipulate the host cytoskeleton to form a scaffold, which could be used by the gliding machinery (Haglund et al. 2010). Gliding seems to be a widespread direct invasion mechanism for different kinds of cells (Sibley et al. 1998; Furusawa et al. 2003; Sibley et al. 2004), thus Cardinium cBtQ1and other Bacteroidetes might use this motor model system to invade new hosts or host tissues. In fact, Cardinium endosymbiont of Ixodes scapularis has been cultivated on insect cell lines and is capable of infecting new cells, even cell lines from different insect species, when they are added to the culture (Morimoto et al. 2006).
The second hypothesis would consider that the gldKLMN operon is not involved in gliding, but it is just required for secretion forming the PorSS system, which was initially described for Porphyromonas gingivalis as a novel secretion system with eight proteins involved (PorK, PorL, PorM, PorN, PorT, PorW, Sov, and PorP) (Sato et al. 2010). Putatively orthologous genes in the gliding system for the first seven are gldK, gldL, gldM, gldN, sprT, sprE, and sprA. The proposed orthologous gene for porP in F. johnsoniae was Fjoh_3477. A similar gene was not detected in either A. asiaticus or Cardinium. Proteins secreted by the PorSS systems are adhesins, as well as some enzymes such as chitinases, and gingipains in F. johnsoniae and P. gingivalis, respectively. Also, proteins secreted by the PorSS secretion system may contain a conserved C-terminal domain (TIGR4131 and 4183) (Sato et al. 2010; McBride and Zhu 2013). However, there were not proteins of Cardinium cBtQ1 with this domain. In the PorSS system, the presence of the protein complex GldKLMN is associated with the generation of the energy required for protein secretion by SprTEA. However, these proteins are not encoded in the genome of Cardinium cBtQ1 (they are also absent in A. asiaticus), and their substitution by the T1SS (RTX system) seems unlikely because T1SS has its own ATP-binding cassette, making the energy production function of the gldKLM unnecessary. This suggests that the PorSS system does not work in Cardinium cBtQ1. Thus, we hypothesized that the retention of the necessary genes for the gliding movement (gldKLMN) and further amplification by the translocation to a multicopy plasmid, together with the acquisition of the RTX TISS is associated to the scattered phenotype of this strain and the ability to glide.
The frequent presence of Cardinium cBtQ1 in B. tabaci biotypes Q and S may be due to its ability to benefit its host (Oliver et al. 2008; Feldhaar 2011; Ferrari and Vavre 2011), possibly related to its motility feature, and/or to the presence of some putative toxin-related genes such as CHV_p018 (low e-value BLAST hit against RTX toxins of H. defensa from A. pisum) and CHV_p021, which could play a role in intercellular competition, intercellular signaling, and insecticidal activity based on the presence of the RHS domain (Koskiniemi et al. 2013). As a mobile endosymbiont, Cardinium cBtQ1 may contact a parasitoid directly and secrete insecticidal toxins near it, could invade the parasitoid tissue, and kill it by secreting unknown toxins or by the cytotoxic effect of lipid A in a nonacclimated host (Furusawa et al. 2003; Caspi-Fluger et al. 2011; Rader et al. 2012). Furthermore, other effects increasing host fitness cannot be excluded, like heat-stress resistance or maybe some advantages conferred by the lipoate supplementation (Moran et al. 2008; Moya et al. 2008). However, fitness may only be improved in some environments or climate conditions.
In conclusion, we have reported the genome of Cardinium cBtQ1 endosymbiont of B. tabaci. Comparative genomics and ancestors’ reconstruction of gene content have shed light on the drastic reduction in many functional categories that have taken place since its free-living ancestor up to the present. The loss of several cofactors and amino acid biosynthetic capabilities, retained in its close relative Cardinium cEper1, rules out an important role in host nutrition and suggests a relationship with its establishment in B. tabaci and its endosymbionts. The genome is still very dynamic, with many active transposable elements and rearrangements. On the basis of genomic data, we propose that Cardinium cBtQ1 has retained the minimal gliding core machinery present in its ancestors, which in combination with the acquired RTX secretion system might be used to move inside its host or invade new hosts.
