

Abstract

The blockade of aberrant hedgehog (Hh) signaling has shown promise for therapeutic intervention in cancer. A cell-based phenotypic high-throughput screen was performed, and the lead structure (1) was identified as an inhibitor of the Hh pathway via antagonism of the Smoothened receptor (Smo). Structure−activity relationship studies led to the discovery of a potent and specific Smoothened antagonist N-(6-((2S,6R)-2,6-dimethylmorpholino)pyridin-3-yl)-2-methyl-4′-(trifluoromethoxy)biphenyl-3-carboxamide (5m, NVP-LDE225), which is currently in clinical development.
Keywords: Hedgehog signaling pathway, Smoothened, medulloblastoma
Smoothened (Smo) is a 7-pass transmembrane protein that functions as the key activator of the hedgehog (Hh) signaling pathway.1 While Hh signaling is tightly controlled during cellular proliferation, differentiation, and embryonic morphogenesis,2,3 aberrant pathway activation has been linked to tumorigenesis in several cancers.4−6 This pathway can be stimulated by one of three Hh ligands: sonic hedgehog (Shh), Indian hedgehog (Ihh), or desert hedgehog (Dhh). Upon binding of Hh ligand to the 12-pass transmembrane protein Patched (Ptch), the repression of Smo by Ptch is relieved. Activated Smo then initiates a downstream signaling cascade leading to the activation of transcription factors of the Gli family. Strong genetic evidence (mutations in Ptch, Smo, SUFU, or Gli) links up-regulated pathway activity to tumor formation in cancers such as basal cell carcinoma and medulloblastoma.7−9 Furthermore, Hh signaling has been shown to play a role in tumorigenesis of various other cancers such as pancreatic, prostate, lung, colorectal, bladder, and ovarian. Thus, inhibition of Hh signaling is an attractive approach for anticancer therapy.10,11
Cyclopamine (Figure 1), a naturally occurring alkaloid, was the first Smo antagonist to be reported in the literature.12 It has been shown to inhibit Hh signaling and to induce the remission of medulloblastoma in a transgenic mouse model.9 Several other synthetic small molecule Smo antagonists have been described in recent years.10,11 The Smo inhibitor GDC-0449 (Figure 1), recently reported by Genentech, has shown promising phase I clinical results in advanced basal cell carcinoma patients.13 A cyclopamine derivative IPI-926 (Figure 1) has also entered clinical development recently. Herein, we report our efforts in this area, which led to the discovery of N-(6-((2S,6R)-2,6-dimethylmorpholino)pyridin-3-yl)-2-methyl-4′-(trifluoromethoxy)biphenyl-3-carboxamide (5m, NVP-LDE225), a potent and selective Smo antagonist currently in phase I clinical trials.
Figure 1.

Structures of cyclopamine, GDC-0449, and IPI-926.
A high-throughput cell-based screen of in-house diversity combinatorial libraries (10K compounds generated by solid-phase synthesis), using a reporter gene assay in a mouse cell line (TM3) stably transfected with a Hh-responsive Gli luciferase construct and stimulated by Shh protein, led to the identification of a class of biphenyl carboxamides, such as compound 1 (Figure 2), as Hh signaling inhibitors. To confirm Smo as the target for these screening hits, a Gli IC50 shift assay was employed.14 A known Smo agonist, Ag1.5,15 was used at two different concentrations (1 and 25 nM) to induce Hh pathway activation in the reporter gene assay. A shift to a higher IC50 at the higher concentration of Ag1.5 was expected for a competitive inhibitor. Compound 1 resulted in IC50 values of 0.17 and 1.13 μM, respectively, under these conditions, corresponding to a 6.5-fold IC50 shift, suggesting its direct interaction with the Smo receptor. Fluorescence binding assays, with both human and mouse Smo, via competition of BODIPY-cyclopamine were also employed in our lead optimization effort to ensure cross-reactivity. A systematic study of structure−activity relationships (SARs) was carried out in three regions (A−C) of compound 1 (Figure 2).
Figure 2.

Structure of 1, a screening hit.
The general synthetic route used to prepare these compounds is illustrated in Scheme 1. It primarily involved two transformations, amide bond formation and Suzuki cross-coupling, from commercially or readily available starting materials. This versatile combination of reaction sequences allowed for the rapid investigation of SARs in all three regions. The key SAR results for select analogues within this series are summarized in Table 1.
