

ABSTRACT
Cytokinesis is the final stage in cell division. Although integrins can regulate cytokinesis, the mechanisms involved are not fully understood. In this study, we demonstrate that integrin-regulated ERK (extracellular signal-related kinase) and RSK (p90 ribosomal S6 kinase) signaling promotes successful cytokinesis. Inhibiting the activation of ERK and RSK in CHO cells by a mutation in the integrin β1 cytoplasmic tail or with pharmacological inhibitors results in the accumulation of cells with midbodies and the formation of binucleated cells. Activation of ERK and RSK signaling by the expression of constitutively active RAF1 suppresses the mutant phenotype in a RSK-dependent manner. Constitutively active RSK2 also restores cytokinesis inhibited by the mutant integrin. Importantly, the regulatory role of the RSK pathway is not specific to CHO cells. MCF-10A human mammary epithelial cells and HPNE human pancreatic ductal epithelial cells exhibit a similar dependence on RSK for successful cytokinesis. In addition, depriving mitotic MCF10A cells of integrin-mediated adhesion by incubating them in suspension suppressed ERK and RSK activation and resulted in a failure of cytokinesis. Furthermore, inhibition of RSK or integrins within the 3D context of a developing salivary gland organ explant also leads to an accumulation of epithelial cells with midbodies, suggesting a similar defect in cytokinesis. Interestingly, neither ERK nor RSK regulates cytokinesis in human fibroblasts, suggesting cell-type specificity. Taken together, our results identify the integrin–RSK signaling axis as an important regulator of cytokinesis in epithelial cells. We propose that the proper interaction of cells with their microenvironment through integrins contributes to the maintenance of genomic stability by promoting the successful completion of cytokinesis.
KEY WORDS: Integrins; Cytokinesis; ERK, RSK
INTRODUCTION
Cytokinesis is the final step in cell division that ensures the proper segregation of genetic and cytoplasmic material between daughter cells (Green et al., 2012). Cytokinesis is initiated when the assembly and constriction of the actomyosin contractile ring drives the ingression of the cleavage furrow, which is regulated by the localized activation of RhoA and supported by membrane remodeling mediated by vesicular trafficking (Green et al., 2012; Neto and Gould, 2011). The central spindle is assembled during anaphase and later serves as the framework for the formation of the midbody. The midbody is an intercellular bridge connecting the two presumptive daughter cells and consists of microtubules together with the components and regulators of the abscission machinery (Green et al., 2012; Neto and Gould, 2011).
Integrins are α-β heterodimeric adhesion receptors that bind components of the extracellular matrix and together with growth factor receptors are required for adhesion-dependent cells to progress through G1 and to enter S phase (Streuli, 2009). Growing evidence implicates integrins in the regulation of cytokinesis (Aszodi et al., 2003; Högnäs et al., 2012; Kittler et al., 2007; Reverte et al., 2006; Thullberg et al., 2007); however, the molecular mechanisms involved in integrin-regulated cytokinesis are not well understood. Previous studies from our laboratory demonstrate that a tyrosine-to-alanine substitution in the membrane-proximal NPIY motif (Y783A) in the integrin β1 cytoplasmic domain (β1 tail) that suppresses integrin activity also inhibits cytokinesis (Reverte et al., 2006). Constitutive activation of the YA (Y783A) mutant rescued cytokinesis (Reverte et al., 2006), but did not restore stress fibers and focal adhesions (Nieves et al., 2010). Thus, the ability to form focal adhesions and stress fibers is not required for successful cytokinesis, at least not in CHO cells. In the current study, we used this mutant integrin to explore the role of integrin-regulated MEK–ERK signaling in the regulation of cytokinesis. Our data indicate that cells adhered by the mutant integrin are inhibited in ERK activation and downstream RSK signaling. We show that pharmacological inhibition of MEK or RSK leads to cytokinesis failure when CHO cells are adhered by wild-type (WT) integrins and that the expression of constitutively active Raf or RSK rescues the mutant phenotype. Integrins also regulate cytokinesis in the human MCF10A mammary epithelial cell line through a RSK-dependent pathway. This pathway also plays an important role in cytokinesis in HPNE, a human pancreatic epithelial cell line as well as in murine salivary gland epithelial cells during gland morphogenesis ex vivo. Our results demonstrate a novel role for integrin-regulated ERK and RSK signaling in promoting cytokinesis and suggest that integrins do so by regulating key events at the midbody necessary for abscission. Accumulating evidence links cytokinesis failure to tumorigenesis (Fujiwara et al., 2005; Galipeau et al., 1996; Ganem et al., 2007; Högnäs et al., 2012; Jonsdottir et al., 2012; Lv et al., 2012), suggesting a possible role for integrins as tumor suppressors owing to their ability to promote successful cytokinesis and thereby contribute to genomic stability.
RESULTS
Tyrosine to alanine (YA) mutation in the integrin β1 tail results in a delay in cytokinesis and the accumulation of binucleated cells
To characterize the mechanism by which integrins regulate cytokinesis, we used CHO cell lines stably expressing chimeric integrins containing either the wild-type β1 tail (WT) or the Y783A mutant β1 tail (YA) in the context of the αIIb-5-β3-1 heterodimeric chimeric integrins (Reverte et al., 2006). We isolated the function of recombinant integrins by adhering cells to fibrinogen in CCM1, a serum-free growth-promoting medium, as previously described (Colello et al., 2012; Nieves et al., 2010; Reverte et al., 2006). To assay cytokinesis, mitotic cells, isolated by mitotic shake off, were plated on fibrinogen-coated coverslips and their progression through cytokinesis was assessed at 30 minute intervals by immunofluorescence microscopy (Fig. 1A,B). The results indicated that 80% of cells adhered by the WT integrin successfully completed cytokinesis by 1.5 hours (Fig. 1B). By contrast, 80% of YA cells were delayed, as indicated by the persistence of midbodies at 1.5 hours (Fig. 1B). In addition, by 3 hours, ∼20% of YA cells were binucleated with two centrosomes, indicating failed cytokinesis (Fig. 1B and data not shown). Statistical analysis indicated that there were significantly more YA cells with midbodies at 1.5 hours and significantly more YA cells that failed cytokinesis at 3 hours compared with WT (supplementary material Table S1).
Fig. 1.

The YA mutant integrin delays and inhibits cytokinesis; activating the mutant integrin rescues these processes. (A,B) Mitotic cells expressing WT or YA mutant integrins were collected and replated on fibrinogen in CCM1 for the indicated times. (A) Cells were fixed with glutaraldehyde, stained for α-tubulin to visualize the microtubules (green) and for DNA (blue). Images were obtained using wide-field microscopy. (B) Plotted is the mean percentage of WT or YA cells in metaphase or anaphase (white) with midbodies (blue), successfully completed cytokinesis (green) or failed cytokinesis (red) from 0.5 hours to 3 hours with or without 2 mM manganese (Mn2+). (C) Mitotic cells expressing WT or YA mutant integrins were replated on fibronectin in CCM1 and analyzed at 3 hours for completion of cytokinesis. Plotted is the mean percentage of cells that completed or failed cytokinesis. (B,C) One hundred cells were analyzed from each of three independent experiments (see supplementary material Table S1 for statistical analysis). Scale bar: 10 µm.
To demonstrate that effects on cytokinesis were specifically due to defects in integrin function, we activated the mutant integrin by replating mitotic cells in the presence of manganese (Mn2+), an extracellular activator of integrins (Mould et al., 2002). Activation of the mutant integrin rescued the cytokinesis defects (Fig. 1B and supplementary material Table S1). Additionally, when mitotic cells expressing YA mutant integrins were adhered to fibronectin by their endogenous α5β1 integrins, YA cells successfully completed cytokinesis (Fig. 1C). Thus, wild-type integrin function promotes timely progression through the midbody stage and successful cytokinesis.