Supplementary Material
Supplementary file S1 is available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
F.J.S., A.L., A.M., D.S.-G., E.Z.F., F.V., and L.M. conceived the study. F.J.S., A.L., A.M., and D.S.-G. designed the experiments. D.S.-G. and F.B. performed the experiments. D.S.-G. and F.J.S. generated the data. D.S.-G., F.J.S., and A.L. analyzed the data. D.S.-G., F.J.S., and A.L. wrote the paper. F.J.S., A.L., A.M., D.S.-G., F.B., P.-A.R.-F., E.Z.F., F.V., and L.M. critically revised the paper. This work was supported by grants BFU2012-39816-C02-01 (cofinanced by FEDER funds and Ministerio de Economía y Competitividad, Spain) to A.L., Prometeo/2009/092 (Conselleria d’Educació, Generalitat Valenciana, Spain) to A.M., and EU COST Action FA0701. These results have been achieved within the framework of the 1st call on Mediterranean agriculture carried out by ARIMNet, with funding from MOARD (IL), ANR (FR), INIA (ES), NAGREF-DEMETER (GR), and GDAR (TR). D.S.-G. is a recipient of a contract from Prometeo 92/2009. P.-A.R.-F. is a recipient of a grant from the European Research Council under the European Community's Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement no. [247073]10 SISYPHE. The authors gratefully acknowledge Martha S. Hunter and Massimo Giorgini for the Encarsia samples, and Mariana Reyes-Prieto and Alejandro Manzano-Marín for bioinformatic advice. They also acknowledge the SCSIE from the Universitat de València for sequencing and microscopy support. Part of this work was performed by F.J.S. during a sabbatical stay at the Uppsala Universitet.
Literature Cited
- Baumann P. Biology bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu Rev Microbiol. 2005;59:155–189. doi: 10.1146/annurev.micro.59.030804.121041. [DOI] [PubMed] [Google Scholar]
- Belda E, Moya A, Bentley S, Silva FJ. Mobile genetic element proliferation and gene inactivation impact over the genome structure and metabolic capabilities of Sodalis glossinidius, the secondary endosymbiont of tsetse flies. BMC Genomics. 2010;11:449. doi: 10.1186/1471-2164-11-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigliardi E, et al. Ultrastructure of a novel Cardinium sp. symbiont in Scaphoideus titanus (Hemiptera: Cicadellidae) Tissue Cell. 2006;38:257–261. doi: 10.1016/j.tice.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Braun TF, Khubbar MK, Saffarini DA, McBride MJ. Flavobacterium johnsoniae gliding motility genes identified by mariner mutagenesis. J Bacteriol. 2005;187:6943–6952. doi: 10.1128/JB.187.20.6943-6952.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JK, Frohlich DR, Rosell RC. The sweetpotato or silverleaf whiteflies: biotypes of Bemisia tabaci or a species complex? Annu Rev Entomol. 1995;40:511–534. [Google Scholar]
- Byrne DN, Bellows TS. Whitefly biology. Annu Rev Entomol. 1991;36:431–457. [Google Scholar]
- Caspi-Fluger A, et al. Rickettsia “in” and “out”: two different localization patterns of a bacterial symbiont in the same insect species. PLoS One. 2011;6:e21096. doi: 10.1371/journal.pone.0021096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Barro PJ, Liu S-S, Boykin LM, Dinsdale AB. Bemisia tabaci: a statement of species status. Annu Rev Entomol. 2011;56:1–19. doi: 10.1146/annurev-ento-112408-085504. [DOI] [PubMed] [Google Scholar]