Scheme 1. General Synthetic Scheme.

Reagents and conditions: (a) Anilines or aminopyridines, HATU, Et3N, DMF. (b) Boronic acids, cat. Pd(PPh3)4, Na2CO3, DME−H2O.
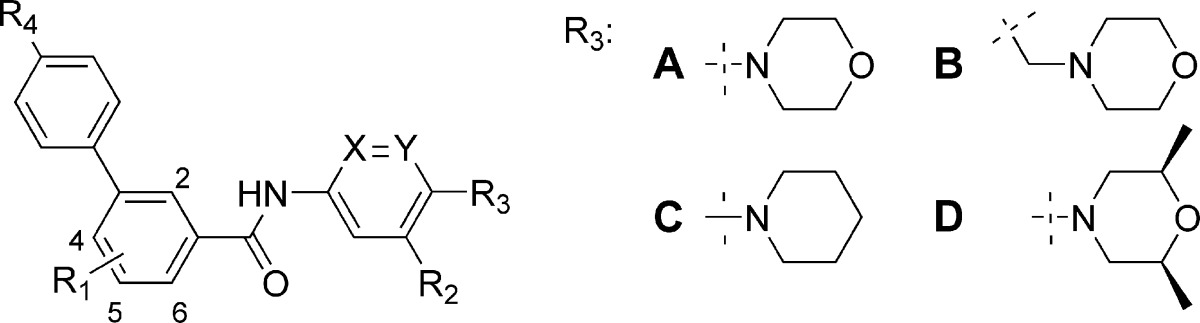
Table 1. IC50 Values for Select Compounds in the TM3-Gli-Luc IC50 Shift Assay and Mouse (m) and Human (h) Smo Fluorescence Binding Assays.
| IC50 (nM) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | X | Y | 1 nM Ag1.5ab | 25 nM Ag1.5ab | mBdgac | hBdgac | |
| 5a | 4-Me | H | A | CN | CH | CH | 43 | 409 | ||
| 5b | 4-Me | F | A | CN | CH | CH | 17 | 256 | ||
| 5c | 4-Me | CF3 | A | CN | CH | CH | 22 | 246 | ||
| 5d | 4-Me | H | A | CN | CH | N | 14 | 271 | 80 | 124 |
| 5e | 4-Me | H | B | CN | CH | CH | 247 | 4385 | ||
| 5f | 4-Me | H | C | CN | CH | N | 14 | 334 | ||
| 5g | 4-Me | H | C | CN | N | CH | 751 | 3914 | ||
| 5h | 4-Me | H | D | CN | CH | N | 5 | 71 | 5 | 12 |
| 5i | 2-Me | H | D | CN | CH | N | 0.6 | 7.3 | 0.5 | 2 |
| 5j | 2-Cl | H | D | CN | CH | N | 0.7 | 14 | ||
| 5k | 2-Me | H | D | OMe | CH | N | 0.6 | 10 | ||
| 5l | 2-Me | H | D | CF3 | CH | N | 0.8 | 7.9 | 3.1 | 11 |
| 5m | 2-Me | H | D | OCF3 | CH | N | 0.6 | 8 | 1.3 | 2.5 |
See the Supporting Information for detailed assay descriptions.
Reporter gene assay in a TM3 luciferized cell line with activation of the pathway using varying concentrations of the Hh agonist Ag1.5.
Smo binding assay utilizing the displacement of BODIPY-cyclopamine.
Our initial focus was on exploring modifications to region A, which in compound 1 features an electron-rich 1,4-diaminophenyl moiety. Replacement of this moiety was desirable as it could potentially form quinone-like reactive intermediates upon metabolic activation, thereby leading to promiscuous toxicities. To mitigate this risk, several different approaches were employed aiming to reduce the electron density of this moiety. The introduction of electron-withdrawing groups at the R2 position, such as −F (5b) and −CF3 (5c), led to marginal increases in activity. The replacement of phenyl with a more electron-deficient pyridine (compound 5d) was also tolerated. However, an attempt to move the morpholine nitrogen one carbon atom away from its direct attachment to the phenyl ring was unsuccessful (compound 5e). At the R3 position, a piperidinyl substituent produced comparable activity to morpholino (compounds 5f vs 5d). Interestingly, the position of the nitrogen in the pyridine moiety was critical for the inhibitory activity. The regioisomeric pyridinyl analogue (compound 5g) led to a significant loss of activity. An appreciable enhancement of activity was achieved upon introduction of cis-dimethyl groups onto the morpholine ring (compound 5h).