Suppression of integrin-regulated MEK–ERK signaling inhibits cytokinesis
Previous studies have implicated ERK in the regulation of cytokinesis in some cell types (Kasahara et al., 2007). ERK activity is inhibited when cells are adhered by the YA mutant integrin and can be restored by activating the mutant integrin with Mn2+ (Fig. 2A). Therefore, we asked whether defects in MEK–ERK signaling contribute to defects in cytokinesis in our system. For these experiments, logarithmically growing WT cells were treated with the MEK inhibitor U0126 or DMSO alone for 2 hours to inhibit ERK signaling (Fig. 2B). Mitotic cells were isolated and adhered to fibrinogen by WT integrins for the indicated period of time in the presence or absence of the inhibitor and then analyzed for progression through cytokinesis (Fig. 2B). The results indicate that inhibition of MEK–ERK signaling prolongs cytokinesis. At 1.5 hours, more than 50% of treated cells still had midbodies compared with 20% of untreated cells, and by 3 hours, 15% of treated cells had failed cytokinesis, whereas most of the untreated cells had successfully completed cytokinesis (Fig. 2B). Importantly, these differences are significant and not inhibitor specific (supplementary material Table S2). Finally, to confirm that the rescue of the cytokinesis defect in YA cells treated with Mn2+ was dependent on ERK, logarithmically growing WT and YA cells were treated with MEK inhibitor U0126 or DMSO for 2 hours to inhibit ERK signaling. Mitotic cells were isolated and adhered to fibrinogen for the times indicated in the presence or absence of Mn2+ and U0126 and then analyzed for progression through cytokinesis (Fig. 2C,D). At 3 hours, 20% of WT and 30% of YA cells treated with Mn2+ and U0126 had failed cytokinesis (Fig. 2C,D). Thus, the rescue of cytokinesis by Mn2+ is ERK dependent.
Fig. 2.

MEK–ERK signaling regulates cytokinesis in CHO cells. (A) ERK activation in cells adhered by WT or mutant integrins with or without 2 mM Mn2+ was analyzed by western blot using phospho-specific antibodies for activated ERK. A representative blot is shown on the left. The right panel shows the average pERK levels normalized to total ERK ± s.d. from three independent experiments. (B) Pharmacological inhibition of MEK results in cytokinesis failure. Logarithmically growing WT cells were treated with 10 µM U0126 for 2 hours. Mitotic cells were isolated and replated on fibrinogen with U0126 for the indicated times. Cells remaining on the plate after shake-off were analyzed for activated ERK. A representative blot and quantification of the average pERK levels normalized to total ERK from three independent experiments are shown on the left. The right panel shows the percentage of cells in metaphase or anaphase (white), with midbodies (blue), successfully completed cytokinesis (green) or failed cytokinesis (red) at the indicated times. (C,D) Logarithmically growing WT and YA cells were treated with 10 µM U0126 for 2 hours. Mitotic cells were isolated and replated on fibrinogen with 2 mM Mn2+ and U0126 for 3 hours. Percentage of cells with Mn2+ and/or U0126 that failed cytokinesis at 3 hours is shown. Representative blots and analysis are shown for activated and total ERK. One hundred individual cells were analyzed from each of three independent experiments in B–D. Values are means ± s.d. *P<0.05, **P<0.005.
RSK functions downstream of ERK to promote cytokinesis
One well-known ERK substrate is the p90 ribosomal S6 kinase (RSK) protein (Anjum and Blenis, 2008; Romeo et al., 2012; Yoon and Seger, 2006). Therefore, we determined whether RSK regulates cytokinesis. RSK activation was compared in cells adhered by WT and YA integrins. Results showed that RSK activation was inhibited when cells were adhered by the mutant integrin, but not when YA cells were adhered to fibrinogen by their endogenous α5β1 integrins (Fig. 3A). To examine the role of RSK in cytokinesis, mitotic cells were isolated from logarithmically growing WT cells that were either pre-treated with the RSK-specific inhibitors BI-D1870 or SL-0101 (Nguyen, 2008; Smith et al., 2005) for 2 hours or treated at shake-off with BI-D1870 and adhered on fibrinogen. The results indicate that 70–80% of cells treated with inhibitor (pre-treatment or at shake-off) showed a persistence of midbodies at 1.5 hours, compared with 50% in control-treated cells (Fig. 3C). Additionally, there was a significant accumulation (15%) of binucleated cells in all treatments compared with 2.5% in the control (Fig. 3D). To identify the time frame of RSK activity required for cytokinesis, we performed a time course of RSK inhibition (Fig. 3E,F). Inhibiting RSK during the first 30 minutes after shake-off had no effect on the outcome of cytokinesis. However, the inhibition of RSK for the first 60 minutes after shake-off was sufficient to significantly delay cytokinesis and result in binucleation (Fig. 3E,F). Additionally, delay and failure of cytokinesis were observed when the RSK inhibitor was added 30 minutes after shake-off (Fig. 3E,F). These results indicate that RSK activity is required at a time after mitotic shake-off, which is consistent with a role for RSK in promoting abscission.
Fig. 3.

RSK signaling regulates cytokinesis in CHO cells. (A) RSK activation is inhibited by the YA mutant. Cells were adhered to fibronectin or fibrinogen by WT or YA mutant integrins for 1 hour in CCM1. RSK activation was determined by western blotting using phospho-specific antibodies to activated RSK. A representative blot is shown on the left. Mean pRSK levels normalized to total RSK ± s.d. from three independent experiments is shown on the right. (B–D) Pharmacological inhibition of RSK results in failed cytokinesis. Mitotic cells were isolated from logarithmically growing WT cells and replated on fibrinogen and were either pre-treated with 1 µM BI-D1870 (Pre-BI-D1870) or 30 µM SL-0101 (Pre-SL-0101) for 2 hours or treated with 1 µM BI-D1870 (BI-D1870) only at shake-off. (B) Levels of p-GSK3β (Ser9) with inhibitor were analyzed as a read out for RSK inhibition from the cells remaining on the plate after shake-off. (C,D) Methanol-fixed cells were stained for α-tubulin and DNA and were assayed for cytokinesis. Plotted is the mean percentage of cells ± s.d. with midbodies at 1.5 hours or binucleated cells at 3 hours. (E,F) Mitotic cells were isolated from logarithmically growing WT cells and replated on fibrinogen and were treated with 1 µM BI-D1870 for the times indicated. Methanol-fixed cells were stained for α-tubulin, PRC1 and DNA and were assayed for cytokinesis. Plotted is the mean percentage of cells ± s.d. with midbodies at 1.5 hours or binucleated cells at 3 hours. One hundred cells were analyzed from each of three independent experiments shown in C–F. *P<0.05, **P<0.005.
Raf is an upstream regulator of MEK–ERK signaling. Therefore, we determined whether experimentally activating MEK–ERK and RSK by the expression of a conditionally activated RAF1 mutant was sufficient to rescue cytokinesis when cells were adhered by YA mutant integrins. For this purpose, we used an adenovirus that expressed GFP alone as a control or an adenovirus that expressed GFP together with the ΔRaf-1:ER fusion protein, which is activated by the addition of tamoxifen. Mitotic cells were isolated from logarithmically growing WT and YA cells infected with the indicated virus with or without tamoxifen and replated onto fibrinogen for the indicated times (Schematic shown in Fig. 4A). Activation of ΔRaf-1:ER dramatically upregulated the activation of ERK in cells adhered by either WT or mutant integrins (Fig. 4B). To determine whether RSK was required for activated ΔRaf1:ER to rescue cytokinesis, mitotic cells infected with the adenoviruses were isolated with or without tamoxifen and replated on fibrinogen-coated coverslips with or without BI-D1870 and assayed for progression through cytokinesis. Cells remaining on the plate after shake-off were assayed for activation of ERK and RSK (Fig. 4C). Tamoxifen-activated ΔRaf1:ER upregulated ERK and RSK activation (Fig. 4C), rescued the midbody delay and promoted successful cytokinesis in cells adhered by the YA integrin (Fig. 4D and E). At 1.5 hours, significantly fewer cells adhered by the YA integrin, expressing GFP with ΔRaf1:ER (22.9%) were connected by midbodies compared with cells expressing GFP alone (63.4%) or GFP with ΔRaf1:ER and BI-D1870 (64.4%) (Fig. 4D). At 3 hours, only 10.4% of cells adhered by the YA integrin expressing GFP with ΔRaf1:ER failed cytokinesis compared with 25.5% of cells expressing GFP alone and 27.1% cells of expressing GFP with ΔRaf1:ER and BI-D1870 (Fig. 4E). Thus, our studies support the conclusion that integrin-regulated ERK signaling in CHO cells facilitates timely progression through cytokinesis by a RSK-dependent pathway.
