

Abstract
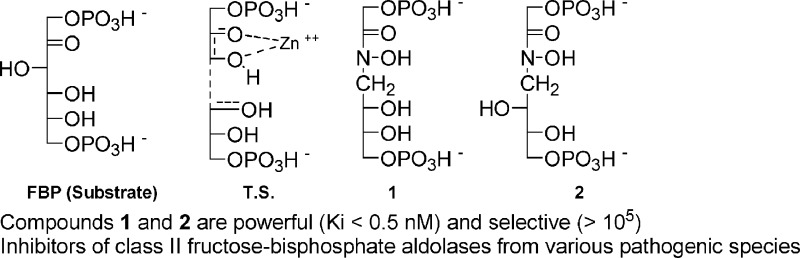
We hereby describe the rationale synthesis and biochemical evaluation of the most powerful and selective inhibitors of class II fructose bis-phosphate aldolases so far reported. These inhibitors are of potential therapeutic interest, since the class II enzyme is present exclusively in microorganisms (among which many pathogenic species) and is absent from man, plants, and animals.
Keywords: Selective inhibitors, antibiotics, microbial enzymes, metallo-enzymes, fructose bis-phosphate aldolase, glycolysis
Bacterial and fungal infections due to multidrug-resistant microorganisms represent a health problem of major concern.1,2 Old known diseases of terrifying reputation like tuberculosis or plague are re-emerging (one-third of the entire world population is believed to be infected by a latent form of tuberculosis).3 Recent and so far limited epidemics of plague have been reported.4 Together with an alarming increase in nosocomial diseases observed in hospitals,5 the present situation clearly shows that there is an urgent need for a rapid elaboration of new antibiotics. This needs the identification of new selective microbial targets.
Class II fructose bis-phosphate aldolase (FBA) could be one of these new therapeutic targets, since it is present exclusively in microorganisms and absent in mammalians (man) who utilize the class I enzyme. This assumption is further supported by the inability to knock out the class II FBA genes of Streptomyces and Escherichia coli, an indication that even when both classes are present (like in E. coli), class II FBA is essential.6,7 This class of enzymes is present in many pathogenic species among which include bacteria, like Mycobacterium tuberculosis (agent of tuberculosis), Yersinia pestis (plague), Mycobacterium leprae (leprosy), Helicobacter pylori (an agent of stomach ulcer and the only known bacteria that causes cancers), and Pseudomona aeruginosa (nosocomial severe respiratory diseases); yeasts, like Candida albicans (an opportunistic yeast responsible for candidiases); parasites, like Giardia lamblia; and many others including agents of potential use in bioterrorism.
Because the two classes have very different structures and mechanisms (Figure 1), it may seem easy to prepare strong inhibitors of the (microbial) class II FBA having only a weak activity on (human) class I. Indeed, this had been suggested a long time ago by Lewis and Lowe8 who reported the synthesis of phosphoglycolo-hydroxamic acid (PGH), a first strong inhibitor of FBA, although missing a sufficient selectivity for the class II enzyme, and even for FBA.8,9 In fact, PGH is probably a strong inhibitor of any enzyme using DHAP as a substrate. Moreover, PGH is relatively unstable in aqueous solutions, releasing (toxic) hydroxylamine, which handicaps its possible use in vivo.
Figure 1.
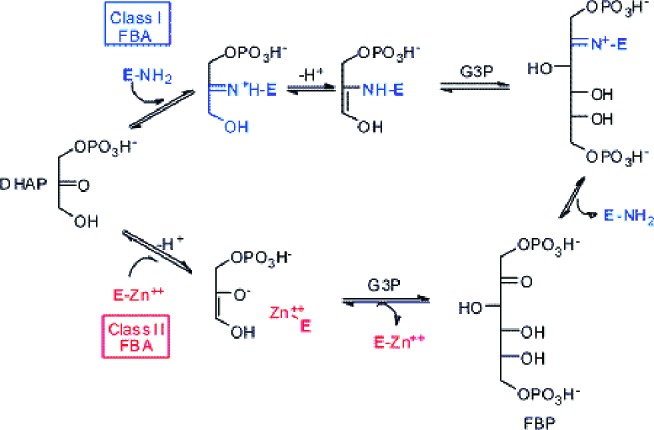
Mechanisms of class I (human) and class II (microbial) FBP aldolases.
Since then, the idea of preparing inhibitors of class II FBA as potential new antibiotics has received very limited attention. Surprisingly, in the same time, the preparation and evaluation of dozens of nonselective inhibitors of FBA have been reported.10
We reasoned that an “ideal” selective inhibitor should have the following characteristics:
-
−
Two phosphoryl groups at a proper distance from each other. This should be longer than in the substrate FBP, to mimic the supposed transition state of the reaction (TS in Figure 2).