Substituents on the central phenyl ring (region B) played an important role in modulating Hh signaling inhibitory activity. Early SARs suggested that a small hydrophobic group, such as −Me (compound 5a), was favored at the 4-position. A breakthrough in increasing potency was realized upon migrating the methyl group from the 4- to the 2-position (compound 5i). Similarly, a 2-chloro substituent gave comparable activity (compound 5j). A detailed survey of substitutions in region C led to the identification of a few preferred substituents at the R4-position, including −CN (5a−j), −OMe (5k), −CF3 (5l), and −OCF3 (5m). As our primary assays to evaluate these compounds used a mouse cell line (TM3), select compounds were tested in fluorescence binding assays with both mouse and human Smo to ensure desired cross-reactivity between species, through competition of BODIPY-cyclopamine, a fluorescent-labeled cyclopamine derivative. We were encouraged by the observation that the IC50 values for mouse and human Smo were highly consistent for the tested compounds.
In a traditional lead optimization approach, pharmaceutical properties, in vitro ADMET and in vivo pharmacokinetics (PKs), were evaluated for compounds meeting the potency and selectivity criteria. On the basis of overall favorable properties, compound 5m (NVP-LDE225) was chosen for further in vitro and in vivo testing.
Compound 5m is highly bound to mouse, rat, and human plasma proteins (>99%) and moderately bound to dog and monkey plasma proteins (77 and 85%, respectively). The compound has high permeability in the PAMPA assay (calculated dose fraction absorbed estimated to be 90.8% in man). Compound 5m shows good oral bioavailability ranging from 69 to 102% in preclinical species when dosed in solution (Table 2). The systemic plasma clearance (CL) is low (mouse, rat, and monkey) to moderate (dog) relative to respective hepatic blood flow, while the volume of distribution (Vss) is moderate (mouse and rat) to high (dog and monkey) as compared to total body water. The mean residence time (MRT) ranges from 3.04 to 6.81 h. The ability to penetrate the blood−brain barrier is considered favorable where the brain/plasma AUC0-inf ratio is moderate (0.57). Therefore, good exposures are predicted in medulloblastoma, a type of brain tumor, which is a potential clinical indication for Smo antagonists.
Table 2. In Vivo Pharmacokinetics among Speciesa.
| CL (mL/min/kg) | Vss (L/kg) | MRT (h) | F (%) | |
|---|---|---|---|---|
| mouse | 10.4 | 1.9 | 3.04 | 69 |
| rat | 6.0 | 1.5 | 4.17 | 83 |
| dog | 17.1 | 6.99 | 6.81 | 101 |
| monkey | 14.8 | 3.4 | 3.83 | 102 |
The compound was administered as a solution in PEG300/5% dextrose in water (75:25 v/v) for mouse and rat or in 20% Captisol for dog and monkey.
Compound 5m is a weak base with a measured pKa of 4.20 and exhibits relatively poor aqueous solubility. In contrast to high oral bioavailability in rats when administered as a solution in PEG300/dextrose water, only marginal drug exposure (oral bioavailability 3%) was obtained when a crystalline free base was dosed as a suspension in 0.5% sodium carboxymethyl cellulose. To increase the drug oral exposure, a stable crystalline diphosphate salt with an improved dissolution rate was developed. Following oral administration in the same suspension formulation, the absolute oral bioavailability was increased to 48%.
Ptch+/−p53−/− mice, which have been shown to spontaneously develop medulloblastomas,9,16,17 have been effectively used to evaluate in vivo antitumor efficacy of Smo antagonists, both directly in the transgenic model16 and in allograft models derived from the Ptch+/−p53−/− medulloblastoma tumors.9,14,18,19 Given that 5m displayed both a favorable PK profile across preclinical species and brain exposure upon oral administration, we elected to assess its antitumor efficacy using both subcutaneous and orthotopic Ptch+/−p53−/− medulloblastoma allograft models.