Fig. 4.

Activated Raf-1 rescues ERK and RSK signaling and cytokinesis. (A) Schematic of experimental methodology. (B,C) Activating ΔRaf-1:ER with tamoxifen rescues ERK signaling in cells adhered by the mutant integrin. Logarithmically growing cells were infected for 2 hours in serum-free F12 medium with adenoviruses directing the expression of either GFP or GFP-tagged ΔRaf-1:ER. Serum-containing F12 medium and 5 µM tamoxifen was added for 16 hours. (B) Mitotic cells were replated on fibrinogen in CCM1 with 5 µM tamoxifen for 1 hour. Shown is a representative western blot used to assay ERK activation in cells adhered by WT or mutant integrins. (C) Representative blots used to confirm ERK and RSK activation. Note that in A, lysates were analyzed from cells that were specifically adhered in CCM1 by either WT or the YA mutant integrin. In C, lysates were analyzed from WT and YA cells that were growing logarithmically in serum-containing medium under conditions where endogenous integrins are engaged and which mask differences caused by the mutant integrin (Reverte et al., 2006). (D,E) Mitotic cells were isolated and plated on fibrinogen with 1 µM BI-D1870 for the indicated times. Methanol-fixed cells were stained for α-tubulin and DNA, and assayed for cytokinesis. One hundred individual cells were analyzed from each of three independent experiments. Plotted is the mean percentage of WT cells and GFP-positive YA cells with midbodies at 1.5 hours (D) and binucleated cells at 3 hours (E) ± s.d. **P<0.005.
Activated RSK partially rescues cytokinesis in YA cells
To determine whether the activation of RSK is sufficient to rescue cytokinesis inhibited by the mutant integrin, YA cells were transfected with the constitutively active RSK2 mutant, HA-RSK2-Y707A (Poteet-Smith et al., 1999). Mitotic cells were isolated from logarithmically growing cells and plated on fibrinogen to assay progression through cytokinesis. Cells remaining on the plate were assayed for RSK activation. HA-RSK2-Y707A-transfected YA cells dramatically upregulated RSK phosphorylation and signaling (Fig. 5A). HA-RSK2-Y707A expression rescued the midbody delay and binucleation phenotype in cells adhered by the mutant integrin (Fig. 5B,C). At 1.5 hours, significantly fewer cells adhered by the YA integrin, expressing HA-RSK2-Y707A (35.1%) were connected by midbodies compared with untransfected YA cells (54.8%) (Fig. 5B). At 3 hours, only 18.8% of HA-RSK2-Y707A-expressing cells adhered by the YA integrins were binucleated compared with 30.7% untransfected YA cells (Fig. 5C). It appears that the expression of this RSK construct only partially rescues the cytokinesis defect in contrast to the ΔRaf1:ER construct, which efficiently rescued the cytokinesis defect in YA cells (Fig. 4). These results indicate that although RSK is required downstream of activated Raf, it might not be sufficient to rescue cytokinesis in YA cells, suggesting the involvement of other Raf–ERK targets.
Fig. 5.
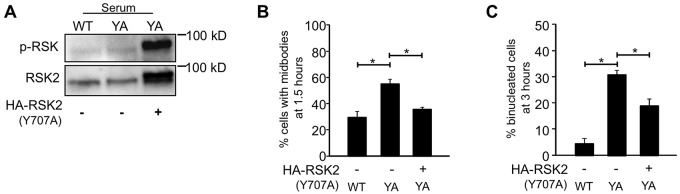
Activated RSK rescues cytokinesis in YA cells. (A) Representative blots showing relative levels of RSK activation in untransfected WT and YA cells, and HA-RSK2-Y707A transfected YA cells. (B,C) Mitotic cells were isolated and plated on fibrinogen for the indicated times. Formaldehyde-fixed cells were stained for α-tubulin, HA and DNA and assayed for cytokinesis. One hundred individual cells were analyzed from each of three independent experiments. Only HA-positive cells were analyzed in the transfected sample. Plotted is the mean percentage of cells with midbodies at 1.5 hours (B) and binucleated cells at 3 hours (C) ± s.d. *P<0.05.
The integrin–RSK signaling pathway promotes cytokinesis in epithelial cells, but not in fibroblasts
We determined whether the requirement for RSK signaling in promoting cytokinesis is specific to CHO cells. Because CHO cells are epithelial in origin, we examined the role of integrin-regulated ERK–RSK signaling in cytokinesis in the human mammary epithelial cell line MCF-10A. For these experiments, mitotic MCF10A cells were isolated from logarithmically growing cultures and either replated on a laminin-rich matrix (Homan et al., 2002; Langhofer et al., 1993) or incubated in suspension and the progression through cytokinesis was analyzed at 2.5 and 5 hours. Cells adhered to laminin-rich matrices exhibited robust activation of ERK and RSK; however, ERK and RSK activity were dramatically downregulated in suspended MCF10A cells at both 2.5 and 5 hours (Fig. 6A). Furthermore, suspended MCF10A cells showed a delayed midbody phenotype (56.4%) and an accumulation of binucleated cells (44.4%) compared with cells that were attached (22.1% and 10.2%, respectively) (Fig. 6B,C)
Fig. 6.

RSK signaling regulates cytokinesis in MCF10A cells. (A–C) Mitotic cells were isolated from logarithmically growing MCF10A cells and either adhered (A) on a laminin-rich matrix or incubated in suspension (S) in CCM1 for the times indicated. (A) Shown are representative western blots for the analysis of ERK and RSK activation. (B,C) The suspended cells were plated on poly-lysine for 5 minutes, methanol-fixed and stained for α-tubulin and DNA and then assayed for cytokinesis. Plotted is the mean percentage of cells with midbodies at 2.5 hours (B) or binucleated cells at 5 hours (C) ± s.d. (D–F) Mitotic MCF10A cells were replated on a laminin-rich matrix in CCM1 with 1 µM BI-D1870 for the times indicated. (D) RSK activation in MCF10A cells with BI-D1870 was assayed by western blotting. A representative blot is shown. (E,F) Methanol-fixed cells were stained for α-tubulin and DNA and assayed for cytokinesis. Plotted is the mean percentage of cells with midbodies at 2.5 hours (E) or cells that were binucleated at 5 hours (F) ± s.d. (G,H) MCF10A cells treated with siRNAs. Knockdown of RSK1 and RSK2 was determined by western blotting (G). Shown are representative blots. (H) Methanol-fixed cells were stained for α-tubulin, lamin B1 and DNA and assayed for cytokinesis. One hundred individual cells were analyzed from each of two independent experiments in B–H. Mean percentage of binucleated cells ± s.d. is plotted. *P<0.05, **P<0.005, ***P<0.0005.
