

Abstract

Phosphoinositide 3-kinase α (PI3Kα) is a critical regulator of cell growth and transformation, and its signaling pathway is the most commonly mutated pathway in human cancers. The mammalian target of rapamycin (mTOR), a class IV PI3K protein kinase, is also a central regulator of cell growth, and mTOR inhibitors are believed to augment the antiproliferative efficacy of PI3K/AKT pathway inhibition. 2,4-Difluoro-N-{2-(methyloxy)-5-[4-(4-pyridazinyl)-6-quinolinyl]-3-pyridinyl}benzenesulfonamide (GSK2126458, 1) has been identified as a highly potent, orally bioavailable inhibitor of PI3Kα and mTOR with in vivo activity in both pharmacodynamic and tumor growth efficacy models. Compound 1 is currently being evaluated in human clinical trials for the treatment of cancer.
Keywords: GSK2126458, phosphoinositide 3-kinase α, mammalian target of rapamycin, PI3K/AKT pathway
The phosphoinositide 3-kinase (PI3K) signaling pathway is activated in a broad spectrum of human cancers.1 Activation of this pathway often occurs indirectly by the activation of receptor tyrosine kinases or the inactivation of the phosphotase and tensin homologue (PTEN) tumor suppressor.2,3 Direct activation of PI3K has been demonstrated with the discovery of several activating mutations in the PIK3CA gene itself, the gene that encodes the p110α catalytic subunit of PI3Kα.4 Several of the mutations found in PIK3CA have been shown to increase the lipid kinase activity of PI3Kα, induce activation of signaling pathways, and promote transformation of cells both in vitro and in vivo.5−7 Furthermore, the PI3K pathway is the most commonly mutated signaling pathway in human cancers.8,9
The PI3K family of enzymes is comprised of 15 lipid kinases with distinct substrate specificities, expression patterns, and modes of regulation.10 In particular, PI3Kα has emerged as an attractive target for cancer therapeutics, and several PI3K inhibitors are currently under evaluation in human clinical trials, including BEZ235 (Novartis), GDC-0941 (Genentech), PX-866 (ProlX), and XL765 (Exelixis).11,12 We describe herein the discovery of 2,4-difluoro-N-{2-(methyloxy)-5-[4-(4-pyridazinyl)-6-quinolinyl]-3-pyridinyl}benzenesulfonamide (GSK2126458, 1, Figure 1), a highly potent and selective inhibitor of class I PI3Ks and the mammalian target of rapamycin (mTOR). Compound 1 is being evaluated in a phase I, open-label, dose-escalation study in subjects with solid tumors or lymphoma.
Figure 1.

GlaxoSmithKline PI3K clinical compounds.
GSK1059615 (2),13 our first PI3K clinical compound, recently entered a dose-escalation study in patients with refractory malignancies. In follow-up studies, we sought to identify a second inhibitor with improved potency, selectivity, and pharmacokinetic (PK) profiles. Key to our approach for achieving the desired levels of PI3K activity was to pursue structure-based design utilizing crystallography of the more amenable PI3Kγ as a surrogate protein.14−17 The inhibitor-bound crystal structure of 2 in PI3Kγ indicated that the thiazolidinedione (TZD) ring formed an interaction with the catalytic lysine (Lys833) within the ATP-binding pocket. However, the structure also showed that larger groups could potentially be accommodated. We reasoned that filling the empty space in the enzyme pocket could lead to inhibitors with improved potency and potentially selectivity, and this was the basis for the initial strategy to identify alternates to the TZD moiety.
Synthesis of these derivatives began with conversion of 6-bromo-4-chloroquinoline (3) to the corresponding 4-iodo intermediate 4, followed by installation of the 4-pyridyl group under standard palladium-catalyzed cross-coupling conditions to provide quinoline 5 (Scheme 1). Various aryl (Ar) groups were then attached to the 6-position of the quinoline core using an in situ borylation and palladium-catalyzed cross-coupling to furnish the desired analogs 6.18,19
Scheme 1. Synthesis of TZD Replacement Analogs.