In the subcutaneous Ptch+/−p53−/− medulloblastoma allograft mouse model, 5m demonstrated dose-related antitumor activity after 10 days of oral administration of a suspension of the diphosphate salt (Figure 3). At a dose of 5 mg/kg/day qd, 5m significantly inhibited tumor growth, corresponding to a T/C value of 33% (p < 0.05 as compared to vehicle controls). When dosed at 10 and 20 mg/kg/day qd, 5m afforded 51 and 83% regression, respectively. Mouse Gli1 mRNA expression levels were utilized as a pharmacodynamic marker to link the antitumor effects of 5m to inhibition of Hh pathway activity in this model. Tumors were harvested at different time points following a single dose of 5m at 5, 10, or 20 mg/kg, and levels of Gli1 mRNA expression were analyzed by real-time polymerase chain reaction (PCR) (Figure 4). As illustrated in Figure 4, a dose- and time-dependent inhibition of Gli1 mRNA levels was observed in the Ptch+/−p53−/− medulloblastoma allograft model. Gli1 mRNA inhibition correlated with tumor and plasma exposure of 5m. Interestingly, a time delay was observed between achieving peak concentrations of drug and maximum inhibition of Gli1 levels, presumably due to the time required to inhibit downstream Gli1 transcription and the Gli1 mRNA turnover time.
Figure 3.

Antitumor activity upon treatment with 5m diphosphate salt or vehicle in a Ptch+/−p53−/− medulloblastoma subcutaneous allograft model in nude mice. Treatment started on day 8 postimplantation (5 million cells/animal). Compound 5m was administered po at 5 (filled triangle), 10 (open triangle), and 20 (filled diamond) mg/kg/day qd for 13 days total. All doses are expressed as free base equivalents. Vehicle control (open square) of 5m: 0.5% methylcellulose and 0.5% Tween 80 in water. All treatment groups consisted of eight animals. The vehicle group was taken down 7−9 days after treatment due to excessive tumor size (greater than 10% of mouse body weight). The body weight change observed was <±5% for all treated groups.
Figure 4.

Gli1 mRNA inhibition (open circle), tumor PK (filled squares), and plasma PK (filled triangles) in Ptch+/−p53−/− medulloblastoma model after treatment with 5m. Plasma and tumors were harvested at 4, 8, 16, and 24 h after a single oral dose of 5m at 5, 10, or 20 mg/kg. Gli1 mRNA levels were analyzed by real-time PCR and normalized to β-actin expression. Data shown are percent inhibition relative to vehicle-treated control tumors. Compound 5m concentrations were determined in plasma and tumor by LC-MS/MS. N = 3 per time point.
An orthotopic Ptch+/−p53−/− medulloblastoma allograft model was also established by implanting tumor cells harvested from Ptch+/−p53−/− mice stereotaxically into the frontal cortex of nude mice. Treatment was initiated on day 17 following tumor implantation in mice demonstrating established tumors as determined by MRI imaging. Treated animals were dosed at 40 mg/kg/day po bid, and tumor size was assessed by MRI after 4 days of dosing (Figure 5). As illustrated in Figure 5, the established tumors grew significantly in the vehicle-treated animals over the 4 day treatment period (+1230 ± 210% as compared to baseline). In contrast, 5m treatment clearly slowed tumor growth relative to vehicle-treated animals (+124 ± 37% as compared to baseline). A separate study performed with a contrast agent demonstrated that the blood−brain barrier remained intact in this model following implantation of tumor cells.20 Together, these studies suggest that 5m successfully penetrates the blood−brain barrier in tumor-bearing animals and results in tumor growth inhibition after 4 days of treatment.
Figure 5.

Antitumor activity in an orthotopic Ptch+/−p53−/− medulloblastoma allograft model in nude mice upon treatment with 5m diphosphate salt dosed at 40 mg/kg/day po bid or vehicle at equal dose volume. Athymic nude mice were implanted with 100000 tumor cells 17 days before the start of dosing. Daily treatment (vehicle or 5m) was initiated on day 0. Eight animals were enrolled into each group. MRI scans were performed at baseline and day 4 postinitiation of treatment. The bar graph represents the group mean ± SEM, with crosses indicating data from individual animals. At each time point, a representative 3D rendering and paired MRI are shown of an animal representative of the group mean. To enable longitudinal comparisons, the selected MRI is from the same animal over time point.