To determine whether RSK signaling is required for cytokinesis in MCF10A cells, mitotic cells were isolated and plated on a laminin-rich matrix with and without BI-D1870. At 2.5 hours, a significant percentage of cells treated with the inhibitor (43.8%) were delayed in the midbody stage compared with control DMSO-treated cells (22.8%). At 5 hours, there was an accumulation of binucleated cells in inhibitor-treated (33.2%) compared with control (8.0%) cells (Fig. 6D–F). When cytokinesis is assayed using cells isolated by mitotic shake-off, the majority of MCF10A cells progress through cytokinesis without cell–cell contacts. To determine the effect of cell density on the requirement for RSK signaling, proliferating MCF10A cells at relatively high density were cultured with and without BI-D1870, and 25 mitotic MCF10A cells from each control and BI-D1870-treated samples were imaged by time-lapse microscopy as they progressed through cytokinesis. The results indicate that 95% of the DMSO-treated MCF10A cells completed cytokinesis successfully between 2.5 and 4.5 hours. By contrast, 23% of the BI-D1870-treated cells were delayed at the midbody stage and 41% ultimately failed cytokinesis. To confirm that RSK regulates cytokinesis, we used an siRNA approach to inhibit RSK expression. The treatment of MCF10A cells with siRNA targeting RSK1, RSK2 or a combination of both inhibited the expression of the respective RSK isoform(s) (Fig. 6G) and resulted in a significant accumulation of binucleated cells compared with the cells treated with a non-targeting siRNA (Fig. 6H). These results suggest that both RSK1 and RSK2 play a role in the regulation of cytokinesis supporting the idea that integrin signaling through ERK and RSK contributes to the timely and successful completion of cytokinesis.
We next asked whether other cells required ERK–RSK signaling for cytokinesis. We assayed cytokinesis in primary human foreskin fibroblasts (HFFs) in the presence and absence of the ERK-specific inhibitor PD184352. Our results indicate that inhibiting ERK signaling (supplementary material Fig. S1A) in primary fibroblasts does not affect progression through cytokinesis. At 1.5 hours, similar percentages of dividing cells had midbodies: 78.67±4.5% (mean ± s.d.) control cells had midbodies compared with 78±5.3% of cells treated with PD184352 (supplementary material Fig. S1B). At 3 hours, both control and treated cells had successfully completed cytokinesis. This was further corroborated in an immortalized human fibroblast cell line, BJT (Hahn et al., 1999), where inhibition of ERK signaling with PD184352 did not affect the progression and successful completion of cytokinesis (data not shown). We also used siRNA to inhibit RSK expression in HFF cells to examine whether RSK played a role in regulating cytokinesis in these cells. Treatment of cells with non-targeting siRNA or siRNA targeting RSK1, RSK2 and a combination of both did not significantly affect the progression through cytokinesis in three independent experiments. Similar percentages of binucleated cells were observed in all the siRNA treatments: 1.8±1.9% (s.d.) non-targeting compared to 2.9±0.96% in RSK1, 3.1±0.2% in RSK2 and 3.4±2.7% in RSK1 and RSK2. Thus, activation of ERK and RSK does not promote cytokinesis in primary and immortalized human foreskin fibroblasts, suggesting that the regulatory function of this pathway is a feature of epithelial cells.
We also tested the role of RSK signaling in the human pancreatic epithelial cell line, HPNE. Since mitotic cells were not easily isolated from logarithmically growing cultures of HPNE cells, we used a different experimental approach. We treated logarithmically growing cultures with BI-D1870, and using time-lapse microscopy, imaged 50 mitotic HPNE cells from each control and BI-D1870-treated samples (supplementary material Fig. S2A,B). The results indicate that 96% of the control DMSO-treated HPNE cells completed cytokinesis successfully between 1.5 and 2.5 hours. By contrast, 52% of the BI-D1870-treated cells were delayed at the midbody stage and by 5 hours (end of filming), 30% had failed cytokinesis. Interestingly, the BI-D1870-treated cells that did not fail cytokinesis exhibited a significant delay in the completion of cytokinesis and required between 2.15 and 3.5 hours to complete cytokinesis.
Last, we determined whether RSK played a role in regulating cytokinesis during tissue morphogenesis as a means to probe the potential significance of this pathway in vivo. For this purpose, we chose to examine the branching morphogenesis of the murine submandibular salivary gland, which requires robust cell proliferation in addition to cleft initiation and progression and can be studied using whole organ ex vivo cultures (Daley et al., 2009). Embryonic day 13 submandibular salivary glands (E13 SMGs) were isolated and cultured for 24 hours and then incubated in culture medium with BI-D1870 for 8 hours. At this time, the α6 integrin was expressed on the surface of epithelial cells throughout the developing gland (Fig. 7A) as previously described (Kadoya and Yamashina, 1993). To identify cells connected by midbodies we used the established midbody markers α-tubulin, which localizes to both sides of the midbody bridge and PRC1, which localizes to the central midbody ring (Green et al., 2012). When we compared glands with and without the inhibitor, we found that there was a significant increase in the number of epithelial cells connected by midbodies in the inhibitor-treated glands (Fig. 7C,D), whereas there was no significant difference in the number of metaphase or anaphase cells in control and treated glands (Fig. 7C,E). Furthermore, when dissociated glands were replated onto laminin matrices, we found that 11.25±0.7% of cells expressing integrin α6 from BI-D1870-treated glands were binucleated compared with 0.49±0.7% of cells expressing α6 from DMSO-treated glands. This set of experiments corroborates the idea that epithelial cells require RSK signaling for timely progression through cytokinesis. Notably, we did not detect mesenchymal cells in mitosis or with midbodies with or without the inhibitor. Thus, conclusions of the effects of RSK inhibition in fibroblasts during salivary gland morphogenesis cannot be made from these experiments.
Fig. 7.

Cells with midbodies accumulate in explant cultures of mouse embryonic salivary glands inhibited for RSK signaling. (A–E) Submandibular salivary glands from day 13 mouse embryos were grown as explants in culture for 24 hours and treated with 3 µM BI-D1870 for 8 hours before being fixed and stained for analysis by confocal microscopy. (A) A confocal z-slice through a salivary gland showing α6 integrin staining in all the cells in the epithelial compartment (proacinar buds and ducts) of the gland. The asterisks and filled triangles identify the proacinar and duct portions of the gland respectively. An enlarged image of a confocal z-slice through a proacinar end bud is shown on the right. (B) A confocal z-slice through an end bud of the salivary gland stained for α-tubulin (red), PRC1 (green) and DNA (blue). Arrowheads indicate mitotic cells. Arrows indicate midbodies that are shown magnified in the insets. (C) A representative western blot of whole gland lysates with BI-D1870 probed for p-GSK3β as a readout of RSK activity. (D) Plotted are the number of cells in telophase or with midbodies per 1000 cells in DMSO and BI-D1870-treated glands from two independent experiments (n = 30 buds). (E) Number of mitotic cells (metaphase and anaphase) per 1000 cells in glands with BI-D1870 from two independent experiments (n = 30 buds). (F,G) Submandibular salivary glands from day 12 mouse embryos were grown as explants in culture for 24 hours with 25 µg/ml control rat IgG or anti-integrin α6 (GoH3) before being fixed and stained for analysis by confocal microscopy. (F) Number of cells in telophase or with midbodies per 1000 cells from two independent experiments (n = 40 buds). (G) Number of mitotic cells per 1000 cells from two independent experiments (n = 40 buds). *P<0.05. Scale bars: 10 µm.
We also tested whether integrin function was required for cytokinesis during salivary gland morphogenesis. Because the integrin α6 subunit was expressed on the surface of epithelial cells of the developing gland, but not by mesenchymal cells (Fig. 7A), we inhibited the α6 integrin with function-blocking antibodies. Submandibular salivary glands from day 12 mouse embryos were grown as explants in culture for 24 hours with 25 µg/ml control or α6 function-blocking antibodies. We found a significant increase in the number of epithelial cells connected by midbodies in the antibody-treated glands compared with control glands (Fig. 7F). There was no significant difference in the number of metaphase or anaphase cells in control and treated glands (Fig. 7G), although cleft formation was delayed (not shown) as previously reported in α6-integrin-inhibited salivary gland organ explants (Kadoya et al., 1995; Sakai et al., 2003). This experiment corroborates the idea that epithelial cells in the developing salivary gland require integrin function for timely progression through cytokinesis.
