

Abstract
The reduced protein expression of SIRT6 tumor suppressor is involved in tumorigenesis. The molecular mechanisms underlying SIRT6 protein downregulation in human cancers remain unknown. Using a proteomic approach, we have identified the ubiquitin-specific peptidase USP10, another tumor suppressor, as one of the SIRT6-interacting proteins. USP10 suppresses SIRT6 ubiquitination to protect SIRT6 from proteasomal degradation. USP10 antagonizes the transcriptional activity of the c-Myc oncogene through SIRT6, as well as p53, to inhibit cell cycle progression, cancer cell growth, and tumor formation. To support this conclusion, we detected significant reductions in both USP10 and SIRT6 protein expression in human colon cancers. Our study discovered crosstalk between two tumor-suppressive genes in regulating cell cycle progression and proliferation and showed that dysregulated USP10 function promotes tumorigenesis through SIRT6 degradation.
Reduced expression and loss-of-function mutation of tumor suppressor genes are common molecular mechanisms that contribute to tumor development, progression, and metastasis. The sirtuin family histone deacetylase member SIRT6 was recently shown to be a tumor suppressor and reduced SIRT6 expression has been detected in human primary cancers (Sebastian et al., 2012). SIRT6 functions as a tumor suppressor through multiple molecular mechanisms. Early studies showed that SIRT6 is a chromatin-bound factor that maintains genomic stability (Mostoslavsky et al., 2006). It localizes to telomeres in human cells and controls cellular senescence and telomere structure by deacetylating histone H3 lysine 9 (H3K9) (Michishita et al., 2008). SIRT6 also promotes DNA end resection by deacetylating CtBP-interacting protein (Kaidi et al., 2010). SIRT6 is also important in suppressing gene transcription of transcription factors, such as HIF-1a and c-myc, or it is recruited to chromatin by transcription factors, such as NF-κB and AP-1, thus deacetylating histone H3K9 to regulate gene transcription (Kawahara et al., 2009; Sebastian et al., 2012; Tasselli and Chua, 2012; Zhong et al., 2010). Therefore, SIRT6 exerts its functions through multiple molecular mechanisms.
It has been reported that the expression of SIRT6 can be regulated at both the transcriptional and post-transcriptional levels. The transcription factors FOXO3a and NRF1 directly bind to the SIRT6 promoter and positively regulate expression of SIRT6, which, in turn, negatively regulates glycolysis (Kim et al., 2010). At the post-transcriptional level, the AP-1 family transcription factor c-Fos induces SIRT6 transcription to suppress the anti-apoptotic activity of survivin by reducing histone H3K9 acetylation and NF-κB activation (Min et al., 2012). A recent study has suggested that microRNA-34a/b targets SIRT6 to regulate fatty acid metabolism and insulin signaling, indicating that post-transcriptional regulation is involved in SIRT6 expression (Davalos et al., 2011). However, the factors that regulate SIRT6’s biological functions at the post-translational levels have not been identified.
USP10 is a member of the mammalian ubiquitin-specific peptidases (USP). Although its biological functions remain largely unknown, evidence suggests that USP10 might suppress tumors by reversing Mdm2-induced p53 nuclear export and degradation (Jochemsen and Shiloh, 2010; Yuan et al., 2010). Hence, USP10 suppresses tumor cell growth in cells with wildtype p53. More recently, it was shown that Beclin1, a tumor suppressor that is frequently lost in human cancers, controls the protein stability of USP10 as well as USP13, thus regulating their deubiquitinating activities. Since USP10 mediates the deubiquitination of p53, regulation of the deubiquitination activity of USP10 by Beclin1 likely plays an important role in tumor suppression (Jochemsen and Shiloh, 2010; Liu et al., 2011; Yuan et al., 2010).
By using a proteomic approach, we identified USP10 as an interaction partner of SIRT6. This finding implies that they form a novel regulatory mechanism. In fact, further studies indicated that USP10 is a SIRT6-specific deubiquitinase that suppresses tumor growth by antagonizing transcriptional activity of the oncogene c-myc. Suppression of USP10 expression promotes human colon cancer cell growth and tumor formation through proteasomal degradation of SIRT6. Of note, we further discovered that human colon cancer tissues had reduced protein expression of both USP10 and SIRT6 compared with adjacent normal tissues. These studies reveal a novel molecular mechanism underlying impaired SIRT6 protein expression in tumorigenesis.
Results
Identification of a highly specific SIRT6 interactome
To determine the molecular mechanisms underlying the impaired protein expression of the tumor suppressor SIRT6 in tumorigenesis, we used a proteomic approach and identified SIRT6 interaction proteins from HCT116 cells, as we recently reported (Lin et al., 2012). Briefly, whole-cell lysate from Flag-SIRT6 expressing cells was subjected to immunoprecipitation with anti-Flag-conjugated agarose beads after extensive precleaning. The beads were washed and eluted with 3× Flag peptide. A small fraction of the eluent was subjected to SDS-PAGE and silver staining; multiple bands, including a strong SIRT6 protein band, were visualized (Fig. 1A). The rest of eluents were analyzed by mass spectrometry after trypsin digestion. As indicated in Fig. 1B, 26 proteins have been identified by this approach, many of which are involved in a variety of biological functions, including signal transduction, RNA processing, ubiquitination and proteolysis, metabolism, and nuclear acid binding (Fig. 1C), indicating that SIRT6 is likely involved in different aspects of biological functions. We then validated SIRT6 interactors by co-immunoprecipitation (co-IP) and western blotting. Of the 26 candidate interactors, 16 were confirmed to be SIRT6-specific interacting proteins (Fig. S1) and four are the known SIRT6-interacters, including PARP1 (Mao et al., 2011), histones H3, H4b and H1FX (Kawahara et al., 2009; Michishita et al., 2008; Zhong et al., 2010). More interestingly, G3BP2, a previously known USP10-interacting protein (Matsuki et al., 2013; Ward et al., 2011), was detected in the SIRT6-pulldown products, suggesting that SIRT6 forms a complex with USP10 and G3BP2. Whereas some candidates, such as MAP3K7, were confirmed as true SIRT6 interactors with only a single peptide detected by mass spectrometry, no more than two peptides were detected for all five false-positive candidates (Fig. 1B, Fig. S1 & S2). Those results showing a positive correlation of the peptide numbers indicate that our proteomic approach revealed, with confidence, true SIRT6 interactors.
Fig. 1. Identification of SIRT6 interactome.