Conditions: (a) 2 M HCl, ether; then NaI, EtCN, reflux. (b) 4-Pyridylboronic acid, Pd(PPh3)4, 2 M aqueous K2CO3, dioxane, 100 °C. (c) Pinacolato(diboron), PdCl2(dppf)-CH2Cl2, KOAc, dioxane, 100 °C; then ArBr, 2 M aqueous Na2CO3, 110 °C.
Inhibition of PI3Kα was measured using a continuous read time-resolved fluorescence resonance energy transfer displacement assay.20 The analogs were also evaluated in a PI3Kα-driven mechanistic cellular assay, which measured the ability of the compounds to decrease intracellular phosphorylation of AKT at S473 (pAKT-S473) in T47D and BT474 cancer cells. Replacement of the TZD with a simple phenyl group (6a) resulted in a dramatic loss in potency (Table 1). The activity was much less attenuated for both pyridine 6b and indazole 6c, although neither showed much improvement in the cellular assay relative to 6a. Molecular modeling overlays indicated that the pyridyl and indazolyl nitrogens of 6b and 6c, respectively, were most likely making distinct interactions with the enzyme. We therefore merged the two heterocycles to form azaindazole 6d, which provided a substantial boost in biochemical and cellular potency. Cocrystallization of 6d with PI3Kγ confirmed that the nitrogen at the 2-position of the indazole forms a hydrogen bond with Lys833 and that the pyridyl nitrogen interacts with a conserved active site water molecule (Figure 2).
Table 1. TZD Replacement SAR.
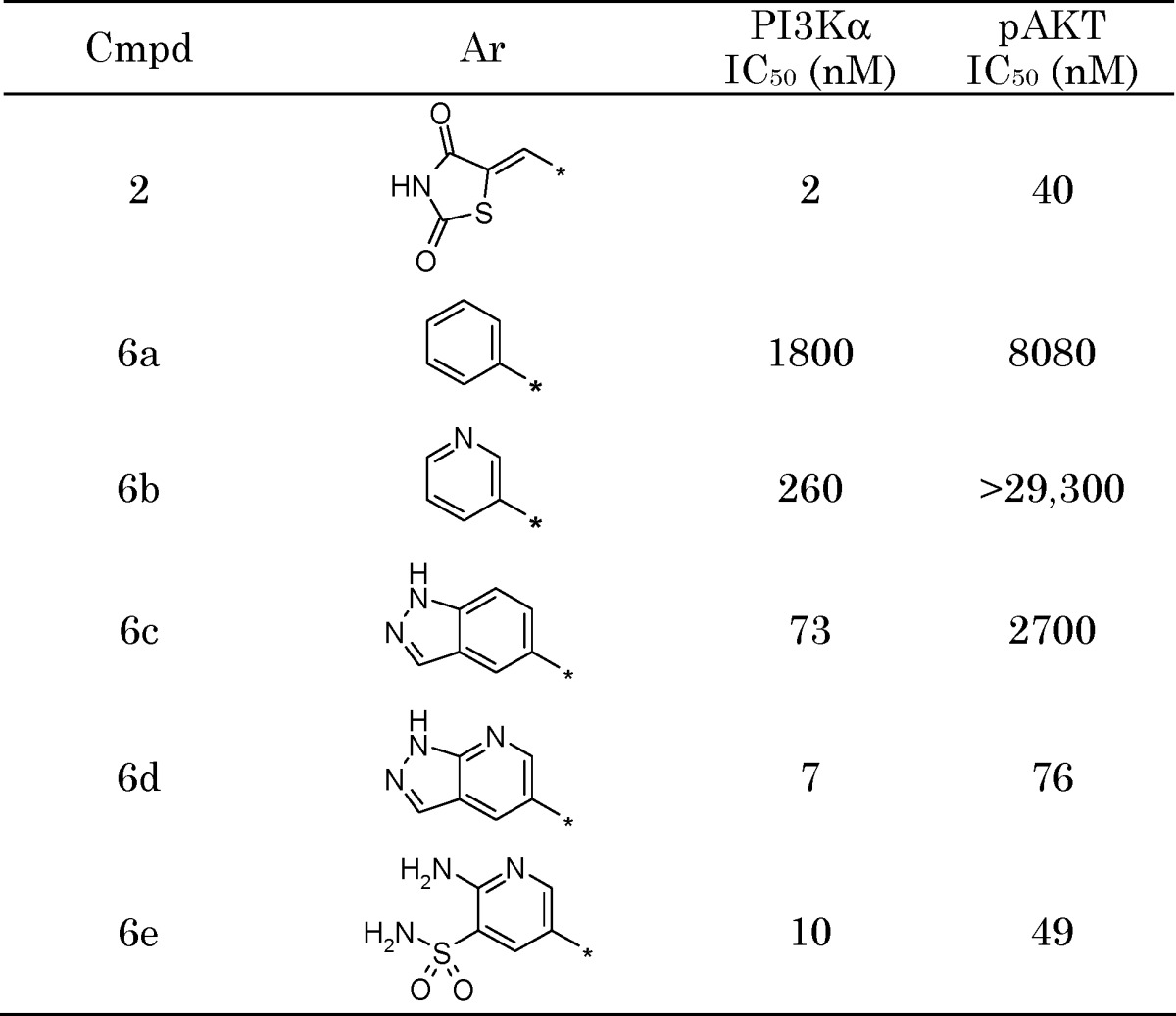 |
Figure 2.