- Degnan PH, Yu Y, Sisneros N, Wing RA, Moran NA. Hamiltonella defensa, genome evolution of protective bacterial endosymbiont from pathogenic ancestors. Proc Natl Acad Sci U S A. 2009;106:9063–9068. doi: 10.1073/pnas.0900194106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duron O. Lateral transfers of insertion sequences between Wolbachia, Cardinium and Rickettsia bacterial endosymbionts. Heredity (Edinb) 2013;2:1–8. doi: 10.1038/hdy.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlund A, Ek K, Breitholtz M, Gorokhova E. Antibiotic-induced change of bacterial communities associated with the copepod Nitocra spinipes. PLoS One. 2012;7:e33107. doi: 10.1371/journal.pone.0033107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson HP, Anderson DE, Osawa M. FtsZ in bacterial cytokinesis: cytoskeleton and force generator all in one. Microbiol Mol Biol Rev. 2010;74:504–528. doi: 10.1128/MMBR.00021-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldhaar H. Bacterial symbionts as mediators of ecologically important traits of insect hosts. Ecol Entomol. 2011;36:533–543. [Google Scholar]
- Ferrari J, Vavre F. Bacterial symbionts in insects or the story of communities affecting communities. Philos Trans R Soc Lond B Biol Sci. 2011;366:1389–1400. doi: 10.1098/rstb.2010.0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa G, Yoshikawa T, Yasuda A, Sakata T. Algicidal activity and gliding motility of Saprospira sp. SS98-5. Can J Microbiol. 2003;49:92–100. doi: 10.1139/w03-017. [DOI] [PubMed] [Google Scholar]
- Gil R, et al. Massive presence of insertion sequences in the genome of SOPE, the primary endosymbiont of the rice weevil Sitophilus oryzae. Int Microbiol. 2008;11:41–48. [PubMed] [Google Scholar]
- Gillespie JJ, et al. A Rickettsia genome overrun by mobile genetic elements provides insight into the acquisition of genes characteristic of an obligate intracellular lifestyle. J Bacteriol. 2012;194:376–394. doi: 10.1128/JB.06244-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb Y, et al. Inherited intracellular ecosystem: symbiotic bacteria share bacteriocytes in whiteflies. FASEB J. 2008;22:2591–2599. doi: 10.1096/fj.07-101162. [DOI] [PubMed] [Google Scholar]
- Gruwell ME, Wu J, Normark BB. Diversity and phylogeny of Cardinium (Bacteroidetes) in armored scale insects (Hemiptera: Diaspididae) Ann Entomol Soc Am. 2009;102:1050–1061. [Google Scholar]
- Gueguen G, et al. Endosymbiont metacommunities, mtDNA diversity and the evolution of the Bemisia tabaci (Hemiptera: Aleyrodidae) species complex. Mol Ecol. 2010;19:4365–4378. doi: 10.1111/j.1365-294X.2010.04775.x. [DOI] [PubMed] [Google Scholar]
- Gupta RS, Lorenzini E. Phylogeny and molecular signatures (conserved proteins and indels) that are specific for the Bacteroidetes and Chlorobi species. BMC Evol Biol. 2007;7:71. doi: 10.1186/1471-2148-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy L, Kultima JR, Andersson SGE. genoPlotR: comparative gene and genome visualization in R. Bioinformatics. 2010;26:2334–2335. doi: 10.1093/bioinformatics/btq413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund CM, Choe JE, Skau CT, Kovar DR, Welch MD. Rickettsia Sca2 is a bacterial formin-like mediator of actin-based motility. Nat Cell Biol. 2010;12:1057–1063. doi: 10.1038/ncb2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison CP, Douglas AE, Dixon AFG. A rapid method to isolate symbiotic bacteria from aphids. J Invertebr Pathol. 1989;53:427–428. [Google Scholar]
- Horn M, et al. Members of the Cytophaga-Flavobacterium-Bacteroides phylum as intracellular bacteria of acanthamoebae: proposal of “Candidatus Amoebophilus asiaticus.”. Environ Microbiol. 2001;3:440–449. doi: 10.1046/j.1462-2920.2001.00210.x. [DOI] [PubMed] [Google Scholar]
- Huson DH, Mitra S, Ruscheweyh H-J, Weber N, Schuster SC. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011;21:1552–1560. doi: 10.1101/gr.120618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrell KF, McBride MJ. The surprisingly diverse ways that prokaryotes move. Nat Rev Microbiol. 2008;6:466–476. doi: 10.1038/nrmicro1900. [DOI] [PubMed] [Google Scholar]