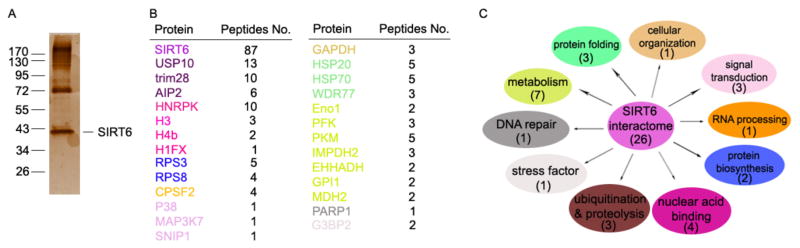
(A) Flag-SIRT6 pull-down products from HCT116 cells were separated by SDS-PAGE and visualized by silver staining. (B) SIRT6-interacting proteins were identified by mass spectrometry. The total peptide numbers of each protein are indicated. (C) Classification of SIRT6-associated proteins. The pathways matched to the candidates in (B) with the same colors.
USP10 interacts with SIRT6
The ubiquitin-specific peptidase USP10, a tumor suppressor that often has low expression in human cancers (Yuan et al., 2010), was confirmed as a SIRT6 interaction partner (Fig. 1D/a). Because USP10 is a deubiquitinase that can protect its interaction partners from ubiquitin-mediated degradation (Draker et al., 2011; Yuan et al., 2010), we speculated that the reduction of SIRT6 protein might be a functional consequence of USP10 reduction in human cancers. To test this hypothesis, we validated the specificities of USP10 and SIRT6 interactions. As indicated in Fig. 2A, USP10 interacts with SIRT6 but not with any of the other six sirtuin family proteins. Conversely, SIRT6 specifically interacts with USP10 but not with USP22 or USP47 (Fig. 2B). Moreover, the specific interaction between the endogenous USP10 and SIRT6 was confirmed in HCT116 colon cancer cells because SIRT6 protein was detected in the immunoprecipitates with anti-USP10 antibody, but not with the control mouse IgG (Fig. 2C). Therefore, we identified USP10 as a specific interacting protein of SIRT6.
Fig. 2. USP10 specifically interacts with and deubiquitinates SIRT6.

(A) USP10 specifically interacts with SIRT6. The expression plasmid of Myc-USP10 was transiently transfected with each of the indicated Flag-SIRTuins into HCT116 cells. Their interactions were examined by co-IP with anti-Flag Abs and by western blotting with anti-Myc Abs (top panel), and the same membranes were reprobed with anti-Myc (2nd panel). The protein expression levels of SIRT1-7 in whole-cell lysates were confirmed by western blotting (bottom panel). (B) SIRT6 interaction with each of the indicated USPs was analyzed as in (A). (C) The interaction of endogenous USP10 and SIRT6 in human colon cancer cells. Cell lysates were pre-cleaned and then subjected to immunoprecipitation with anti-USP10 specific Abs using normal mouse IgG as a control. The bound SIRT6 was determined by western blotting using anti-SIRT6 Abs (top panel). The protein expression levels of USP10 (middle panel) and SIRT6 (bottom) in the whole cell lysate were confirmed by western blotting. (D) Domain structures of USP10 and its truncated mutants. USP10 protein has a conserved catalytic domain in its C-terminus. (E) HA-SIRT6 plasmid was cotransfected with USP10 or with each of its truncated mutants. Their interactions were determined as described in (A). (F) Domain structures of SIRT6 and its truncated mutants. SIRT6 protein has a conserved HDAC domain or SIRTuin/SIRT domain, indicated in red. (G) Myc-USP10 plasmid was cotransfected with Flag-tagged SIRT6 or each of its truncated mutants. Their interaction was determined as described in (A). (H) Flag-SIRT6 and HA-ubiquitin expression plasmid DNA was cotransfected with USP10, USP/CA mutant, or USP22 into HCT116 cells. SIRT6 ubiquitination in transiently transfected cells was analyzed by co-IP with anti-Flag Abs and western blotting with anti-HA Abs (top panel). The protein expression levels of SIRT6 were confirmed by western blotting (middle panel). (I) Ubiquitinated Flag-SIRT6 proteins in transiently transfected HCT116 cells were pulled down by anti-Flag-conjugated beads, followed by incubation with purified GST-USP10, GST-USP10/CA, or GST-USP22 proteins. SIRT6 ubiquitination levels were determined by western blotting with anti-HA (top panel), and SIRT6 and GST or GST fusion proteins were confirmed by western blotting (bottom two panels). (J) HCT116 colon cancer cells were transfected with shRNA specifically against USP10 or USP22 using a commercial control shRNA (ctrl). The effects of USP10 or USP22 knockdown on SIRT6 ubiquitination were determined as described in (G). Tubulin was used as loading control.
Next, we mapped the regions of USP10 that mediate its interaction with SIRT6 by generating truncated mutants (Fig. 2D). USP10 protein carries a C-terminal C19 peptidase domain and an N-terminal regulatory region. Co-IP and western blotting analysis revealed that the N-terminal regulatory domain of USP10 is required for its interaction with SIRT6, as deletion of this region completely abolished its interaction with SIRT6. In contrast, expression of the USP10 N-terminal 1–205 fragment alone is sufficient to pull SIRT6 protein down (Fig. 2E). Similarly, truncated mutation analyses showed that the C-terminus of SIRT6 is required for its interaction with USP10 (Fig. 2F & 2G).
USP10 negatively regulates SIRT6 ubiquitination
Because USP10 suppresses the ubiquitination of its interacting proteins, which protects them from degradation (Draker et al., 2011; Yuan et al., 2010), we analyzed the effect of USP10 expression on SIRT6 ubiquitination. We detected SIRT6 protein ubiquitination in transiently transfected HCT116 cells (Fig. 2H, lane 1). Co-expression of wildtype USP10 with SIRT6 significantly inhibited its ubiquitination (Fig. 2H, lane 2). In contrast, the deubiquitinase catalytically inactive USP10/CA mutant failed to suppress SIRT6 ubiquitination (Fig. 2H, lane 3). USP10-mediated SIRT6 deubiquitination is highly specific, as co-expression of USP22, which was recently shown to be a SIRT1-specific deubiquitinase (Lin et al., 2012), did not have any effect on SIRT6 ubiquitination (Fig. 2H, lane 4). The suppression of SIRT6 ubiquitination by the deubiquitinase activity of USP10 was further confirmed by using an in vitro deubiquitination assay. Incubation of ubiquitinated SIRT6 with a purified GST-USP10 fusion protein, but not with the GST-USP10/CA mutant or the GST-USP22 fusion protein, inhibited SIRT6 ubiquitination (Fig. 2I). Conversely, suppression of USP10 by shRNA-mediated knockdown in HCT116 cells resulted in elevated SIRT6 ubiquitination. As a control, SIRT6 ubiquitination was not affected by shRNA specific to USP22 (Fig. 2J). Together with the fact that USP10 interacts with SIRT6, our studies indicate that USP10 is a specific deubiquitinase of SIRT6.