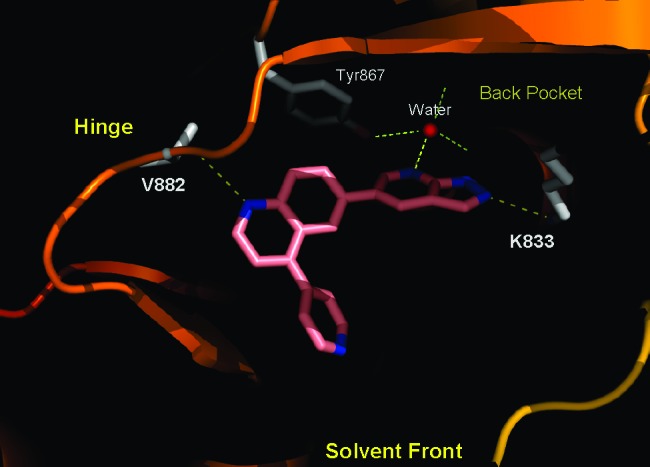
X-ray cocrystal structure of p110γ with 6d. The protein is shown in the solid ribbon. Selected residues are shown in gray. Figures 2 and 3 were prepared using PyMOL.21
Although 6d exhibited a promising activity profile, the compound displayed very low aqueous solubility and a poor PK profile, characterized by high clearance, a short half-life, and a lack of oral bioavailability. Opening of the pyrazole portion of the azaindazole to give 2,3,5-trisubstituted pyridine analogs led to the identification of pyridylsulfonamide 6e, which retained the gains made in potency and showed increased solubility. However, no improvement in the PK profile was noted.
We next examined the structure-activity relationship (SAR) of the sulfonamide moiety and found that arylsulfonamides (e.g., 6f) exhibited oral exposure in rats with no loss of inhibitory activity (Table 2). Removal of the 2-amino group to yield 6g further increased exposure ∼3-fold. Reversal of the sulfonamide connectivity (6h) led to an increase in biochemical and cellular potency, while maintaining the improved oral exposure.
Table 2. Selected Pyridylsulfonamide Analog SARa.