- Karlsson FH, Ussery DW, Nielsen J, Nookaew I. A closer look at bacteroides: phylogenetic relationship and genomic implications of a life in the human gut. Microb Ecol. 2011;61:473–485. doi: 10.1007/s00248-010-9796-1. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur SJ, et al. TolC-Dependent secretion of an Ankyrin repeat-containing protein of Rickettsia typhi. J Bacteriol. 2012;194:4920–4932. doi: 10.1128/JB.00793-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns DB. Bright insight into bacterial gliding. Science. 2007;315:773–774. doi: 10.1126/science.1138995. [DOI] [PubMed] [Google Scholar]
- Kitajima EW, et al. In situ observation of the Cardinium symbionts of Brevipalpus (Acari: Tenuipalpidae) by electron microscopy. Exp Appl Acarol. 2007;42:263–271. doi: 10.1007/s10493-007-9090-1. [DOI] [PubMed] [Google Scholar]
- Koskiniemi S, et al. Rhs proteins from diverse bacteria mediate intercellular competition. Proc Natl Acad Sci U S A. 2013;110:7032–7037. doi: 10.1073/pnas.1300627110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5 doi: 10.1186/gb-2004-5-2-r12. R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartillot N, Lepage T, Blanquart S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics. 2009;25:2286–2288. doi: 10.1093/bioinformatics/btp368. [DOI] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhartová I, et al. RTX proteins: a highly diverse family secreted by a common mechanism. FEMS Microbiol Rev. 2010;34:1076–1112. doi: 10.1111/j.1574-6976.2010.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo N, Casiraghi M, Salati E, Bazzocchi C, Bandi C. How many Wolbachia supergroups exist? Mol Biol Evol. 2002;19:341–346. doi: 10.1093/oxfordjournals.molbev.a004087. [DOI] [PubMed] [Google Scholar]
- Maddison WP, Maddison DR. Mesquite: a modular system for evolutionary analysis. 2011 Version 2.75 [cited 2014 Apr 21]. Available from: http://mesquiteproject.org. [Google Scholar]
- Manzano-Marín A, Lamelas A, Moya A, Latorre A. Comparative genomics of Serratia spp.: two paths towards endosymbiotic life. PLoS One. 2012;7:e47274. doi: 10.1371/journal.pone.0047274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauriello EMF, et al. Bacterial motility complexes require the actin-like protein, MreB and the Ras homologue, MglA. EMBO J. 2010;29:315–326. doi: 10.1038/emboj.2009.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride MJ. Cytophaga-flavobacterium gliding motility. J Mol Microbiol Biotechnol. 2004;7:63–71. doi: 10.1159/000077870. [DOI] [PubMed] [Google Scholar]
- McBride MJ, Zhu Y. Gliding motility and Por secretion system genes are widespread among members of the phylum bacteroidetes. J Bacteriol. 2013;195:270–278. doi: 10.1128/JB.01962-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot T, Shaevitz JW, Hartzell PL, Zusman DR. Evidence that focal adhesion complexes power bacterial gliding motility. Science. 2007;315:853–856. doi: 10.1126/science.1137223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, McCutcheon JP, Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 2008;42:165–190. doi: 10.1146/annurev.genet.41.110306.130119. [DOI] [PubMed] [Google Scholar]
- Morimoto S, Kurtti TJ, Noda H. In vitro cultivation and antibiotic susceptibility of a Cytophaga-like intracellular symbiont isolated from the tick Ixodes scapularis. Curr Microbiol. 2006;52:324–329. doi: 10.1007/s00284-005-0349-7. [DOI] [PubMed] [Google Scholar]
- Moya A, Guirao P, Cifuentes D, Beitia F, Cenis JL. Genetic diversity of Iberian populations of Bemisia tabaci (Hemiptera: Aleyrodidae) based on random amplified polymorphic DNA-polymerase chain reaction. Mol Ecol. 2001;10:891–897. doi: 10.1046/j.1365-294x.2001.01221.x. [DOI] [PubMed] [Google Scholar]