USP10 protects SIRT6 protein from proteasome-mediated degradation
Ubiquitination promotes protein degradation, which can be reversed by ubiquitin-specific peptidases. Our discovery that USP10 is a deubiquitinase of SIRT6 implies that USP10 might regulate SIRT6 protein stability. Indeed, gain of USP10 functions by transient transfection resulted in elevated SIRT6 protein expression and prolonged half-life of SIRT6 proteins in HCT116 cells (Fig. 3A & 3B). In contrast, mRNA expression levels of SIRT6 were not affected by USP10 expression (Fig. 3C). Expression of a deubiquitinase catalytically inactive USP10/CA mutant failed to protect SIRT6 from degradation, indicating that SIRT6 stabilization requires ubiquitin-specific peptidase activity by USP10 (Fig. 3D & 3E). Conversely, shRNA-mediated knockdown of USP10 decreased the endogenous SIRT6 protein levels without affecting SIRT6 mRNA levels (Fig. 3F–H). As a negative control, suppression of USP22 expression did not affect SIRT6 protein stability (Fig. 3F). Moreover, we found that treatment of cells with the proteasome-specific inhibitor MG132 protected SIRT6 from degradation (Fig. 3I), suggesting that SIRT6 protein degradation occurs through the proteasomal pathway. Taken together, our results indicate that USP10 is a SIRT6-specific deubiquitinase that protects SIRT6 protein from ubiquitin-mediated degradation through the proteasomal pathway. The same conclusion was further confirmed using another colon cancer cell line RKO that expresses a wildtype p53 gene. As shown in Fig. S3, over-expression of USP10 but not its CA mutant prolonged SIRT6 half-life. In contrast, knockdown of USP10 but not USP22 in RKO cells promoted SIRT6 degradation.
Fig. 3. USP10 protects SIRT6 protein from degradation.

(A&B) USP10 expression plasmids or empty vectors were transfected into HCT116 cells. The transfected cells were treated with cycloheximide (CHX) for different times. The protein levels in the treated cells were determined by western blotting using anti-SIRT6 (top panel) and anti-Flag Abs (middle panel). Tubulin was used as a loading control (bottom panel) (A). The band intensities of SIRT6 proteins were quantified, and their relative levels are shown in (B). (C) A fraction of cells from (A) was prepared in parallel for total RNA extraction. The mRNA levels of both USP10 and SIRT6 were determined by real-time PCR. Their relative levels are indicated. The error bar represents the SEM of triplicate experiments. *P<0.05, two-tailed Student t test. (D & E) USP10 or USP10/CA mutant plasmids were transfected into HCT116 cells. SIRT6 protein stabilities in the transiently transfected HCT116 cells was examined as described in (A & B). (F & G) HCT116 cells were transfected with control shRNA or with shRNA specifically against USP10 or USP22. SIRT6 protein stabilities were analyzed as described in (A) (top panels). The expression levels of USP10 (2nd panel) and USP22 (3rd panel) were confirmed by western blotting using Tubulin as a loading control (bottom panel) (F). The relative expression levels of SIRT6 protein are indicated (G). (H) The relative mRNA levels of SIRT6, USP10, and USP22 in cells used in panels F and G were analyzed by real-time PCR. The error bars represent the SEM of triplicate experiments. *P < 0.05, two-tailed Student t test. (I) USP10 or USP10/CA mutant plasmids were transfected into HCT116 cells and treated with the proteasome inhibitor MG132 as indicated 48 hours after transfection. The protein levels of SIRT6 (top panel) and USP10 (middle panel) were determined by western blotting using Tubulin as loading control (bottom panel).
USP10 inhibits c-Myc transcriptional activity
Recent studies have shown that one of the molecular mechanisms by which SIRT6 carries out its tumor-suppressive function is through suppressing the transcriptional activity of the c-Myc oncogene (Sebastian et al., 2012). Thus, we determined whether USP10 functions as a tumor suppressor through SIRT6-mediated c-Myc inactivation. In fact, we demonstrated that USP10 knockdown in human colon cancer cells leads to elevated expression of c-Myc target genes, including the proliferating cell nuclear antigen (PCNA) (Gazitt et al., 1993), cell division control protein 2 (cdc2), cyclin A1 (CCNA1), and cdc25c (Charollais et al., 1990; Dalal et al., 1999; Furukawa et al., 1990), presumably because USP10 knockdown resulted in increased c-Myc transcriptional activity. We then reasoned that, if USP10 suppresses c-Myc transcriptional activity through SIRT6 stabilization, knockdown of SIRT6 expression should achieve a similar level of increase in c-Myc target gene expression in human colon cancer cells. To our surprise, SIRT6 gene suppression resulted in a lower level of increase in c-Myc target gene expression than that of USP10 knockdown, implying that additional factors may contribute to USP10-mediated c-Myc suppression. To support this notion, over-expression of SIRT6 only partially suppressed the elevated c-Myc target gene expression caused by USP10 gene knockdown (Fig. 4A). It has been shown that SIRT6 can be recruited to the chromatin by specific transcription factors to suppress gene transcription through antagonizing histone acetylation at the specific promoter regions (Kawahara et al., 2009; Zhong et al., 2010). Indeed, we detected the elevated promoter binding of SIRT6 in cells with USP10 over-expression (Fig. 4B). As a consequence, the levels of the lysine 9 acetylation in histone H3 protein (H3K9) at the promoters of c-Myc targets were significantly reduced (Fig. 4C). Collectively, our results indicate that USP10 suppresses c-Myc transcriptional activity partially through SIRT6.
Fig. 4. USP10 inhibits c-Myc transcriptional activity.

(A) Total RNA from HCT116 cells expressing indicated plasmids or combination were extracted (sh-USP10 & sh-SIRT6: sh-RNA-mediated knockdown; USP10 & SIRT6: over-expression). The level of each indicated c-Myc target gene was analyzed by real-time PCR using β-actin as an internal control. The error bars represent the SEM of triplicate experiments. (B) USP10 expression plasmids or empty vectors were transfected into HCT116 cells. 48 hours later, the transfected cells were cross-linked with 1% formaldehyde for 10 minutes, and lysed with SDS lysis buffer. ChIP assay was performed according to the manufacturer’s protocol by ChIP with anti-SIRT6 antibody. QPCR was then performed with c-Myc target gene primers. The experiment was performed in triplicate, and data were represented as the means ± S.D. The asterisks indicate a statistically significant difference (*, p<0.05). (C) c-Myc target gene promoter sequences were amplified from genomic DNA pulled down by ac-H3K9 CHIP and analyzed as in (A). The asterisks indicate a statistically significant difference (*, p<0.05; **, p<0.01). (D) HCT116 cells were transfected with USP10 or with SIRT6 expression plasmids or both, or with their specific shRNAs as indicated. Three days after transfection, cell cycle progression was determined by propidium iodide staining followed by flow cytometry. The percentage of cells in each indicated cell cycle stage is shown.