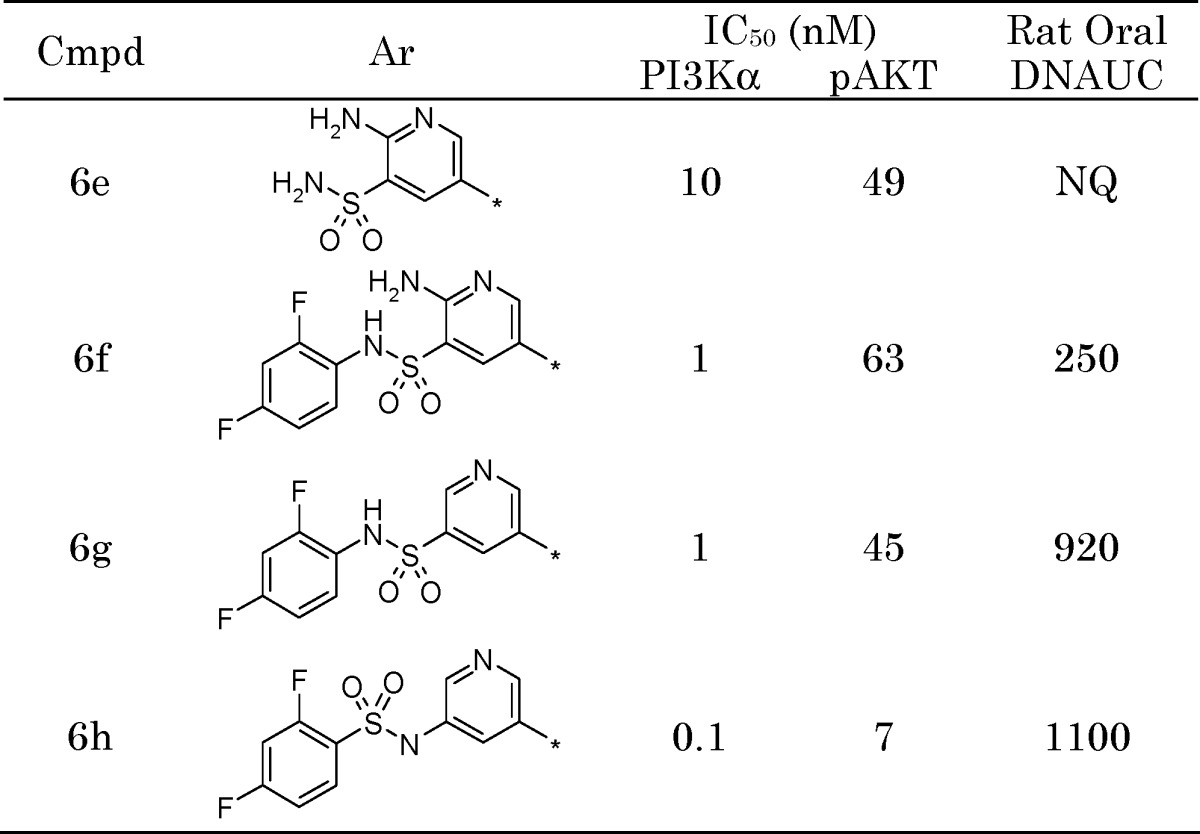 |
Units: Dose-normalized area under the curve (DNAUC) (ng h mL−1 mg−1 kg−1); NQ, not quantifiable.
Reintroduction of a small substituent at the 2-position of the pyridine (e.g., methoxy, methyl, halogen, etc.) resulted in a significant boost in both enzyme and cellular potency (data not shown). We reasoned that this was due to the substituent filling unoccupied space deep within the enzyme pocket, as well as a potential effect on the orientation of the neighboring sulfonamide moiety. Further efforts showed that the incorporation of a pyridazine at the 4-position of the quinoline resulted in a moderate improvement in the CYP inhibition profile as compared to the pyridine (data not shown).
These studies eventually led to the identification of 1, an extraordinarily potent inhibitor of PI3Kα (p110α/p85α) with low picomolar activity (PI3Kα IC50 = 0.04 nM). In biochemical assays, compound 1 is significantly more potent than 2 (PI3Kα IC50 = 2 nM) and is the most potent PI3Kα inhibitor reported to date. In comparison with other clinical PI3K inhibitors, 1 is ∼100-fold more potent than BEZ235 (IC50 = 6 nM)22 and GDC-0941 (IC50 = 9 nM)23 and ∼1000-fold more active than XL-765 (IC50 = 39 nM).11,12 Importantly, 1 is also a low picomolar inhibitor of the common activating mutants of p110α (E542K, E545K, and H1047R) found in human cancer (Table 3). Similar to the other reported PI3K inhibitors, 1 is also active against the other class I PI3K isoforms (β, γ, and δ).