DISCUSSION
The importance of cell adhesion for successful cytokinesis has been recognized for over a decade (Ben-Ze'ev and Raz, 1981; Orly and Sato, 1979; Winklbauer, 1986). A direct role for integrins in the regulation of cytokinesis was later demonstrated in both in vivo and in vitro. The first was the demonstration that conditional deletion of integrin β1 in chondrocytes in developing mice leads to cytokinesis failure and chondrodysplasia (Aszodi et al., 2003). Later, cell culture studies showed that suppression of integrin function either by a mutation in the integrin β1 tail or by the inhibition of integrin expression with siRNA caused cytokinesis failure (Kittler et al., 2007; Reverte et al., 2006). Furthermore, defective β1 integrin recycling was also shown to inhibit cytokinesis further supporting the importance of integrins in promoting successful cytokinesis (Pellinen et al., 2008). Our current study identifies a novel and cell-type-specific role for integrin-regulated RSK signaling in the regulation of cytokinesis.
Multiple substrates have been identified for RSK, including both cytoplasmic and nuclear proteins (Anjum and Blenis, 2008; Romeo et al., 2012). GSK3β is one of the known RSK substrates. GSK-3β phosphorylates several microtubule-binding proteins to regulate microtubule dynamics (Etienne-Manneville, 2013). RSK phosphorylates and inhibits GSK-3β activity. Thus, RSK by targeting GSK-3β, could regulate cytokinesis by impacting microtubule function.
Filamin A is the only RSK substrate that has been reported to localize to the midbody (Mondal et al., 2012; Nunnally et al., 1980; Playford et al., 2006). Recent studies indicate that filamin A recruits BRCA2 to the midbody, where it promotes the midbody localization of components of the ESCRT complex (Mondal et al., 2012). Thus, it is tempting to speculate that integrin-regulated, RSK-dependent phosphorylation of filamin A promotes cytokinesis by localizing BRCA2 to the midbody. However, we do not know whether the phosphorylation of filamin A regulates its function at the midbody; furthermore, the role of BRCA2 in cytokinesis is not universally accepted (Lekomtsev et al., 2010).
Interestingly, RSK has been shown to suppress integrin activity by phosphorylating filamin A (Gawecka et al., 2012; Vial and McKeown-Longo, 2012). It has been proposed that RSK2 functions in a feedback loop to modulate integrin activity during cell migration with integrins activating RSK, which feeds back to negatively regulate integrin activity (Gawecka et al., 2012). One could hypothesize that a similar RSK2-dependent modulation of integrin activity could occur during cytokinesis. A recent study using HeLa cells demonstrated that tension at the midbody prolonged cytokinesis, whereas the release of tension induced abscission (Lafaurie-Janvore et al., 2013). Tension at the midbody could be promoted by integrin and release of tension by the downregulation of integrin activity. In this regard, it is interesting to note that in our CHO cell model, locking integrins in an activated conformation either by treating cells with Mn2+ or with activating antibodies promoted cytokinesis and prevented the accumulation of cells delayed in cytokinesis (Reverte et al., 2006) (Fig. 1).
The completion of cytokinesis is regulated by the recruitment of the abscission machinery to the midbody, including components of the exocyst and ESCRT complexes (Neto and Gould, 2011). The exocyst complex targets vesicular trafficking to the midbody in preparation for abscission. Components of the ESCRT complex mediate abscission upon the depolymerization and/or severing of microtubules remaining in the midbody. We propose that RSK signaling (directly or indirectly) regulates the localization or activity of components of these complexes and thereby promotes efficient and successful cytokinesis. Our analysis of the time frame when RSK signaling is required to promote cytokinesis is consistent with this idea. Based on the known RSK substrates, it is possible that RSK phosphorylates multiple substrates to promote abscission. RSK activity might target midbody components, microtubules and/or integrin activity. Our RSK siRNA results in MCF10A cells suggest that both RSK1 and RSK2 isoforms regulate cytokinesis. It will be interesting to explore whether these isoforms play distinct roles in the regulation of cytokinesis.
ERK is a known activator of RSK. We recently identified a novel role for integrins in promoting microtubule nucleation from the interphase centrosome through an ERK-dependent pathway (Colello et al., 2012). However, we do not know whether this regulation involves RSK signaling. Interestingly, although ERK promotes microtubule nucleation from the interphase centrosome in both CHO cells and HFFs (Colello et al., 2012), the suppression of ERK signaling inhibits cytokinesis in CHO cells, but not HFFs. These results suggest that the effects of ERK inhibition on microtubule nucleation and cytokinesis are separable. It is also possible that the level of microtubule nucleation that occurs in ERK inhibited HFFs is sufficient to support microtubule function during cytokinesis.
Other laboratories have implicated ERK in the regulation of abscission (Kasahara et al., 2007). They proposed that in HeLa S3 cells, ERK becomes activated by Src signaling in early mitosis and is then trafficked to the midbody by a Rab11-dependent mechanism, where it signals abscission. Our studies have identified RSK as an important downstream target of ERK in the regulation of cytokinesis in CHO cells. Thus, it is possible that RSK is activated by ERK at the midbody. Future studies will examine whether active RSK localizes to the midbody.
Others have shown that activation of Raf-1 can activate signaling pathways independent of MEK–ERK (Pearson et al., 2000). RSK-dependent regulation of integrin activity occurs by ERK-dependent and independent mechanisms (Gawecka et al., 2012; Vial and McKeown-Longo, 2012). We found that the inhibition of either ERK or RSK led to a similar phenotype in CHO cells, which suggests that ERK and RSK are functioning together in the same pathway in these cells. Consistent with this notion, ΔRaf1:ER leads to the activation of both ERK and RSK (Fig. 4). Nonetheless it is important to note that we have not ruled out the possibility that RSK regulates cytokinesis in MCF10A, HPNE and salivary gland epithelial cells by an ERK-independent mechanism, because we have not tested the requirement for ERK in the cell types.
Our results, together with findings from others (Kasahara et al., 2007), indicate that some pathways regulate cytokinesis in a cell-type-dependent manner. We found that RSK promotes cytokinesis in various types of epithelial cells without affecting integrin surface expression (supplementary material Fig. S3), including murine salivary gland epithelial cells during ex vivo morphogenesis, but not in human fibroblasts. Unfortunately, we cannot make conclusions regarding embryonic fibroblasts associated with salivary gland morphogenesis because we did not detect mesenchymal cells in mitosis or cytokinesis with or without RSK inhibitor in our analysis. Kasahara and colleagues indicated that HeLa (ovarian cancer), A431 (squamous cell cancer) and Cos-1 (monkey kidney fibroblastic-like) cells required MEK–ERK signaling for cytokinesis, whereas SYF fibroblasts, MCF-7 (breast cancer) and HCT116 (colon cancer) do not. In light of our findings, it would be interesting to compare the sensitivity of these cell lines to RSK inhibition.
In summary, our data indicate that both adhesion-dependent and -independent mechanisms support the completion of cytokinesis. We have shown that RSK signaling promotes cytokinesis downstream of integrins in epithelial cells in culture, that RSK and α6 integrins regulate cytokinesis during tissue morphogenesis ex vivo and that RSK regulates cytokinesis in a cell-type-specific manner. Others have shown that cytokinesis failure can lead to aneuploidy and tumorigenesis and that tetraploid cells are present at early stages of tumors from different origins (Fujiwara et al., 2005; Galipeau et al., 1996; Ganem et al., 2007; Högnäs et al., 2012; Jonsdottir et al., 2012; Lv et al., 2012), suggesting that the proper interaction of cells with components of the ECM might contribute to maintaining genomic stability by promoting successful cytokinesis. More studies are needed to understand the significance of cell-type requirements for specific signaling pathways, the availability of redundant pathways, as well as integrin-dependent regulation of cytokinesis both in the context of developmental processes and tumorigenesis.