-
−
One chemical function able to strongly bind the zinc ion present in the active site of class II FBA (and absent in class I).
-
−
Two hydroxyl groups in charge of mimicking the alcohol functions present at C-3 and C-4 (threo) or at C-4 and C-5 (erythro) of FBP.
Figure 2.

Fischer representations of fructose bis-phosphate (FBP; substrate of FBA), of the supposed transition state of the reaction catalyzed by a class II FBA (TS) and of the designed inhibitors 1 and 2.
This logically led us to prepare the two compounds 1 and 2 represented in Figure 2. As compared to PGH, the two compounds bearing an N-alkylated hydoxamic acid are also much more stable in water. The syntheses of the two inhibitors, summarized in Schemes 1 and 2, are based on classical organic reactions and are fully reported in the Supporting Information.
Scheme 1. Synthesis of (rac)-Inhibitor 1.

(a) DIBALH, 2 equiv, toluene, 0 °C. (b) BnONH2, 2 equiv, MeOH, 65 °C. (c) ClCOCH2OAc, MeOH, NET3, 0 °C. (d) MeOH/NET3/H2O, room temperature. (e) mCPBA, 2 equiv, CH2Cl2, solid Na2HPO4, 0 °C. (f) P(OBn)2N(iPr)2, 4 equiv, imidazole, triazole, AcCN, room temperature, and then tBuOOH. (g) H2/Pd/C, MeOH, NET3, 2 equiv. (h) H2O.
Scheme 2. Synthesis of (rac)-inhibitor 2.

(a) MsCl, 0.15 equiv, NEt3, dioxanne, 0 °C. (b) BnONH2, 2 equiv, MeOH, 65 °C. (c) ClCOCH2OAc, MeOH, NET3, 0 °C. (d) MeOH/NET3/H2O, room temperature. (e) Pd (Lindlar) 10%, EtOH, room temperature. (f) mCPBA, 2 equiv, CH2Cl2, solid Na2HPO4, 0 °C. (g) P(OBn)2N(iPr)2, 4 equiv, imidazole, triazole, AcCN, room temperature, and then tBuOOH. (h) H2/Pd/C, MeOH, NET3, 2 equiv. (h) H2O.
Compounds 1 and 2 were characterized as single products, showing that the key steps, epoxidation and hydrolysis of the intermediate epoxides, were stereospecific reactions. The conditions for the final debenzylation in the presence of two molar equivalents of triethylamine were finally selected after several trials. In the absence of base or in the presence of 4 equiv or more of a base, mixtures of products were obtained, probably as a consequence of some rearrangement. We were somewhat surprised by the spontaneous opening of the intermediate epoxides in pure water, leading to 1 and 2 without the addition of any acidic catalyst. Most probably, a participation of one of the phosphate groups in an intramolecular acid catalysis occurs.
We next took advantage of the availability of recombinant class II FBAs derived from four pathogenic microorganisms to test our products in vitro on these enzymes. By comparison, commercial aldolase from rabbit muscle (RMA), very close to the human enzyme (98% identity), was taken as representative of class I FBA to check the selectivity of the inhibitors. Results of the biochemical assays are given in Table 1.
Table 1. Biochemical Estimation of Compounds 1 and 2 on Class I and Class II FBA from Various Origins.
| compd |
Km or Ki (selectivity × 103) b |
||||
|---|---|---|---|---|---|
| FBAa | RM | H.p | Y.p | C.a | M.tb |
| FBP | 17 ± 1 μM | 37 ± 2.2 μM | 55 ± 3.3 μM | 100 ± 6 μM | 11 ± 0.7 μM |
| 1 | 55 ± 3.3 μM | 18 ± 1.1 nM (6.6) | 4.5 ± 0.3 nM (39.4) | 0.45 ± 0.03 nM (717) | 0.35 ± 0.02 nM (101.3) |
| 2 | 100 ± 6 μM | 20 ± 1.2 nM (10.9) | 3.5 ± 0.2 nM (92.5) | 10 ± 0.6 nM (58.8) | 0.47 ± 0.03 nM (137.7) |
RM, rabbit muscle; H.p, H. pylori; Y.p, Y. pestis; C.a, C. albicans; and M.tb, M. tuberculosis.
Expressed as Km/Ki class II/Km/Ki class I.