Table 3. Biochemical Activity of 1.
| p110 isoform app Ki (nM) |
p110α mutant app Ki (nM) |
|||||
|---|---|---|---|---|---|---|
| α | β | δ | γ | E542K | E545K | H1047R |
| 0.019 | 0.13 | 0.024 | 0.06 | 0.008 | 0.008 | 0.009 |
A cocrystal structure of PI3Kγ in complex with 1 shows the inhibitor bound in the ATP-binding site of the enzyme (Figure 3). The structure was determined to 2.7 Å resolution and shows, like 6e, that the pyridyl nitrogen forms a key hydrogen bond with the conserved water molecule. The sulfonamide interacts with Lys833, making a strong charged interaction. On the basis of the pKa of the sulfonamide-NH (6.56),24 ∼87% of the moiety exists in its deprotonated form at physiological pH. This charged interaction may help to explain the superior potency of 1 as compared to the other reported PI3K inhibitors. In addition, the difluorophenyl group fills a hydrophobic region in the back pocket of the enzyme, while the quinoline nitrogen forms an interaction with the hinge (Val882).
Figure 3.
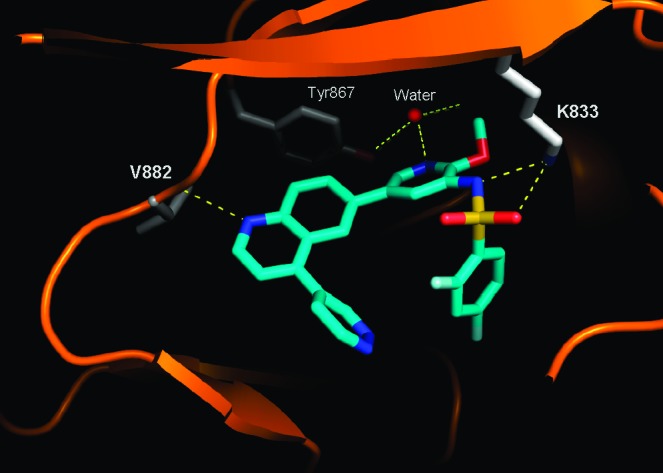
X-ray cocrystal structure of p110γ with GSK2126458 (1).
Compound 1 shows excellent selectivity over protein kinases (>10,000-fold vs >240 kinases evaluated) with the notable exception of the class IV PI3K family. mTOR, a class IV PI3K protein kinase, is a central regulator of cell growth and exists in two functional complexes, mTORC1 and mTORC2.25 mTORC2 is proposed to regulate AKT S473 phosphorylation, and its inhibition is believed to augment the antiproliferative efficacy of a PI3K inhibitor by dual inhibition of the PI3K/AKT pathway.26 The kinase domain of mTOR is homologous to the p110α catalytic subunit of the class I PI3Ks,27 and 1 is a potent inhibitor of both mTOR complexes with subnanomolar activity (Table 4). Compound 1 is also a potent inhibitor of the class IV PI3 kinase, DNA-PK (IC50 = 0.28 nM).
Table 4. mTOR Complex Activity of GSK2126458 (1).
| mTORC1 app Ki (nM) | mTORC2 app Ki (nM) |
|---|---|
| 0.18 | 0.3 |
In mechanistic cellular assays, 1 caused a significant reduction in the levels of pAKT-S473 with remarkable potency (Table 5). Consistent with its activity against both PI3Kα and mTOR, 1 also inhibits phosphorylation of AKT-T308 and p70S6K at low nanomolar concentrations (data not shown). Compound 1 induces a G1 cell cycle arrest and inhibits cell proliferation in a large panel of cell lines, including T47D and BT474 breast cancer lines.
Table 5. Mechanistic and Functional Cellular Activity of 1.
| pAKT-S473 IC50 (nM) |
growth IC50 (nM) |
||
|---|---|---|---|
| T47D | BT474 | T47D | BT474 |
| 0.41 | 0.18 | 3 | 2.4 |
The PK profile of 1 was studied in four preclinical species (mouse, rat, dog, and monkey). The compound showed low blood clearance and good oral bioavailability (Table 6). In addition, 1 had minimal potential to inhibit the human cytochrome P450 isoforms (IC50 > 25 μM vs CYPs 3A4, 1A2, 2C9, 2C19, and 2D6).
Table 6. Preclinical PK Profile of 1a.
| iv dosing |
oral dosing |
||||
|---|---|---|---|---|---|
| species | Clb | Vdss | T1/2 | DNAUC | % F |
| mouse | 10 | 1.0 | 2.1 | 1100 | 100 |
| rat | 2.3 | 1.1 | 6.2 | 6100 | 81 |
| dog | 5.8 | 0.7 | 1.3 | 2400 | 80 |
| monkey | 3.6 | 0.8 | 3.5 | 2300 | 49 |
Units: Clb, mL min−1 kg−1; Vdss, L kg−1; T1/2, h; and DNAUC, ng h mL−1 mg−1 kg−1.