- Moya A, Peretó J, Gil R, Latorre A. Learning how to live together: genomic insights into prokaryote-animal symbioses. Nat Rev Genet. 2008;9:218–229. doi: 10.1038/nrg2319. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, et al. Prevalence of Cardinium bacteria in planthoppers and spider mites and taxonomic revision of “Candidatus Cardinium hertigii” based on detection of a new Cardinium group from biting midges. Appl Environ Microbiol. 2009;75:6757–6763. doi: 10.1128/AEM.01583-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakane D, Sato K, Wada H, McBride MJ, Nakayama K. Helical flow of surface protein required for bacterial gliding motility. Proc Natl Acad Sci U S A. 2013;110:11145–11150. doi: 10.1073/pnas.1219753110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan B, et al. Myxobacteria gliding motility requires cytoskeleton rotation powered by proton motive force. Proc Natl Acad Sci U S A. 2011;108:2498–2503. doi: 10.1073/pnas.1018556108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan B, et al. Flagella stator homologs function as motors for myxobacterial gliding motility by moving in helical trajectories. Proc Natl Acad Sci U S A. 2013;110:E1508–E1513. doi: 10.1073/pnas.1219982110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki EP. 2009 Structural RNA homology search and alignment using covariance models [Ph.D. thesis]. [Saint Louis]: Washington University, School of Medicine. [Google Scholar]
- Oakeson KF, et al. Genome degeneration and adaptation in a nascent stage of symbiosis. Genome Biol Evol. 2014;6:76–93. doi: 10.1093/gbe/evt210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver KM, Campos J, Moran NA, Hunter MS. Population dynamics of defensive symbionts in aphids. Proc Biol Sci. 2008;275:293–299. doi: 10.1098/rspb.2007.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver KM, Degnan PH, Burke GR, Moran NA. Facultative symbionts in aphids and the horizontal transfer of ecologically important traits. Annu Rev Entomol. 2010;55:247–266. doi: 10.1146/annurev-ento-112408-085305. [DOI] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- Park J, et al. Identification of biotypes and secondary endosymbionts of Bemisia tabaci in Korea and relationships with the occurrence of TYLCV disease. J Asia Pac Entomol. 2012;15:186–191. [Google Scholar]
- Penz T, et al. Comparative genomics suggests an independent origin of cytoplasmic incompatibility in Cardinium hertigii. PLoS Genet. 2012;8:e1003012. doi: 10.1371/journal.pgen.1003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. Vienna (Austria): the R Foundation for Statistical Computing; 2011. R: a language and environment for statistical computing. [Google Scholar]
- Rader BA, Kremer N, Apicella MA, Goldman WE, McFall-Ngai MJ. Modulation of symbiont lipid A signaling by host alkaline phosphatases in the squid-vibrio symbiosis. MBio. 2012;3i:e00093–12. doi: 10.1128/mBio.00093-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JA, Latorre A, Sabater-Muñoz B, Moya A, Moran NA. Sidestepping secondary symbionts: widespread horizontal transfer across and beyond the Aphidoidea. Mol Ecol. 2003;12:1061–1075. doi: 10.1046/j.1365-294x.2003.01780.x. [DOI] [PubMed] [Google Scholar]
- Santos-Garcia D, et al. Complete genome sequence of “Candidatus Portiera aleyrodidarum” BT-QVLC, an obligate symbiont that supplies amino acids and carotenoids to Bemisia tabaci. J Bacteriol. 2012;194:6654–6655. doi: 10.1128/JB.01793-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, et al. A protein secretion system linked to bacteroidetes gliding motility and pathogenesis. Proc Natl Acad Sci U S A. 2010;107:276–281. doi: 10.1073/pnas.0912010107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz-Esser S, Penz T, Spang A, Horn M. A bacterial genome in transition—an exceptional enrichment of IS elements but lack of evidence for recent transposition in the symbiont Amoebophilus asiaticus. BMC Evol Biol. 2011;11:270. doi: 10.1186/1471-2148-11-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz-Esser S, et al. The genome of the amoeba symbiont “Candidatus Amoebophilus asiaticus” reveals common mechanisms for host cell interaction among amoeba-associated bacteria. J Bacteriol. 2010;192:1045–1057. doi: 10.1128/JB.01379-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley LD, Hâkansson S, Carruthers VB. Gliding motility: an efficient mechanism for cell penetration. Curr Biol. 1998;8:R12–R14. doi: 10.1016/s0960-9822(98)70008-9. [DOI] [PubMed] [Google Scholar]