Km/Ki values >12000 and up to 220000 are consistent with the two compounds behaving as transition state analogue inhibitors of class II FBA.11 Alternatively, one can consider 1 and 2 as efficient bisubstrate type inhibitors; the main factor responsible for the inhibitory power is the presence of the hydroxamate function. Indeed, the moderate improvement in the inhibitory properties of 1 and 2 (40-fold) as compared to our previous product 4 (Table 2) can be attributed to a better adjustment of the “glyceraldehyde-phosphate” part of the inhibitor. The two compounds were found to give pure competitive inhibition patterns. At the same time, they are only weak substrate analogue inhibitors of the class I enzyme RAMA (Km/Ki < 0.31). The selectivity for class II FBA, arbitrarily but logically calculated as Km/Ki class II/Km/Ki class I, reaches unprecedented values of up to 7.1 × 105, which validates a posteriori the design of these inhibitors. The relative stereochemistries of 1 and 2 seem of importance only in the case of the C. albicans enzyme. Differences in Ki and selectivity values were found between enzymes from various origins, in spite of the high analogy observed between class II FBAs of known structure.12−15 This is probably due to subtle differences in accommodation of the substrate and inhibitors within the active site, which will be highlighted only after crystallization of the enzyme−inhibitor complexes. This work is currently under progress.
Table 2. Inhibitory Properties of Various Inhibitors against M. tuberculosis FBA.
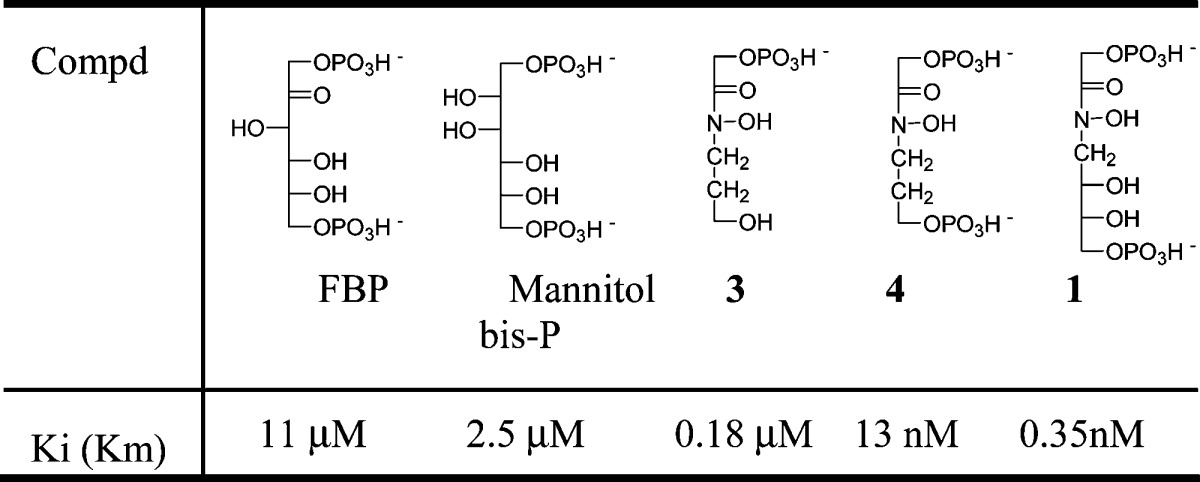 |
The most relevant structural characteristics for a compound to be a good inhibitor of class II FBA can be deduced from a survey of the inhibitory properties of our previously prepared products,12,16 as compared to 1 (Table 2): As expected, the presence of the hydroxamate function and of two phosphoryl groups like in FBP is crucial. The distance between the two phosphate groups in compound 4 is obviously in good accordance with the one found in substrate FBP. By increasing this distance and adding two hydroxyl groups as in 1, the affinity for the enzyme is increased by a factor of roughly 40 (or 80, considering that 1 is racemic), indicating that this last compound is closer to the transition state than 4.
As dreaded, however, the compounds were inactive on cultivated pathogens (Y. pestis, M. tuberculosis, and C. albicans). The presence of phosphate groups very polar and sensitive to phosphatases is obviously harmful to the bactericidal activity. The derivatization of our inhibitors as enzymolabile lipophilic prodrugs is thus under progress.
In conclusion, we have designed inhibitors of FBP aldolases with unprecedent selectivities and affinities for the class II (zinc-dependent) enzymes. Because this class is present exclusively in microorganisms (among which a number of pathogens), they could lead to the elaboration of a new class of antibiotics.
Acknowledgments
We are grateful to Prof. Jurgen Sygusch (Université de Montreal) for the kind supply of H. pylori FBA, Dr. Mary Jackson (Colorado State University) for the kind supply of M. tb and Y. pestis FBAs, and Dr. Jean-Michel Bruneau (Novexel, Romainville, France) for the kind supply of C. albicans FBA.