The oncogene c-Myc is a transcription factor that promotes tumor cell cycle progression by regulating the expression levels of genes that are involved in cell cycle progression (Charollais et al., 1990; Charollais and Mester, 1988; Dalal et al., 1999; Ely et al., 1987; Furukawa et al., 1990; Gazitt et al., 1993; Kelly et al., 1983). We then determined whether USP10 and SIRT6 act as tumor suppressors by inhibiting the cell cycle progression of cancer cells. As expected, ectopic expression of either USP10 or SIRT6 led to a significant reduction in the percentage of cells in the G2/M and S phases, and co-expression of both USP10 and SIRT6 achieved synergistic suppression of colon cancer cell cycle progression (Fig. 4D). Conversely, knockdown of either USP10 or SIRT6 facilitated cell cycle progression by increasing the percentage of cells at S and G2/M phases. By knockdown of both USP10 and SIRT6, we also confirmed their synergy in promoting cell cycle progression (Fig. 4D). Because one of the molecular mechanisms by which SIRT6 achieves its tumor suppressive functions is through c-Myc inhibition (Sebastian et al., 2012), our results suggest that USP10 inhibits the c-Myc oncogene, at least partially, through SIRT6 stabilization.
USP10 suppresses c-Myc transcriptional activity through SIRT6 and p53
Our results in Fig. 4 imply that additional factors are involved in USP10-mediated c-Myc suppression. It has been reported that one of the molecular mechanisms underlying USP10 tumor-suppressive functions is that it protects p53 from degradation (Yuan et al., 2010), raising the possibility that USP10 also antagonizes c-Myc transcriptional activity through p53. To test this, we compared the c-Myc-suppressing activities of USP10 in p53 wildtype and p53−/− HCT116 human colon cancer cells. As shown in Fig. 5A, the expression levels of c-Myc target gene were elevated in p53−/− HCT116 cells compared to HCT116 cells carrying a wildtype p53 gene, confirming previous studies showing that p53 antagonizes c-Myc transcriptional activity (Aguda et al., 2011; Golomb et al., 2012; Ho et al., 2005; Kress et al., 2011; Rochlitz et al., 1995; Wang et al., 2010). Expression of USP10 significantly suppressed c-Myc transcriptional activity in p53 wildtype and p53−/− HCT116 cells, but the levels of USP10-mediated suppression in c-Myc target gene transcription in p53-null cells were partially reversed. These results indicate that p53 is downstream of USP10 in suppressing c-Myc transcriptional activity, but additional factors, one of which presumably is SIRT6 as indicated from our results in Fig. 4A, are involved in USP10-mediated c-Myc suppression. To support this speculation, we discovered that knockdown of USP10 expression resulted in dramatic elevations in the transcription of c-Myc target genes (Fig. 5A). Notably, USP10 knockdown largely diminished the differences in c-Myc target gene expression levels between wildtype and p53−/− HCT116 cells, indicating that loss of USP10 releases the suppressive activity of both SIRT6 and p53 to c-Myc target gene transcription. To further prove the principle that USP10 suppresses c-Myc transcriptional activity through both SIRT6 and p53, we tested whether knockdown of SIRT6 rescues this USP10 suppressive activity. Indeed, knockdown of SIRT6 in p53+/+ cells partially reversed the suppressive activity of USP10. In contrast, the suppressive activity of USP10 to c-Myc transcriptional activity was completely reversed by SIRT6 knockdown in p53-null cells (Fig. 5A), providing a direct evidence that SIRT6 is involved in c-Myc suppression in the absence of p53.
Fig. 5. Inhibition of c-Myc activity by USP10 depends on both SIRT6 and p53.

(A) Wildtype or p53−/− HCT116 cells stably expressing the control shRNA or specific to USP10 (sh-USP10) or SIRT6 (sh-SIRT6) or with USP10 expression plasmid at each indicated combination. Total RNA was extracted from and the levels of c-Myc target genes were analyzed by real-time PCR using β-actin as an internal control. (B) Total RNA was extracted from wildtype or SIRT6−/− MEF cells expressing indicated plasmids or combination of plasmids and the levels of c-Myc target genes were analyzed as in (A). (C) HCT116 cells were transfected with indicated combinations of plasmids. Three days after transfection, the cell cycle stage was determined by PI staining and flow cytometry. The percentage of cells in each cell cycle stage is indicated.
We then used SIRT6-null mouse embryonic fibroblasts (MEFs) to prove the principle that SIRT6 is another downstream factor in USP10-mediated c-Myc suppression. Similar to that in p53−/− cells, a significant increase in c-Myc transcriptional activity in SIRT6-deficient MEFs was detected (Fig. 5B), confirming SIRT6 as a c-Myc suppressor (Sebastian et al., 2012). Consistent with the results shown in Fig. 5A, USP10 knockdown resulted in a dramatic increase in c-Myc target gene transcription both in wildtype and in SIRT6−/− MEF cells and differences in c-Myc target gene expression levels between the wildtype and in SIRT6−/− MEF cells are largely reduced. Conversely, expression of USP10 significantly suppressed c-Myc target gene transcription both in wildtype MEFs, and the USP10 suppressive activity of c-Myc target gene expression was partially reversed in SIRT6−/− MEFs (Fig. 5B), further suggesting that other factors, such as p53, are involved in USP10-mediated c-Myc suppression. To support this, p53 knockdown in SIRT6−/− MEFs resulted in a significant increase in c-Myc target gene transcription, which is to a similar level as that of USP10 knockdown. In addition, p53 knockdown partially abolished USP10 over-expression-mediated suppression in c-Myc target gene expression in SIRT6+/+ MEFs, and p53 knockdown in SIRT6−/− MEFs fully reversed USP10-mediated suppression of c-Myc targets expression (Fig. 5B). Based on the results from experiments using both p53-null HCT116 cells and SIRT6-null MEFs, we conclude that USP10 regulates c-Myc transcriptional activity through both p53 and SIRT6.
Furthermore, USP10 knockdown-mediated cell cycle progression was partially rescued by either SIRT6 or p53 co-expression. A complete rescue was achieved by p53 and SIRT6 co-expression in the USP10 knocked down cells (Fig. 5C). Collectively, these studies indicate that one of the molecular mechanisms by which USP10 achieves tumor suppression is through potentiating both SIRT6- and p53-mediated suppression of c-myc.