MATERIALS AND METHODS
Cell culture
Chinese hamster ovary (CHO K1) cell lines stably expressing αIIb-5β3-1 recombinant chimeric integrins containing the WT or the Y783A (YA) β1 tail (Reverte et al., 2006) and HFFs were maintained as previously described (Colello et al., 2010). The immortalized human mammary epithelial cell line MCF10A was maintained as previously published (Debnath et al., 2003). The immortalized human pancreatic nestin-expressing cell line (hTERT-HPNE, ATCC) was maintained according to ATCC guidelines. For cytokinesis experiments, cells were replated in the serum-free growth-promoting medium CCM1 (Hyclone) as indicated.
Antibodies and other reagents
The following antibodies were purchased from Santa Cruz Biotechnology Inc.: mouse anti-α-tubulin (DM1α); rabbit anti-PRC1; rabbit anti-Raf-1; rabbit anti-RSK1; and goat anti-RSK2. The following antibodies were purchased from Cell Signaling: mouse anti-p-ERK1/2; rabbit anti-p-RSK (Ser380); rabbit anti-p-GSK3β; and mouse anti-pan-GSK3β. Mouse anti-pan-ERK was from BD Transduction Laboratories; rabbit anti-panRSK was from R and D Systems; mouse anti-HA antibody was from Covance; rat IgG, whole molecule was from Pierce, and rat anti-integrin-alpha-6 (GoH3) was from BD Biosciences. Alexa-Fluor-conjugated secondary antibodies were obtained from Invitrogen. The Cy2- and Cy3-conjugated F(ab′)2 fragments from Jackson ImmunoResearch were used as secondary antibodies for salivary gland experiments. DNA was stained using Hoechst 33342 from Invitrogen or Draq5 from Cell Signaling. The MEK-specific inhibitors U0126 and PD98059 were from Cell Signaling and PD 184,354 (CI-1040) was from Santa Cruz Biotechnology. RSK-specific inhibitor BI-D1870 was from Enzo Lifesciences and SL-0101 was from Toronto Research Chemicals. Tamoxifen was purchased from Krackeler Scientific. The constitutively active RSK2 plasmid pKH3-RSK2 Y707A was previously described (Poteet-Smith et al., 1999).
Cytokinesis assay
Loosely attached mitotic cells were gently shaken off from logarithmically growing cultures and replated in CCM1 onto fibrinogen- or fibronectin-coated coverslips and incubated at 37°C as indicated. Cells were fixed and stained and then analyzed for progression through cytokinesis by immunofluorescence microscopy. To compare cytokinesis success in adhered and suspended cells, mitotic MCF10A cells were isolated and either plated on a laminin-5-rich matrix (Homan et al., 2002) for 2.5 and 5 hours or left in suspension with intermittent shaking for 2.5 and 5 hours. The suspended cells were then plated on poly-lysine for 5 minutes and then analyzed as above. Because mitotic HPNE cells were difficult to isolate, effects of RSK inhibition on cytokinesis were determined by time-lapse imaging. Cells in metaphase were identified in logarithmically growing cultures by phase-contrast microscopy (see supplementary material Fig. S2) and then followed by time-lapse microscopy from metaphase to the completion of cytokinesis. Complete separation of daughter cells was scored as successful and formation of binucleated cells was scored as failed cytokinesis. The delay at the midbody stage was measured from the time of appearance of the midbody ring until the cells completed or failed cytokinesis.
Western blotting
Western blotting was used to assay the activation of ERK and RSK and to monitor the expression of recombinant proteins and RSK isoforms following siRNA knockdown. Cells were lysed in mRIPA buffer containing phosphatase (Fisher Scientific) and protease inhibitor cocktails (Krackeler Scientific) and equal amounts of protein (25–30 µg) were separated by SDS-PAGE and transferred to nitrocellulose for antibody analysis.
Microscopy
Microscopy was performed using a Nikon inverted TE2000-E microscope equipped with phase contrast and epifluorescence, a digital CoolSNAP HQ camera, a Prior ProScanII motorized stage, a C1 confocal system (Nikon) and a NIS-Elements acquisition and analysis software. Live imaging was performed in an environmental chamber maintained at 37°C with 5% CO2. Images were taken every 5 minutes for 3 hours or 5 hours as indicated and processed using NIS-Elements software.
Generation of ΔRaf-1:ER-expressing adenovirus
An acceptor cassette with Xho1 and BamH1 restriction sites on the 5′ and 3′ ends, respectively, was amplified by PCR and cloned into pAdtrack-GFP, which was then genetically recombined with pAdEasy, as previously described (He et al., 1998; Meadows et al., 2001) to generate the ‘acceptor’ vector for Cre-mediated recombination, pAdeasy-ACC. A BamH1 fragment containing ΔRaf-1:ER was isolated from pBabe-puro-ΔRaf-1:ER (a generous gift from Martin McMahon, UCSF Medical School) and cloned in to the BamH1 site of pDNR3 (Clontech Laboratories Inc.) to generate a ΔRaf-1:ER*-pDNR3 transfer vector, which was then recombined into the pAdeasy-ACC vector using Cre recombinase following the protocols outlined for the Creator System (Clontech Laboratories Inc.).
Transfection
MCF10A cells were transfected with siRNAs as previously published (Colello et al., 2010). Non-targeting, RSK1 and RSK2 siRNA pools were from Dharmacon/Thermo Scientific. One or two rounds of transfection were performed to get optimal knockdown of the RSK proteins in HFF and MCF10A cells respectively. After 3 days, the cells were trypsinized and replated on a laminin-rich matrix and fixed and analyzed for binucleation. CHO cells were transfected with the constitutively active RSK plasmid using the Amaxa cell line nucleofector kit T from Lonza using their optimized protocol for CHO-K1 cells. The following changes were made: 500 µl of warm complete F12 medium was added to the cuvette and the cells were gently transferred to a plate of pre-warmed complete F12 medium (avoiding repeated aspiration of the sample) and incubated at 37°C for 48 hours. Mitotic cells were isolated and replated to assay cytokinesis.
Salivary gland explant culture
Mouse submandibular salivary glands (SMGs) were dissected from timed-pregnant female mice (strain CD-1, Charles River Laboratories) at embryonic day 12 (E12) or 13 (E13) with the day of plug discovery designated as E0, in accordance with protocols approved by the University at Albany IACUC. The SMGs were cultured as described previously (Daley et al., 2009; Sequeira et al., 2012). For experiments using integrin function-blocking antibodies, isolated E12SMGs were preincubated with either 25 µg/ml control antibody or GoH3 anti-integrin α6 antibody for 45 minutes at room temperature before plating. The glands were then incubated in the presence of control or GoH3 antibodies at 37°C for 24 hours. The glands were then fixed in ice-cold methanol for 20 minutes and permeabilized with 0.4% Triton X-100 for 30 minutes with gentle shaking. Following a wash step with PBS-Tween-20, the glands were blocked and stained for immunofluorescence microscopy as described previously (Larsen et al., 2003). For the RSK inhibitor experiments, isolated E13 MSGs were cultured for 24 hours; the medium was replaced on the E13SMGs with medium with or without 3 µM BI-D1870 and the glands were cultured for an additional 8 hours. The glands were then fixed and processed for immunofluorescence microscopy as described above.
To determine whether RSK inhibition resulted in the formation of binucleated cells, 20 glands were isolated from 10 E13 mice in each of two independent experiments. Glands were cultured and incubated with or without inhibitor as described above. We found it is difficult to quantify failed cytokinesis with any confidence by determining the presence of binucleated cells in the context of the 3D gland. Therefore, after inhibitor treatment, glands were dissociated into individual cells as previously described (Sequeira et al., 2013; Wei et al., 2007). These cells were plated in DMEM/F12 containing 10% FBS onto laminin-rich matrix, gently centrifuged to promote their association with the matrix and incubated for 2 hours at 37°C to allow them to adhere and spread. The cells were fixed and stained for DNA, the α6 integrin to identify epithelial cells and lamin B1 to visualize the nuclear membrane. Binucleated cells were then identified microscopically.