- Sibley LD, et al. Intracellular parasite invasion strategies. Science. 2004;304:248–253. doi: 10.1126/science.1094717. [DOI] [PubMed] [Google Scholar]
- Skaljac M, Zanic K, Ban SG, Kontsedalov S, Ghanim M. Co-infection and localization of secondary symbionts in two whitefly species. BMC Microbiol. 2010;10:142. doi: 10.1186/1471-2180-10-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, Moran NA. Endosymbiotic bacteria as a source of carotenoids in whiteflies. Biol Lett. 2012;8:986–989. doi: 10.1098/rsbl.2012.0664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding MD, Prigge ST. Lipoic acid metabolism in microbial pathogens. Microbiol Mol Biol Rev. 2010;74:200–228. doi: 10.1128/MMBR.00008-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spormann AM. Gliding motility in bacteria: insights from studies of Myxococcus xanthus. Microbiol Mol Biol Rev. 1999;63:621–641. doi: 10.1128/mmbr.63.3.621-641.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Sun M, Wartel M, Cascales E, Shaevitz JW, Mignot T. Motor-driven intracellular transport powers bacterial gliding motility. Proc Natl Acad Sci U S A. 2011;108:7559–7564. doi: 10.1073/pnas.1101101108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thao MLL, Baumann P. Evolutionary relationships of primary prokaryotic endosymbionts of whiteflies and their hosts. Appl Environ Microbiol. 2004;70:3401–3406. doi: 10.1128/AEM.70.6.3401-3406.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toft C, Andersson SGE. Evolutionary microbial genomics: insights into bacterial host adaptation. Nat Rev Genet. 2010;11:465–475. doi: 10.1038/nrg2798. [DOI] [PubMed] [Google Scholar]
- Uchiyama I. MBGD: microbial genome database for comparative analysis. Nucleic Acids Res. 2003;31:58–62. doi: 10.1093/nar/gkg109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varani AM, Siguier P, Gourbeyre E, Charneau V, Chandler M. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 2011;12:R30. doi: 10.1186/gb-2011-12-3-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes GR, Bolker B, Lumley T. gplots: various R programming tools for plotting data. 2013 [Google Scholar]
- Wernegreen JJ. Strategies of genomic integration within insect-bacterial mutualisms. Biol Bull. 2012;223:112–122. doi: 10.1086/BBLv223n1p112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JA, Kelly SE, Cockburn SN, Perlman SJ, Hunter MS. Endosymbiont costs and benefits in a parasitoid infected with both Wolbachia and Cardinium. Heredity (Edinb) 2011;106:585–591. doi: 10.1038/hdy.2010.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RAM, Westrop GD, Coombs GH. Two pathways for cysteine biosynthesis in Leishmania major. Biochem J. 2009;420:451–462. doi: 10.1042/BJ20082441. [DOI] [PubMed] [Google Scholar]
- Xie G, et al. Genome sequence of the cellulolytic gliding bacterium Cytophaga hutchinsonii. Appl Environ Microbiol. 2007;73:3536–3546. doi: 10.1128/AEM.00225-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zchori-Fein E, Perlman SJ. Distribution of the bacterial symbiont Cardinium in arthropods. Mol Ecol. 2004;13:2009–2016. doi: 10.1111/j.1365-294X.2004.02203.x. [DOI] [PubMed] [Google Scholar]
- Zchori-Fein E, et al. Characterization of a “Bacteroidetes” symbiont in Encarsia wasps (Hymenoptera: Aphelinidae): proposal of “Candidatus Cardinium hertigii.”. Int J Syst Evol Microbiol. 2004;54:961–968. doi: 10.1099/ijs.0.02957-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.