Downregulation of USP10 leads to enhanced cancer cell proliferation and tumorigenesis
c-Myc is an oncogenic transcription factor that promotes cell cycle progression and tumor growth (Chiswell et al., 1981; Hayward et al., 1981). Therefore, USP10 might antagonize c-Myc activity to inhibit tumor cell growth though SIRT6 and p53. To test this hypothesis, we determined whether suppression of USP10 promotes tumor formation by affecting tumor cell growth. As expected, USP10 over expression inhibited the proliferation of colon cancer cells with either a wildtype p53 or with p53-deficency. On the other hand, shRNA-mediated USP10 knockdown caused more vigorous tumor cell growth of both wildtype and p53-null cells, confirming the tumor-suppressive function of USP10 (Fig. 6A, 6B, S4A & S4B). SIRT6 knockdown in p53-null HCT116 colon cancer cells enhanced their proliferation to a similar level caused by the USP10 knockdown. In contrast, SIRT6 knockdown in either HCT116 and RKO colon cancer cells that carry a wildtype p53 gene resulted in a much lower level of increase in cell proliferation than that of p53-null cell. On the other hand, SIRT6 knockdown partially abolished the suppressive activity in cancer cell growth by USP10 expression in p53 wildtype, but not p53-null colon cancer cells (Fig. 6A, 6B, S4A & S4B). Therefore, USP10 suppresses colon cancer cell growth through both SIRT6 and p53.
Fig. 6. Downregulation of USP10 leads to enhanced cell proliferation and tumorigenesis.

(A & B) The cell proliferation of wildtype or p53−/− HCT116 cells stably expressing indicated plasmids or combination of plasmids were determined either by counting cell numbers (A) or by WST-1 assay (B). The error bar represents the SEM of triplicate experiments. (C & D) Anchorage-independent colony formation of wildtype or p53−/− HCT116 cells stably expressing indicated plasmids or combination of plasmids was determined by soft agar assay. Representative images from three experiments are shown (C). The average number of colonies from three experiments is indicated (D). (E) 2×106 wildtype or p53−/− HCT116 cells stably expressing indicated plasmids or combination of plasmids were injected subcutaneously into nude mice (n=3 per group). Three weeks after injection, mice were euthanized, tumors were isolated and photographed (E) and weighed at the end of the experiment (F). (G) Primary human colon cancer tissues (T) and their adjacent normal colon tissues (N) were used for western blotting analysis of the protein expression levels of USP10, SIRT6, CDC2 and CCNA2. Tubulin was used as loading control. (H) Total RNA was extracted from corresponding colon cancer tissues (T) and their adjacent normal colon tissues (N) in (G) and the mRNA levels of USP10 or SIRT6 were analyzed by real-time PCR. The error bars represent the SEM of triplicate experiments. (I) Our working model of c-Myc and SIRT6 regulation by USP10. Black arrow: known findings; Purple arrow: our finding in this study.
We then used in vitro anchorage-independent colony formation assay and analyzed the role of USP10-mediated c-Myc suppression in tumor formation. USP10 expression in either HCT116 or RKO colon cancer cells carrying wildtype p53 resulted in a dramatic decrease in colony number. Conversely, knockdown of USP10 expression significantly enhanced the colony formation (Fig. 6C & 6D and S4C & S4D). However, the suppression of colon cancer colony formation by gain of USP10 function is partially abrogated by either loss of p53 or SIRT6 gene knockdown (Fig. 6C & 6D). Therefore, USP10 appears to achieve its tumor suppressive function through both SIRT6 and p53. We further confirmed this conclusion using the xenograft model (Fig. 6E & 6F). When nude mice were implanted with HCT116 cells, three weeks later tumors were formed with average sizes of 0.5 g. Loss of p53 doubled the average tumor sizes, confirming p53 is a tumor suppressor. USP10 stable expression in p53+/+HCT116 colon cancer cells reduced the tumor sizes to about 0.3 g in nude mice. Loss of p53 partially abrogated the suppressive activity of USP10 overexpression in tumor formation because the average tumor weights from the USP10-expression p53-null HCT116 cells were 0.7g comparing to 0.3 g by USP10-expressed p53+/+ HCT116 cells. Similarly, knockdown of SIRT6 partially reversed USP10 stable expression-mediated tumor suppression in wildtype but not p53-null HCT116 cells. In addition, knockdown of USP10 expression in wildtype and p53-null HCT116 cells resulted in the elevated tumor formation by increasing the average tumor weight to 1.2 and 1.5 g, respectively. Collectively, our results indicate that USP10 suppress tumorigenesis through a both p53 and SIRT6-dependent manner.
Decreased expression of USP10 and SIRT6 has been reported in human primary cancer tissues (Sebastian et al., 2012; Yuan et al., 2010). Our studies suggest that one of the molecular mechanisms underlying the decreased protein expression of SIRT6 is probably impaired USP10 expression. If so, we expected a positive correlation between the reduced protein expressions of USP10 and SIRT6. To test this hypothesis, freshly frozen human colon cancer tissues and adjacent normal tissue (as controls) were collected. Western blotting showed dramatic reductions in both USP10 and SIRT6 protein expression levels in colon cancer tissues compared to controls, indicating that reduced protein expression of the two tumor-suppressive genes is involved in human colon cancer development. Importantly and as expected, the reductions in USP10 and SIRT6 protein expression in human colon cancers showed a strong positive correlation (Fig. 6G). Next, we analyzed the mRNA expression levels of both USP10 and SIRT6 in human colon cancer samples using their adjacent tissues as controls. Results in Fig. 6H show that SIRT6 mRNA was reduced in two (#2 & #10) out of 10 cancer patients. However, regardless whether SIRT6 mRNA levels are reduced in cancer tissues or not, a positive correlation between the reduced SIRT6 and USP10 protein expression levels was detected (Fig. 6G). These results suggest that SIRT6 protein expression in human colon cancers were regulated at least through both mRNA and protein levels. We also analyzed the mRNA expression levels of USP10 in colon cancer tissue and our results show that USP10 mRNA levels are indistinguishable between cancer and normal tissues, implying that USP10 protein reduction is through a post-mRNA regulatory mechanism. Interestingly, significant reduction in the protein expression levels of c-Myc targets, both CDC2 and CCNA2, were detected in human primary colon cancers (Fig. 6G), further supporting our conclusion that USP10 suppresses tumor through antagonizing c-Myc transcriptional activation.
Based on these findings, we propose a model of USP10’s functions as a tumor suppressor (Fig. 6I). USP10 deubiquitinates and stabilizes SIRT6, which amplifies the tumor-suppressive activity of SIRT6. USP10 also potentially suppresses tumor formation by reversing Mdm2-induced p53 nuclear export and degradation (Yuan et al., 2010). The tumor suppressor USP10 antagonizes the transcriptional activity of the oncogene c-Myc through both SIRT6 and p53 protein stabilization. In contrast, reduction in USP10 expression facilitates SIRT6 and p53 protein degradation through the ubiquitin pathway, resulting in elevated activation of downstream oncogenes, such as c-myc. Therefore, the reduced expression of USP10 is positively associated with human cancer, and this association is presumably involved in promoting tumor development and progression.