Analysis of cytokinesis in salivary glands
Confocal z-stacks were obtained using Nikon EZ-C1 software with a step size of 0.5 µm. Planes were chosen for analysis based on clear resolution of α-tubulin staining of cells in the epithelial compartment of the glands. Midbodies and mitotic figures were identified based on distinct α-tubulin and PRC1 staining (as shown in Fig. 7A,B). Analyses were normalized to cell number as determined by the number of nuclei using the Bitplane scientific software Imaris. Briefly, the chosen planes were cropped to exclude most of the mesenchymal compartment and viewed in SURPASS mode for further analysis. Using the Surfaces option, a contoured image of the epithelial compartment (bud) was masked and separated for analysis from the rest of the image. An automated nuclei count was generated using the Spots option keeping the spot size constant at 4.5 µm for all images. This was checked visually using the ortho-slicer view to ensure that all nuclei were included in the spot count. Numbers of cells in telophase or with midbodies or mitotic (meta/anaphase) cells per 1000 cells were used to plot the graphs shown in the figure.
Statistical analysis
Graphpad Prism software was used to perform statistical analysis on the data generated from the experiments. Where multiple comparisons were made (>two sets of data), a one-way ANOVA with either a Dunnett's test or Bonferroni's test for post-hoc analysis was performed. Where two sets of data were being compared, a Student's t-test was used. P<0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
The authors thank Dr Jane Sottile (University of Rochester) for critically reading the manuscript, Dr Deborah Lannigan (Vanderbilt University) for kindly providing us with the constitutively active RSK2 construct and Debbie Moran (Albany Medical College) for helping us with the preparation of this manuscript.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
S.S.M., B.N., S. Sequeira and S. Sambandamoorthy generated data presented in this manuscript. K.P. generated an essential reagent used in this study. S.S.M., B.N., S. Sequeira, M.L. and S.E.L. contributed to experimental design and data interpretation. The manuscript was written by S.S.M., B.N. and S.E.L. with input from all authors.
Funding
This work was supported by the National Institutes of Health (NIH) [grant number GM51540 to S.E.L., DE022467 and DE02184101 to M.L. and F32DE20980 to S. Sequeira]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.133280/-/DC1
References
- Anjum R., Blenis J. (2008). The RSK family of kinases: emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 9, 747–758 10.1038/nrm2509 [DOI] [PubMed] [Google Scholar]
- Aszodi A., Hunziker E. B., Brakebusch C., Fässler R. (2003). Beta1 integrins regulate chondrocyte rotation, G1 progression, and cytokinesis. Genes Dev. 17, 2465–2479 10.1101/gad.277003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ze'ev A., Raz A. (1981). Multinucleation and inhibition of cytokinesis in suspended cells: reversal upon reattachment to a substrate. Cell 26, 107–115 10.1016/0092-8674(81)90038-6 [DOI] [PubMed] [Google Scholar]
- Colello D., Reverte C. G., Ward R., Jones C. W., Magidson V., Khodjakov A., LaFlamme S. E. (2010). Androgen and Src signaling regulate centrosome activity. J. Cell Sci. 123, 2094–2102 10.1242/jcs.057505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colello D., Mathew S., Ward R., Pumiglia K., LaFlamme S. E. (2012). Integrins regulate microtubule nucleating activity of centrosome through mitogen-activated protein kinase/extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase (MEK/ERK) signaling. J. Biol. Chem. 287, 2520–2530 10.1074/jbc.M111.254128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley W. P., Gulfo K. M., Sequeira S. J., Larsen M. (2009). Identification of a mechanochemical checkpoint and negative feedback loop regulating branching morphogenesis. Dev. Biol. 336, 169–182 10.1016/j.ydbio.2009.09.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J., Muthuswamy S. K., Brugge J. S. (2003). Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268 10.1016/S1046-2023(03)00032-X [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S. (2013). Microtubules in cell migration. Annu. Rev. Cell Dev. Biol. 29, 471–499 10.1146/annurev-cellbio-101011-155711 [DOI] [PubMed] [Google Scholar]
- Fujiwara T., Bandi M., Nitta M., Ivanova E. V., Bronson R. T., Pellman D. (2005). Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 437, 1043–1047 10.1038/nature04217 [DOI] [PubMed] [Google Scholar]
- Galipeau P. C., Cowan D. S., Sanchez C. A., Barrett M. T., Emond M. J., Levine D. S., Rabinovitch P. S., Reid B. J. (1996). 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett's esophagus. Proc. Natl. Acad. Sci. USA 93, 7081–7084 10.1073/pnas.93.14.7081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem N. J., Storchova Z., Pellman D. (2007). Tetraploidy, aneuploidy and cancer. Curr. Opin. Genet. Dev. 17, 157–162 10.1016/j.gde.2007.02.011 [DOI] [PubMed] [Google Scholar]
- Gawecka J. E., Young-Robbins S. S., Sulzmaier F. J., Caliva M. J., Heikkilä M. M., Matter M. L., Ramos J. W. (2012). RSK2 protein suppresses integrin activation and fibronectin matrix assembly and promotes cell migration. J. Biol. Chem. 287, 43424–43437 10.1074/jbc.M112.423046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R. A., Paluch E., Oegema K. (2012). Cytokinesis in animal cells. Annu. Rev. Cell Dev. Biol. 28, 29–58 10.1146/annurev-cellbio-101011-155718 [DOI] [PubMed] [Google Scholar]
- Hahn W. C., Counter C. M., Lundberg A. S., Beijersbergen R. L., Brooks M. W., Weinberg R. A. (1999). Creation of human tumour cells with defined genetic elements. Nature 400, 464–468 10.1038/22780 [DOI] [PubMed] [Google Scholar]
- He T. C., Zhou S., da Costa L. T., Yu J., Kinzler K. W., Vogelstein B. (1998). A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA 95, 2509–2514 10.1073/pnas.95.5.2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Högnäs G., Tuomi S., Veltel S., Mattila E., Murumägi A., Edgren H., Kallioniemi O., Ivaska J. (2012). Cytokinesis failure due to derailed integrin traffic induces aneuploidy and oncogenic transformation in vitro and in vivo. Oncogene 31, 3597–3606 10.1038/onc.2011.527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homan S. M., Martinez R., Benware A., LaFlamme S. E. (2002). Regulation of the association of alpha 6 beta 4 with vimentin intermediate filaments in endothelial cells. Exp. Cell Res. 281, 107–114 10.1006/excr.2002.5643 [DOI] [PubMed] [Google Scholar]
- Jonsdottir A. B., Stefansson O. A., Bjornsson J., Jonasson J. G., Ogmundsdottir H. M., Eyfjord J. E. (2012). Tetraploidy in BRCA2 breast tumours. Eur. J. Cancer 48, 305–310 10.1016/j.ejca.2011.11.008 [DOI] [PubMed] [Google Scholar]
- Kadoya Y., Yamashina S. (1993). Distribution of alpha 6 integrin subunit in developing mouse submandibular gland. J. Histochem. Cytochem. 41, 1707–1714 10.1177/41.11.8409377 [DOI] [PubMed] [Google Scholar]
- Kadoya Y., Kadoya K., Durbeej M., Holmvall K., Sorokin L., Ekblom P. (1995). Antibodies against domain E3 of laminin-1 and integrin alpha 6 subunit perturb branching epithelial morphogenesis of submandibular gland, but by different modes. J. Cell Biol. 129, 521–534 10.1083/jcb.129.2.521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara K., Nakayama Y., Nakazato Y., Ikeda K., Kuga T., Yamaguchi N. (2007). Src signaling regulates completion of abscission in cytokinesis through ERK/MAPK activation at the midbody. J. Biol. Chem. 282, 5327–5339 10.1074/jbc.M608396200 [DOI] [PubMed] [Google Scholar]