In an in vivo setting, 1 exhibited a dose-dependent reduction in pAKT-S473 levels in human BT474 tumors implanted in mice. In the study, designed to measure the magnitude and duration of the pharmacodynamic (PD) response, mice were treated orally with drug, and pAKT levels were determined over the course of 24 h. Following a single 300 μg/kg dose, 1 showed a profound and sustained PD response over the 10 h observation period with pAKT levels returning to those of control by 24 h (Figure 4). Remarkably, the sustained PD response was achieved with very low circulating levels of drug, consistent with the high in vitro potency of 1.
Figure 4.
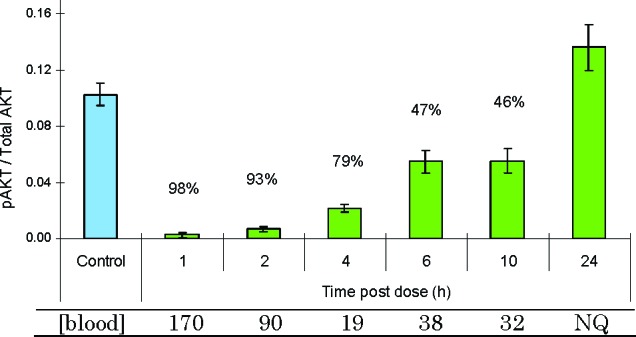
PD effect of 1 in BT474 human tumor xenografts following a single 300 μg kg−1 oral dose. The ratio of pAKT/total AKT was measured and compared to control. [Blood], concentration of drug in the blood in ng mL−1; NQ, not quantifiable.
Compound 1 was also evaluated in a BT474 human tumor xenograft growth efficacy model where mice were administered a single oral dose five times per week for 3 weeks. Consistent with inhibition of the PI3K/AKT/mTOR pathway, the drug exhibited dose-dependent tumor growth inhibition (Figure 5). The top dose (3 mg kg−1) was well-tolerated in the study. As reported previously,13 compound 2 exhibited efficacy in BT474 xenografts following twice daily dosing at 25 mg kg−1. In comparison, compound 1 exhibited similar efficacy at a much lower dose and less frequent administration.28
Figure 5.
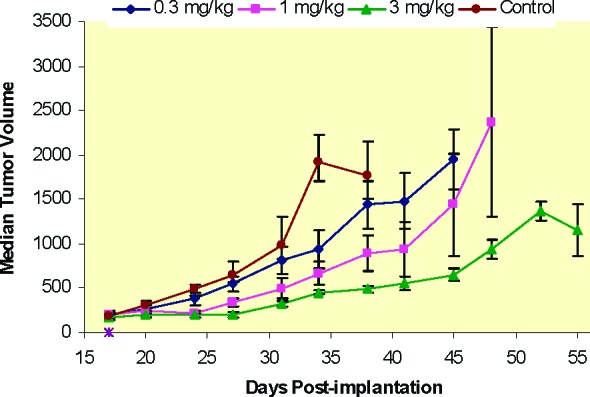
Tumor growth efficacy of 1 in BT474 human tumor xenografts as measured by median tumor volume (cu mm). Mice were dosed once daily for 5 days/week (M−F) for 3 weeks (days 17−21, 24−28, and 31−35). Error bars denote the standard error of the mean (SEM).
In conclusion, we report the discovery of 1, a structurally novel inhibitor of the PI3K/AKT/mTOR signaling pathway with picomolar activity against PI3Kα and mTOR. Compound 1 displays remarkable potency in both mechanistic and antiproliferative cellular assays. Compound 1 also exhibits excellent in vivo activity, highlighted by a sustained PD effect at very low circulating drug levels. Inhibition of the PI3K/AKT/mTOR pathway is expected to have a beneficial effect on cancer therapy, and 1 has been advanced into a phase I, open-label, dose-escalation study in subjects with solid tumors or lymphoma.