Discussion
Our study demonstrated that USP10 suppresses tumor cell growth through potentiating both SIRT6- and p53-mediated suppression of the oncogene c-myc. This conclusion is supported by the following key discoveries: first, USP10 was identified as a SIRT6-interacting protein by using a proteomic approach, and their interaction was confirmed by co-IP and western blotting. Second, USP10 suppresses SIRT6 ubiquitination and protects SIRT6 from proteasome-mediated degradation. Third, USP10 antagonizes c-Myc transcriptional activity and cell cycle progression through both SIRT6 and p53. Fourth, USP10 suppresses human colon cancer cell growth and tumor formation. In addition, we discovered that the reduced USP10 protein expression levels positively correlated with reduction in SIRT6 protein expression in human primary colon cancers, providing a possible molecular explanation for the low SIRT6 level in cancers. These discoveries suggest that crosstalk between two tumor suppressors is involved in regulating cancer development and progression.
Several transcription factors, including FOXO3 and AP-1, have been shown to promote SIRT6 gene expression (Kim et al., 2010; Min et al., 2012). In addition, a recent study suggested that microRNA-34a/b targets SIRT6 to regulate fatty acid metabolism and insulin signaling (Davalos et al., 2011). However, whether and how SIRT6 is regulated at the post-translational level are not known. We showed that SIRT6 is extensively ubiquitinated and quickly degraded in human colon cancer cells. This ubiquitination-mediated degradation is likely through the proteasome, as treatment of cells with the proteasome inhibitor MG132 blocked SIRT6 degradation. The E3 ubiquitin ligases that catalyze SIRT6 ubiquitination are not known. Although a HECT-type E3 ligase, AIP2, was identified as a SIRT6-interacting protein by our proteomic study, its expression did not enhance SIRT6 ubiquitination, thereby excluding the possibility that AIP2 is a ubiquitin ligase of SIRT6. Another possible E3 ubiquitin ligase is the plant homeo-domain (PhD) finger-containing ligase TRIM28, because it was also identified as a SIRT6-interacting protein. However, evidence indicates that the PhD finger of TRIM28 is a ligase of the small ubiquitin-like modifier (Goodarzi et al., 2011; Ivanov et al., 2007; Liang et al., 2011). A recent study has identified CHIP (carboxyl terminus of Hsp70-interacting protein) as an E3 ubiquitin ligase of Sirt6 (Ronnebaum et al., 2013), it will be interesting to study the crosstalk among Sirt6, CHIP and USP10 during tumorigenesis.
We discovered that USP10 is a deubiquitinase of SIRT6 and protects SIRT6 from ubiquitination-mediated degradation in human colon cancer cells. Because the oncogene c-Myc was recently shown to be a substrate of SIRT6 (Sebastian et al., 2012), it was not surprising that we found that USP10 antagonizes c-myc-driven transcriptional activity. USP10 has also been shown to potentiate the functions of p53, a tumor suppressor that inhibits c-Myc expression (Liu et al., 2011). Consistent with these discoveries, we demonstrated that p53 is also involved in USP10-mediated c-Myc suppression, as ectopic USP10 expression partially inhibited c-Myc activity in cancer cells. Therefore, USP10 achieves its tumor-suppressive activity by stabilizing both SIRT6 and p53 proteins to antagonize c-Myc transcriptional activity. This discovery implies that in tumors with loss-of-function mutations in p53 or loss of p53 expression, USP10 suppresses tumor growth through SIRT6-mediated c-Myc suppression. When either the protein expression or the function of USP10 is deregulated, both SIRT6 and p53 proteins lose USP10 protection from ubiquitination-induced degradation. Consequently, the transcriptional activity of c-Myc is elevated, leading to tumor development or promotion of tumor cell growth. Therefore, USP10 achieves its tumor-suppressive functions by protecting two other tumor suppressors, SIRT6 and p53, from ubiquitination-mediated degradation. Interestingly, both SIRT6 and p53 share the same target—the c-Myc oncogene. In addition to c-Myc, it has been shown that Sirt6 achieves its function by suppressing HIF-1α (Zhong et al., 2010). However, our study did not detect any changes in HIF1-α transcriptional activity by USP10 expression under normal culture condition. Therefore, it seems that USP10-mediated SIRT6 stabilization is largely involved in regulating c-Myc transcriptional activity. One speculation is that the USP10 is specifically directed into the Sirt6/c-Myc but not Sirt6/HIF-1a complex. Additional efforts are needed to further delineate the molecular nature of this interesting specificity of USP10 in regulating Sirt6 functions.
Reduced protein expression levels of USP10 or SIRT6 have been shown in human cancers (Jochemsen and Shiloh, 2010; Sebastian et al., 2012; Yuan et al., 2010). We showed here that their reduction is positively associated with human colon cancers. Based on that finding and our discovery that USP10 is a deubiquitinase of SIRT6, we speculate that the reduction of SIRT6 is a consequence of, at least partially, a decrease in USP10 protein expression. Similarly, a positive correlation was found between reduced protein expression levels of USP10 and p53 in human clear cell carcinomas without p53 (Jochemsen and Shiloh, 2010; Yuan et al., 2010). However, the correlation patterns of USP10 with p53, as well as SIRT6 in human cancers with p53 mutations or deletions are not known. Further studies are needed to determine whether cooperative regulation of USP10 and SIRT6 protein expression is associated with p53 expression as well as p53 mutations.
Materials and Methods
Isolation of SIRT6 interactors using a proteomic approach
HCT116 cells were transfected with Flag-tagged SIRT6 expression plasmids. The transfected cells were lysed with RIPA buffer and pre-cleaned by incubating them with agarose beads three times. Flag-tagged SIRT6 proteins were immunoprecipitated with anti-Flag Ab-conjugated agarose, and the immune complex was eluted from the agarose with 100 μM Flag peptide (Sigma Aldrich). A small fraction of eluent was subjected to SDS-PAGE analysis and silver staining using a silver staining kit (Thermo Scientific). The rest of the eluted proteins were digested with trypsin and characterized by mass spectrometry.