- Kittler R., Pelletier L., Heninger A. K., Slabicki M., Theis M., Miroslaw L., Poser I., Lawo S., Grabner H., Kozak K. et al. (2007). Genome-scale RNAi profiling of cell division in human tissue culture cells. Nat. Cell Biol. 9, 1401–1412 10.1038/ncb1659 [DOI] [PubMed] [Google Scholar]
- Lafaurie-Janovore J., Maiuri P., Wang I., Pinot M., Manneville J. B., Betz T., Balland M., Piel M. (2013). ESCRT-III assembly and cytokinetic abscission are induced by tension release in the intercellular bridge. Science 339, 1625–1629 10.1126/science.1233866 [DOI] [PubMed] [Google Scholar]
- Langhofer M., Hopkinson S. B., Jones J. C. (1993). The matrix secreted by 804G cells contains laminin-related components that participate in hemidesmosome assembly in vitro. J. Cell Sci. 105, 753–764 [DOI] [PubMed] [Google Scholar]
- Larsen M., Hoffman M. P., Sakai T., Neibaur J. C., Mitchell J. M., Yamada K. M. (2003). Role of PI 3-kinase and PIP3 in submandibular gland branching morphogenesis. Dev. Biol. 255, 178–191 10.1016/S0012-1606(02)00047-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekomtsev S., Guizetti J., Pozniakovsky A., Gerlich D. W., Petronczki M. (2010). Evidence that the tumor-suppressor protein BRCA2 does not regulate cytokinesis in human cells. J. Cell Sci. 123, 1395–1400 10.1242/jcs.068015 [DOI] [PubMed] [Google Scholar]
- Lv L., Zhang T., Yi Q., Huang Y., Wang Z., Hou H., Zhang H., Zheng W., Hao Q., Guo Z. et al. (2012). Tetraploid cells from cytokinesis failure induce aneuploidy and spontaneous transformation of mouse ovarian surface epithelial cells. Cell Cycle 11, 2864–2875 10.4161/cc.21196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows K. N., Bryant P., Pumiglia K. (2001). Vascular endothelial growth factor induction of the angiogenic phenotype requires Ras activation. J. Biol. Chem. 276, 49289–49298 10.1074/jbc.M108069200 [DOI] [PubMed] [Google Scholar]
- Mondal G., Rowley M., Guidugli L., Wu J., Pankratz V. S., Couch F. J. (2012). BRCA2 localization to the midbody by filamin A regulates cep55 signaling and completion of cytokinesis. Dev. Cell 23, 137–152 10.1016/j.devcel.2012.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mould A. P., Askari J. A., Barton S., Kline A. D., McEwan P. A., Craig S. E., Humphries M. J. (2002). Integrin activation involves a conformational change in the alpha 1 helix of the beta subunit A-domain. J. Biol. Chem. 277, 19800–19805 10.1074/jbc.M201571200 [DOI] [PubMed] [Google Scholar]
- Neto H., Gould G. W. (2011). The regulation of abscission by multi-protein complexes. J. Cell Sci. 124, 3199–3207 10.1242/jcs.083949 [DOI] [PubMed] [Google Scholar]
- Nguyen T. L. (2008). Targeting RSK: an overview of small molecule inhibitors. Anticancer. Agents Med. Chem. 8, 710–716 10.2174/187152008785914770 [DOI] [PubMed] [Google Scholar]
- Nieves B., Jones C. W., Ward R., Ohta Y., Reverte C. G., LaFlamme S. E. (2010). The NPIY motif in the integrin beta1 tail dictates the requirement for talin-1 in outside-in signaling. J. Cell Sci. 123, 1216–1226 10.1242/jcs.056549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnally M. H., D'Angelo J. M., Craig S. W. (1980). Filamin concentration in cleavage furrow and midbody region: frequency of occurrence compared with that of alpha-actinin and myosin. J. Cell Biol. 87, 219–226 10.1083/jcb.87.1.219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orly J., Sato G. (1979). Fibronectin mediates cytokinesis and growth of rat follicular cells in serum-free medium. Cell 17, 295–305 10.1016/0092-8674(79)90155-7 [DOI] [PubMed] [Google Scholar]
- Pearson G., Bumeister R., Henry D. O., Cobb M. H., White M. A. (2000). Uncoupling Raf1 from MEK1/2 impairs only a subset of cellular responses to Raf activation. J. Biol. Chem. 275, 37303–37306 10.1074/jbc.C000570200 [DOI] [PubMed] [Google Scholar]
- Pellinen T., Tuomi S., Arjonen A., Wolf M., Edgren H., Meyer H., Grosse R., Kitzing T., Rantala J. K., Kallioniemi O. et al. (2008). Integrin trafficking regulated by Rab21 is necessary for cytokinesis. Dev. Cell 15, 371–385 10.1016/j.devcel.2008.08.001 [DOI] [PubMed] [Google Scholar]
- Playford M. P., Lyons P. D., Sastry S. K., Schaller M. D. (2006). Identification of a filamin docking site on PTP-PEST. J. Biol. Chem. 281, 34104–34112 10.1074/jbc.M606277200 [DOI] [PubMed] [Google Scholar]
- Poteet-Smith C. E., Smith J. A., Lannigan D. A., Freed T. A., Sturgill T. W. (1999). Generation of constitutively active p90 ribosomal S6 kinase in vivo. Implications for the mitogen-activated protein kinase-activated protein kinase family. J. Biol. Chem. 274, 22135–22138 10.1074/jbc.274.32.22135 [DOI] [PubMed] [Google Scholar]
- Reverte C. G., Benware A., Jones C. W., LaFlamme S. E. (2006). Perturbing integrin function inhibits microtubule growth from centrosomes, spindle assembly, and cytokinesis. J. Cell Biol. 174, 491–497 10.1083/jcb.200603069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo Y., Zhang X., Roux P. P. (2012). Regulation and function of the RSK family of protein kinases. Biochem. J. 441, 553–569 10.1042/BJ20110289 [DOI] [PubMed] [Google Scholar]
- Sakai T., Larsen M., Yamada K. M. (2003). Fibronectin requirement in branching morphogenesis. Nature 423, 876–881 10.1038/nature01712 [DOI] [PubMed] [Google Scholar]
- Sequeira S. J., Soscia D. A., Oztan B., Mosier A. P., Jean-Gilles R., Gadre A., Cady N. C., Yener B., Castracane J., Larsen M. (2012). The regulation of focal adhesion complex formation and salivary gland epithelial cell organization by nanofibrous PLGA scaffolds. Biomaterials 33, 3175–3186 10.1016/j.biomaterials.2012.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequeira S. J., Gervais E. M., Ray S., Larsen M. (2013). Genetic modification and recombination of salivary gland organ cultures. J. Vis. Exp. 71, e50060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. A., Poteet-Smith C. E., Xu Y., Errington T. M., Hecht S. M., Lannigan D. A. (2005). Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res. 65, 1027–1034 [PubMed] [Google Scholar]
- Streuli C. H. (2009). Integrins and cell-fate determination. J. Cell Sci. 122, 171–177 10.1242/jcs.018945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thullberg M., Gad A., Le Guyader S., Strömblad S. (2007). Oncogenic H-Ras V12 promotes anchorage-independent cytokinesis in human fibroblasts. Proc. Natl. Acad. Sci. USA 104, 20338–20343 10.1073/pnas.0706609105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vial D., McKeown-Longo P. J. (2012). Epidermal growth factor (EGF) regulates α5β1 integrin activation state in human cancer cell lines through the p90RSK-dependent phosphorylation of filamin A. J. Biol. Chem. 287, 40371–40380 10.1074/jbc.M112.389577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C., Larsen M., Hoffman M. P., Yamada K. M. (2007). Self-organization and branching morphogenesis of primary salivary epithelial cells. Tissue Eng. 13, 721–735 10.1089/ten.2006.0123 [DOI] [PubMed] [Google Scholar]
- Winklbauer R. (1986). Cell proliferation in the ectoderm of the Xenopus embryo: development of substratum requirements for cytokinesis. Dev. Biol. 118, 70–81 10.1016/0012-1606(86)90074-6 [DOI] [PubMed] [Google Scholar]
- Yoon S., Seger R. (2006). The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24, 21–44 10.1080/02699050500284218 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.