Abbreviations
PI3K, phosphoinositide 3-kinase; mTOR, mammalian target of rapamycin; PTEN, phosphotase and tensin homologue; DNAUC, dose-normalized area under the curve.
Supporting Information Available
Biological assays, biological data, and experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
The coordinates for 1 and 6d have been deposited with the RCSB Protein Data Bank under the accession codes 3L08 (1) and 3L54 (6d).
Supplementary Material
References
- Engelman J. A.; Luo J.; Cantley L. C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [DOI] [PubMed] [Google Scholar]
- Auger K. R.; Serunian L. A.; Soltoff S. P.; Libby P.; Cantley L. C. PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell 1989, 57, 167–175. [DOI] [PubMed] [Google Scholar]
- Cully M.; You H.; Levine A. J.; Mak T. W. Beyond PTEN mutations: The PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer 2006, 6, 184–192. [DOI] [PubMed] [Google Scholar]
- Samuels Y.; Wang Z.; Bardelli A.; Silliman N.; Ptak J.; Szabo S.; Yan H.; Gazdar A.; Powell S. M.; Riggins G. J.; Willson J. K. V.; Markowitz S.; Kinzler K. W.; Vogelstein B.; Velculescu V. E. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [DOI] [PubMed] [Google Scholar]
- Kang S.; Bader A. G.; Vogt P. K. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc. Natl. Acad. Sci. 2005, 102, 802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader A. G.; Kang S.; Vogt P. K. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc. Natl. Acad. Sci. 2006, 103, 1475–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J. J.; Liu Z.; Wang L.; Shin E.; Loda M. F.; Roberts T. M. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc. Natl. Acad. Sci. 2005, 102, 18443–18448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Vogt P. K.; Kang S.; Elsliger M. A.; Gymnopoulos M. Cancer-specific mutations in phosphatidylinositol 3-kinase. Trends Biochem. Sci. 2007, 32, 342–349. [DOI] [PubMed] [Google Scholar]
- Karakas B.; Bachman K. E.; Park B. H. Mutation of the PIK3CA oncogene in human cancers. Br. J. Cancer 2006, 94, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katso R.; Okkenhaug K.; Ahmadi K.; White S.; Timms J.; Waterfield M. D. Cellular function of phosphoinositide 3-kinases: Implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 621–637. [DOI] [PubMed] [Google Scholar]
- Yap T. A.; Garrett M. D.; Walton M. I.; Raynaud F.; de Bono J. S.; Workman P. Targeting the PI3K-AKT-mTOR pathway: Progress, pitfalls, and promises. Curr. Opin. Pharmacol. 2008, 8, 393–412. [DOI] [PubMed] [Google Scholar]
- Kong D.; Yamori T. Advances in development of phosphatidylinositol 3-kinase inhibitors. Curr. Med. Chem. 2009, 16, 2839–2854. [DOI] [PubMed] [Google Scholar]
- Auger K. R.; Luo L.; Knight S. D.; Van Aller G.; Tummino P. J.; Copeland R. A.; Diamond M.; Sutton D.; Lu H.; Oleykowski K.; Sudakin V.; Dhanak D.; Jackson J. R.. GSK1059615: A Novel Inhibitor of Phosphoinositide 3-Kinase for the Treatment of Cancer. EORTC-NCI-AACR International Conference on Molecular Targets and Cancer, Geneva Palexpo, Geneva, Switzerland, October, 2008.
- Walker E. H.; Perisic O.; Ried C.; Stephens L.; Williams R. L. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature 1999, 402, 313–320. [DOI] [PubMed] [Google Scholar]
- Walker E. H.; Pacold M. E.; Perisic O.; Stephens L.; Hawkins P. T.; Wymann M. P.; Williams R. L. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 2000, 6, 909–919. [DOI] [PubMed] [Google Scholar]
- Knight Z. A.; Gonzalez B.; Feldman M. E.; Zunder E. R.; Goldenberg D. D.; Williams O.; Loewith R.; Stokoe D.; Balla A.; Toth B.; Balla T.; Weiss W. A.; Williams R. L.; Shokat K. M. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 2006, 125, 733–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The structure of the p110α/p85α complex has recently been solved. Huang C.-H.; Mandelker D.; Schmidt-Kittler O.; Samuels Y.; Velculescu V. E.; Kinzler K. W.; Vogelstein B.; Gabelli S. B.; Amzel L. M. The Structure of a Human p110α/p85α Complex Elucidates the Effects of Oncogenic PI3Kα Mutations. Science 2007, 318, 1744–1748. [DOI] [PubMed] [Google Scholar]
- Adams N. D.; Burgess J. L.; Darcy M. G.; Donatelli C. A.; Knight S. D.; Newlander K. A.; Ridgers L.; Sarpong M.; Schmidt S. J.. Preparation of quinoline derivatives as PI3 kinase inhibitors. WO2008144463, November 27, 2008.