In vivo and in vitro deubiquitination assay
For the in vivo ubiquitination assay (Lin et al., 2012), cells were lysed with ubiquitination buffer containing 1% SDS and boiled at 95°C for 10 min. The denatured cell lysates were diluted with SDS-negative RIPA buffer to reduce SDS to 0.2% and then subjected to co-IP followed by western blotting with anti-HA or anti-ubiquitin Abs. The in vitro deubiquitination assay was performed as described (Shan et al., 2009; Yuan et al., 2010). Briefly, HCT116 cells were transiently transfected with Flag-SIRT6 and HA-ubiquitin expression plasmids. Ubiquitinated SIRT6 proteins were immunoprecipitated with anti-Flag antibody-conjugated Sepharose (Sigma Aldrich) and eluted with the Flag peptide. The purified ubiquitinated SIRT6 proteins were incubated with GST or GST-USP10 or GST-USP22 proteins in deubiquitination buffer (50 mM Tris-HCl, pH 8.0; 50 mM NaCl; 1 mM EDTA; 10 mM DTT, and 5% glycerol) at 37°C for 2 hours. SIRT6 ubiquitination was detected by western blotting with anti-HA Abs.
Chromatin Immunoprecipitation (ChIP) Assay
HCT116 cells were cross-linked with 1% formaldehyde, and lysed with SDS lysis buffer. Cell lysates were sonicated, and 5% of cell lysate was removed and used to determine the total amount of target DNA in input. Remaining cell lysates were diluted in ChIP dilution buffer. Immunoprecipitation was performed with each of the indicated antibodies (4 μg) at 4 °C overnight. Immune complexes were then mixed with salmon sperm DNA/protein agarose 50% slurry at 4 °C for 1 h. After immunoprecipitates were washed sequentially with low salt buffer, high salt buffer, LiCl wash buffer, and Tris EDTA, DNA-protein complexes were eluted with elution buffer, and cross-linking was reversed. Genomic DNA was extracted using phenol/chloroform, and ethanol-precipitated DNA was resuspended in Tris EDTA. PCR was performed with specific primers (purchased from Qiagen company) according to the manufacturer’s protocol (Millipore).
Cell cycle analysis
HCT116 cells were seeded in a 6-well dish at 1×106 cells per well 1–2 days prior to analysis. The cells were collected and fixed in precooled ethanol and incubated at −20°C overnight. Cells were treated with 100 μg/ml RNAse in PBS, washed, and stained with 5 μg/ml of propidium iodide. After washing with ice-cold PBS twice, cells were analyzed by flow cytometry and Flowjo software.
RNA extraction and real-time PCR analysis of gene expression
Total RNA was extracted using Trizol reagent (Invitrogen, San Diego, CA) as described (Lee et al., 2008). Quantitative real-time RT-PCR was performed using SYBR-Green qPCR master mix (Clontech, San Diego, CA). The β-actin gene was used as a reference for sample normalization. Primers for mouse or human genes, including β-actin, usp22, usp10, and SIRT6, were purchased from RealTime Primers (Elkins Park, PA). A standard amplification protocol was used according to the manufacturer’s instructions. c-Myc target gene primers were: cdc2 forward: 5′-CAGTCTTCAGGATGTGCTTATGC-3′, cdc2 reverse: 5′-GAGGTTTTAAGTCTCTGTGAAGAACTC-3′; PCNA forward: 5′-AGGGCTCCATCCTCAAGAAGG-3′; PCNA reverse: 5′-TGGTGCTTCAAATACTAGCGC-3′; cyclinA2 forward: 5′-GAAGACGAGACGGGTTGCA-3′ CyclinA2 reverse: 5′-AGGAGGAACGGTGACATGCT-3′; Cdc25c forward: 5′-GAACAGGCCAAGACTGAAGC-3′ cdc25c reverse: 5′-GCCCCTGGTTAGAATCTTCC-3′;
Cell proliferation assay
In vitro cell proliferation was measured by using the colorimetric WST-1 assay (Cell proliferation reagent WST-1, Roche Diagnostics). Briefly, 4,000 cells were seeded in a 96-well plate with DMEM containing 10% FBS. Every 24 hours, 10 μl of WST-1 reagent was added to each well followed by incubation for 2 hours. The absorbance at 450 nm was measured using a microplate reader.
Soft agar colony formation assay
HCT116 or RKO cells were suspended at low density (0.75 × 104 cells) in 3 ml of culture medium containing 0.3% agar (USB Corporation, Cleveland, OH) and seeded onto a base layer of 3 ml of 0.6% agar in 60-mm tissue culture dishes. After 3–4 weeks, colonies were stained, photographed, and scored.
Supplementary Material
HIGHLIGHTS.
SIRT6 is identified as a USP10-interactor in human colon cancer cells.
USP10 deubiquitinates and stabilizes SIRT6.
USP10 antagonizes c-Myc transcriptional activity through SIRT6 and p53.
USP10 and SIRT6 protein expressions are reduced in human colon cancers.
Acknowledgments
This work was supported by research grants R01AI079056, R56AI079056, and DP2DK083050.
Footnotes
Disclosure of Conflicts of Interest. The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguda BD, Kim Y, Kim HS, Friedman A, Fine HA. Qualitative network modeling of the Myc-p53 control system of cell proliferation and differentiation. Biophysical journal. 2011;101:2082–2091. doi: 10.1016/j.bpj.2011.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charollais RH, Buquet C, Mester J. Butyrate blocks the accumulation of CDC2 mRNA in late G1 phase but inhibits both the early and late G1 progression in chemically transformed mouse fibroblasts BP-A31. J Cell Physiol. 1990;145:46–52. doi: 10.1002/jcp.1041450108. [DOI] [PubMed] [Google Scholar]
- Charollais RH, Mester J. Resumption of cell cycle in BALB/c-3T3 fibroblasts arrested by polyamine depletion: relation with “competence” gene expression. J Cell Physiol. 1988;137:559–564. doi: 10.1002/jcp.1041370323. [DOI] [PubMed] [Google Scholar]
- Chiswell DJ, Ramsay G, Hayman MJ. Two virus-specific rna species are present in cells transformed by defective leukemia virus OK10. J Virol. 1981;40:301–304. doi: 10.1128/jvi.40.1.301-304.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalal SN, Schweitzer CM, Gan J, DeCaprio JA. Cytoplasmic localization of human cdc25C during interphase requires an intact 14-3-3 binding site. Mol Cell Biol. 1999;19:4465–4479. doi: 10.1128/mcb.19.6.4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos A, Goedeke L, Smibert P, Ramirez CM, Warrier NP, Andreo U, Cirera-Salinas D, Rayner K, Suresh U, Pastor-Pareja JC, et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:9232–9237. doi: 10.1073/pnas.1102281108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draker R, Sarcinella E, Cheung P. USP10 deubiquitylates the histone variant H2A.Z and both are required for androgen receptor-mediated gene activation. Nucleic Acids Res. 2011;39:3529–3542. doi: 10.1093/nar/gkq1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ely CM, Leftwich JA, Chenevix-Trench G, Hall RE, Westin EH. Altered regulation of c-myc in an HL-60 differentiation resistant subclone, HL-60-1E3. Cancer Res. 1987;47:4595–4600. [PubMed] [Google Scholar]