As part of the pharmaceutical developability assessment, compound 5m was run through a series of preclinical safety assays. The IC50 values for 5m for the major human CYP450 drug metabolizing enzymes was greater than 10 μM. In addition, 5m did not exhibit time-dependent CYP inhibition nor induction of CYP3A4, suggesting low potential for drug−drug interactions. In an automated hERG patch clamp assay, it showed an IC50 value of greater than 30 μM. Compound 5m was negative in tests for genotoxicity (Ames and micronucleus tests). The selectivity of compound 5m was evaluated by screening against a large panel of receptors, ion channels, transporters, kinases, and proteases. No appreciable activities (i.e., IC50 <10 μM) were identified, suggesting its low potential for off-target effects.
In summary, a novel chemical series identified via high-throughput screening, the biphenyl-3-carboxamides, was optimized for Smo antagonism, selectivity, safety, and PKs to discover the clinical candidate 5m. Treatment with 5m in a subcutaneous Ptch+/−p53−/− medulloblastoma allograft mouse model led to dose-related tumor growth inhibition, with tumor regression observed in the higher dosing groups. The ability of 5m to penetrate the blood−brain barrier and to inhibit tumor growth in brain was demonstrated in an orthotopic Ptch+/−p53−/− medulloblastoma allograft mouse model. This compound is now in phase I clinical trials, and its clinical PK, efficacy, and safety are currently under evaluation.
Acknowledgments
We thank Lucas Westling for analytical support and Dr. Rosalind Segal and Dr. Andrew Kung (Dana-Farber Cancer Institute, Boston, MA) for providing Ptch+/−p53−/− tumors.
Supporting Information Available
Procedures for the preparation of 5m, analytical data, and procedures and methods for in vitro and in vivo assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Cancer Research Technology, Gower Street, London WC1E 6BT, United Kingdom
Supplementary Material
References
- Stone D. M.; Hynes M.; Armanini M.; Swanson T. A.; Gu Q.; Johnson R. L.; Scott M. P.; Pennica D.; Goddard A.; Phillips H.; Noll M.; Hooper J. E.; de Sauvage F.; Rosenthal A. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 1996, 384, 129–134. [DOI] [PubMed] [Google Scholar]
- McMahon A. P.; Ingham P. W.; Tabin C. J. Developmental roles and clinical significance of hedgehog signaling. Curr. Top. Dev. Biol. 2003, 53, 1–114. [DOI] [PubMed] [Google Scholar]
- Ingham P. W.; McMahon A. P. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [DOI] [PubMed] [Google Scholar]
- di Magliano M. P.; Hebrok M. Hedgehog signaling in cancer formation and maintenance. Nat. Rev. Cancer 2003, 3, 903–911. [DOI] [PubMed] [Google Scholar]
- Rubin L. L.; de Sauvage F. J. Targeting the hedgehog pathway in cancer. Nat. Rev. Drug Discovery 2006, 5, 1026–1033. [DOI] [PubMed] [Google Scholar]
- Theunissen J.-W.; de Sauvage F. J. Paracrine hedgehog signaling in cancer. Cancer Res. 2009, 69, 6007–6010. [DOI] [PubMed] [Google Scholar]
- Hahn H.; Wicking C.; Zaphiropoulous P. G.; Gailani M. R.; Shanley S.; Chidambaram A.; Vorechovsky I.; Holmberg E.; Unden A. B.; Gilles S.; Negus K.; Smyth I.; Pressman C.; Leffell D. J.; Gerrard B.; Goldstein A. M.; Dean M.; Toftgard R.; Chenevix-Trench g.; Wainwright B.; Bale A. E. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [DOI] [PubMed] [Google Scholar]
- Ferretti E.; DeSmaele E.; DiMarcotullio L.; Screptani I.; Gulino A. Hedgehog checkpoints in medulloblastoma: The chromosome 17p deletion paradigm. Trends Mol. Med. 2005, 11, 537–545. [DOI] [PubMed] [Google Scholar]