- Adams N. D.; Chaudhari A. M.; Donatelli C. A.; Knight S. D.; Newlander K. A.; Parrish C. A.; Ridgers L.; Sarpong M. A.. Preparation of quinoline derivatives as PI3 kinase inhibitors. WO2008144464, November 27, 2008.
- Gray A.; Olsson H.; Batty I. H.; Priganica L.; Downes C. P. Nonradioactive methods for the assay of phosphoinositide 3-kinases and phosphoinositide phosphatases and selective detection of signaling lipids in cell and tissue extracts. Anal. Biochem. 2003, 313, 234–245. [DOI] [PubMed] [Google Scholar]
- DeLano W. L.The PyMOL Molecular Graphics System; DeLano Scientific: Palo Alto, CA, 2002. [Google Scholar]
- Our data (shown) were consistent with literature reports. ; Maira S.-M.; Stauffer F.; Brueggen J.; Furet P.; Schnell C.; Fritsch C.; Brachmann S.; Chene P.; De Pover A.; Schoemaker K.; Fabbro D.; Gabriel D.; Simonen M.; Murphy L.; Finan P.; Sellers W.; Garcia-Echeverria C. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [DOI] [PubMed] [Google Scholar]
- Our data (shown) were consistent with literature reports. Folkes A. J.; Ahmadi K.; Alderton W. K.; Alix S.; Baker S. J.; Box G.; Chuckowree I. S.; Clarke P. A.; Depledge P.; Eccles S. A.; Friedman L. S.; Hayes A.; Hancox T. C.; Kugendradas A.; Lensun L.; Moore P.; Olivero A. G.; Pang J.; Patel S.; Pergl-Wilson G. H.; Raynaud F. I.; Robson A.; Saghir N.; Salphati L.; Sohal S.; Ultsch M. H.; Valenti M.; Wallweber H. J. A.; Wan N. C.; Wiesmann C.; Workman P.; Zhyvoloup A.; Zvelebil M. J.; Shuttleworth S. J. The Identification of 2-(1H-Indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a Potent, Selective, Orally Bioavailable Inhibitor of Class I PI3 Kinase for the Treatment of Cancer. J. Med. Chem. 2008, 51, 5522–5532. [DOI] [PubMed] [Google Scholar]
- Spectroscopic determination of the pKa of 1 was done using Sirius GLpKa-D-PAS.
- Shaw R. J.; Cantley L. C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [DOI] [PubMed] [Google Scholar]
- Sarbassov D. D.; Guertin D. A.; Ali S. M.; Sabatini D. M. Phosphorylation and regulation of Akt/PKB by the rictor−mTOR complex. Science 2005, 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B.; Waterfield M. D. Signaling by Distinct Classes of Phosphoinositide 3-Kinases. Exp. Cell Res. 1999, 253, 239–254. [DOI] [PubMed] [Google Scholar]
- For additional in vivo characterization, see the following: Hardwicke M. A.; Lu H.; Luo L.; Diamond M.; Sung C.-M.; Carson J. D.; Plant R.; Gardiner C.; Wang J.; Oleykowski K.; Gontarek R. R.; Van Aller G.; Gilbert S.; Sutton D.; Tummino P. J.; Wooster R.; Knight S. D.; Jackson J. R.; Dhanak D.; Auger K. R.. Biological Characterization of GSK2126458, A Novel and Potent Inhibitor of Phosphoinositide 3-Kinase and the Mammalian Target of Rapamycin (mTOR). EORTC-NCI-AACR International Conference on Molecular Targets and Cancer, Boston, MA, November, 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.