- Furukawa Y, Piwnica-Worms H, Ernst TJ, Kanakura Y, Griffin JD. cdc2 gene expression at the G1 to S transition in human T lymphocytes. Science. 1990;250:805–808. doi: 10.1126/science.2237430. [DOI] [PubMed] [Google Scholar]
- Gazitt Y, Erdos GW, Cohen RJ. Ultrastructural localization and fluctuation in the level of the proliferating cell nuclear antigen and myc oncoproteins in synchronized neuroblastoma cells. Cancer Res. 1993;53:1899–1905. [PubMed] [Google Scholar]
- Golomb L, Bublik DR, Wilder S, Nevo R, Kiss V, Grabusic K, Volarevic S, Oren M. Importin 7 and exportin 1 link c-Myc and p53 to regulation of ribosomal biogenesis. Molecular cell. 2012;45:222–232. doi: 10.1016/j.molcel.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi AA, Kurka T, Jeggo PA. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nature structural & molecular biology. 2011;18:831–839. doi: 10.1038/nsmb.2077. [DOI] [PubMed] [Google Scholar]
- Hayward WS, Neel BG, Astrin SM. Activation of a cellular onc gene by promoter insertion in ALV-induced lymphoid leukosis. Nature. 1981;290:475–480. doi: 10.1038/290475a0. [DOI] [PubMed] [Google Scholar]
- Ho JS, Ma W, Mao DY, Benchimol S. p53-Dependent transcriptional repression of c-myc is required for G1 cell cycle arrest. Molecular and cellular biology. 2005;25:7423–7431. doi: 10.1128/MCB.25.17.7423-7431.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC, Psulkowski E, Fredericks WJ, White DE, Maul GG, et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Molecular cell. 2007;28:823–837. doi: 10.1016/j.molcel.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochemsen AG, Shiloh Y. USP10: friend and foe. Cell. 2010;140:308–310. doi: 10.1016/j.cell.2010.01.034. [DOI] [PubMed] [Google Scholar]
- Kaidi A, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science. 2010;329:1348–1353. doi: 10.1126/science.1192049. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, McCord RA, Ongaigui KC, Boxer LD, Chang HY, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 2009;136:62–74. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly K, Cochran BH, Stiles CD, Leder P. Cell-specific regulation of the c-myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell. 1983;35:603–610. doi: 10.1016/0092-8674(83)90092-2. [DOI] [PubMed] [Google Scholar]
- Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A, Vazquez-Ortiz G, Jeong WI, Park O, Ki SH, et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell metabolism. 2010;12:224–236. doi: 10.1016/j.cmet.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress TR, Cannell IG, Brenkman AB, Samans B, Gaestel M, Roepman P, Burgering BM, Bushell M, Rosenwald A, Eilers M. The MK5/PRAK kinase and Myc form a negative feedback loop that is disrupted during colorectal tumorigenesis. Molecular cell. 2011;41:445–457. doi: 10.1016/j.molcel.2011.01.023. [DOI] [PubMed] [Google Scholar]
- Lee SM, Gao B, Fang D. FoxP3 maintains Treg unresponsiveness by selectively inhibiting the promoter DNA-binding activity of AP-1. Blood. 2008;111:3599–3606. doi: 10.1182/blood-2007-09-115014. [DOI] [PubMed] [Google Scholar]
- Liang Q, Deng H, Li X, Wu X, Tang Q, Chang TH, Peng H, Rauscher FJ, 3rd, Ozato K, Zhu F. Tripartite motif-containing protein 28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J Immunol. 2011;187:4754–4763. doi: 10.4049/jimmunol.1101704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Yang H, Kong Q, Li J, Lee SM, Gao B, Dong H, Wei J, Song J, Zhang DD, et al. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Molecular cell. 2012;46:484–494. doi: 10.1016/j.molcel.2012.03.024. [DOI] [PubMed] [Google Scholar]
- Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai Y, Norberg HV, Zhang T, Furuya T, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223–234. doi: 10.1016/j.cell.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011;332:1443–1446. doi: 10.1126/science.1202723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki H, Takahashi M, Higuchi M, Makokha GN, Oie M, Fujii M. Both G3BP1 and G3BP2 contribute to stress granule formation. Genes to cells: devoted to molecular & cellular mechanisms. 2013;18:135–146. doi: 10.1111/gtc.12023. [DOI] [PubMed] [Google Scholar]
- Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min L, Ji Y, Bakiri L, Qiu Z, Cen J, Chen X, Chen L, Scheuch H, Zheng H, Qin L, et al. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nature cell biology. 2012;14:1203–1211. doi: 10.1038/ncb2590. [DOI] [PubMed] [Google Scholar]
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Rochlitz CF, Heide I, Thiede C, Herrmann R, de Kant E. Evidence for a mutual regulation of p53 and c-myc expression in human colorectal cancer metastases. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 1995;6:981–986. doi: 10.1093/oxfordjournals.annonc.a059094. [DOI] [PubMed] [Google Scholar]
- Ronnebaum SM, Wu Y, McDonough H, Patterson C. The Ubiquitin Ligase CHIP Prevents SirT6 Degradation through Noncanonical Ubiquitination. Mol Cell Biol. 2013;33:4461–4472. doi: 10.1128/MCB.00480-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L, Ram O, Truelove J, Guimaraes AR, Toiber D, et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151:1185–1199. doi: 10.1016/j.cell.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan J, Zhao W, Gu W. Suppression of cancer cell growth by promoting cyclin D1 degradation. Molecular cell. 2009;36:469–476. doi: 10.1016/j.molcel.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasselli L, Chua KF. Cancer: Metabolism in ‘the driver’s seat. Nature. 2012;492:362–363. doi: 10.1038/492362a. [DOI] [PubMed] [Google Scholar]
- Wang B, Xiao Z, Ko HL, Ren EC. The p53 response element and transcriptional repression. Cell cycle. 2010;9:870–879. doi: 10.4161/cc.9.5.10825. [DOI] [PubMed] [Google Scholar]
- Ward AM, Bidet K, Yinglin A, Ler SG, Hogue K, Blackstock W, Gunaratne J, Garcia-Blanco MA. Quantitative mass spectrometry of DENV-2 RNA-interacting proteins reveals that the DEAD-box RNA helicase DDX6 binds the DB1 and DB2 3′ UTR structures. RNA biology. 2011;8:1173–1186. doi: 10.4161/rna.8.6.17836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Luo K, Zhang L, Cheville JC, Lou Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell. 2010;140:384–396. doi: 10.1016/j.cell.2009.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010;140:280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.