- Berman D. M.; Karhadkar S. S.; Hallahan A. R.; Pritchard J. I.; Eberhart C. G.; Watkins D. N.; Chen J. K.; Cooper M. K.; Taipale J.; Olson J. M.; Beachy P. A. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 2002, 297, 1559–1561. [DOI] [PubMed] [Google Scholar]
- Mahindroo N.; Punchihewa C.; Fujii N. Hedgehog-Gli signaling pathway inhibitors as anticancer agents. J. Med. Chem. 2009, 52, 3829–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay M. R.; Nesler M.; Weatherhead R.; Castro A. C. Recent patents for hedgehog pathway inhibitors for the treatment of malignancy. Expert Opin. Ther. Pat. 2009, 19, 1039–1056. [DOI] [PubMed] [Google Scholar]
- Chen J. K.; Taipale J.; Cooper M. K.; Beachy P. A. Inhibition of hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff D. D.; LoRusso P. M.; Rudin C. M.; Reddy J. C.; Yauch R. L.; Tibes R.; Weiss G. J.; Borad M. J.; Hann C. L.; Brahmer J. R.; Mackey H. M.; Lum B. L.; Darbonne W. C.; Marsters J. C. Jr.; de Sauvage F. J.; Low J. A. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 2009, 361, 1164–1172. [DOI] [PubMed] [Google Scholar]
- Miller-Moslin K.; Peukert S.; Jain R. K.; McEwan M. A.; Karki R.; Llamas L.; Yusuff N.; He F.; Li Y.; Sun Y.; Dai M.; Perez L.; Michael W.; Sheng T.; Lei H.; Zhang R.; Williams J.; Bourret A.; Ramamurthy A.; Yuan J.; Guo R.; Matsumoto M.; Vattay A.; Maniara W.; Amaral A.; Dorsch M.; Kelleher J. F. III 1-Amino-4-benzylphthalazines as orally bioavailable smoothened antagonists with antitumor activity. J. Med. Chem. 2009, 52, 3954–3968. [DOI] [PubMed] [Google Scholar]
- Frank-Kamenetsky M.; Zhang X. M.; Bottega S.; Guicherit O.; Wichterle H.; Dudek H.; Bumcrot D.; Wang F. Y.; Jones S.; Shulok J.; Rubin L. L.; Porter J. A. Small-molecule modulators of hedgehog signaling: Identification and characterization of smoothened agonists and antagonists. J. Biol. 2002, 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romer J. T.; Kimura H.; Magdaleno S.; Sasai K.; Fuller C.; Baines H.; Connelly M.; Stewart C. F.; Gould S.; Rubin L. L.; Curran T. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(±)p53(−/−) mice. Cancer Cell 2004, 6, 229–240. [DOI] [PubMed] [Google Scholar]
- Wetmore C.; Eberhart D. E.; Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res. 2001, 61, 513–516. [PubMed] [Google Scholar]
- Robarge K. D.; Brunton S. A.; Castanedo G. M.; Cui Y.; Dina M. S.; Goldsmith R.; Gould S. E.; Guichert O.; Gunzner J. L.; Halladay J.; Jia W.; Khojasteh C.; Koehler M. F. T.; Katkow K.; La H.; LaLonde R. L.; Lau K.; Lee L.; Marshall D.; Marsters J. C.; Murray L. J.; Qian C.; Rubin L. L.; Salphati L.; Stanley M. S.; Stibbard J. H. A.; Sutherlin D. P.; Ubhayaker S.; Wang S.; Wong S.; Xie M. GDC-0449—A potent inhibitor of the hedgehog pathway. Bioorg. Med. Chem. Lett. 2009, 19, 5576–5581. [DOI] [PubMed] [Google Scholar]
- Tremblay M. R.; Lescarbeau A.; Grogan M. J.; Tan E.; Lin G.; Austad B. C.; Yu L.-C.; Behnke M. L.; Nair S. J.; Hagel M.; White K.; Conley J.; Manna J. D.; Alvarez-Diez T. M.; Hoyt J.; Woodward C. N.; Sydor J. R.; Pink M.; MacDougall J.; Campbell M. J.; Cushing J.; Ferguson J.; Curtis M. S.; McGovern K.; Read M. A.; Palombella V. J.; Adams J.; Castro A. C. Discovery of a Potent and Orally Active Hedgehog Pathway Antagonist (IPI-926). J. Med. Chem. 2009, 52, 4400–4418. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for full details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.