Supporting Information Available
Detailed syntheses of compounds 1 and 2, NMR spectra, and enzymatic kinetics data. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by a scholarship to R.D. from the Region Ile de France.
Supplementary Material
References
- Wright G. D.; Sutherland A. D. New strategies for combating multidrug-resistant bacteria. Trends Mol. Med. 2007, 13, 260–267. [DOI] [PubMed] [Google Scholar]
- Alekshun M. N.; Levy S. B. Molecular mechanisms of antibacterial multidrug resistance. Cell 2007, 128, 1037–1050. [DOI] [PubMed] [Google Scholar]
- Maartens G.; Wilkinson R. J. Tuberculosis. Lancet 2007, 370, 2030–2043. [DOI] [PubMed] [Google Scholar]
- Galimand M.; Carniel E.; Courvalin P. Resistance of Yersinia pestis to antimicrobial agents. Antimicrob. Agents Chemother. 2006, 50, 3233–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent J. L. Nosocomial infections in adult intensive-care units. Lancet 2003, 361, 2068–2077. [DOI] [PubMed] [Google Scholar]
- Wehmeier U. F. Molecular cloning, nucleotide sequence and structural analysis of the Streptomyces galbus DSM40480 fda gene: The S. galbus fructose-1,6-bisphosphate aldolase is a member of the class II aldolases. FEMS Microbiol. Lett. 2001, 197, 53–58. [DOI] [PubMed] [Google Scholar]
- Gerdes S. Y.; Scholle M. D.; Campbell J. W.; Balazsi G.; Ravasz E.; Daugherty M. D.; Somera A. L.; Kyrpides N. C.; Anderson I.; Gelfand M. S.; Bhattacharya A.; Kapatral V.; D'Souza M.; Baev M. V.; Grechkin Y.; Mseeh F.; Fonstein M. Y.; Overbeek R.; Barabasi A.-L.; Oltvai Z. N.; Osterman A. L. Molecular cloning, nucleotide sequence and structural analysis of the Streptomyces galbus DSM40480 fda gene: The S. galbus fructose-1,6-bisphosphate aldolase is a member of the class II aldolases. J. Bacteriol. 2003, 185, 5673–5684.13129938 [Google Scholar]
- Lewis D. J.; Lowe G. Phosphoglycolohydroxamic acid. Inhibitor of Class I and II aldolases and triose phosphate isomerase. Potential antibacterial and antifungal agent. J. Chem. Soc., Chem. Commun. 1973, 713–715. [Google Scholar]
- Collins K. D. An activated intermediate analogue. The use of phosphoglycolohydroxamate as a stable analogue of a transiently occurring dihydroxyacetone phosphate-derived enolate in enzymatic catalysis. J. Biol. Chem. 1974, 249, 136–142. [PubMed] [Google Scholar]
- Gefflaut T.; Blonski C.; Perie J.; Willson M. Class I aldolases: substrate specificity, mechanism, inhibitors and structural aspects. Prog. Biophys. Mol. Biol. 1995, 63, 301–340. [DOI] [PubMed] [Google Scholar]
- Wolfenden R. Transition state analogues for enzyme catalysis. Nature 1969, 223, 704–705. [DOI] [PubMed] [Google Scholar]
- Fonvielle M.; Coinçon M.; Daher R.; Desbenoit N.; Kosieradzka K.; Barilone N.; Gicquel B.; Sygusch J.; Jackson M.; Therisod M. Synthesis and biochemical evaluation of selective inhibitors of class II fructose bisphosphate aldolases: Towards new synthetic antibiotics. Chem.—Eur. J. 2008, 14, 8521–8529. [DOI] [PubMed] [Google Scholar]
- Hall D. R.; Leonard G. A.; Reed C. D.; Watt C. L.; Berry A.; Hunter W. N. The crystal structure of Escherichia coli class II fructose-1,6-bisphosphate aldolase in complex with phosphoglycolohydroxamate reveals details of mechanism and specificity. J. Mol. Biol. 1999, 287, 383–394. [DOI] [PubMed] [Google Scholar]
- Galkin A.; Kulakova L.; Melamud E.; Li L.; Wu C.; Mariano P.; Dunaway-Mariano D.; Nash T. E.; Herzberg O. Characterization, kinetics, and crystal structures of fructose-1,6-bisphosphate aldolase from the human parasite, Giardia lamblia. J. Biol. Chem. 2007, 282, 4859–4867. [DOI] [PubMed] [Google Scholar]
- Izard T; Sygusch J. Induced fit movements and metal cofactor selectivity of class II aldolases. Structure of thermus aquaticus fructose-1,6-bisphosphate aldolase. J. Biol. Chem. 2004, 279, 11825–11833. [DOI] [PubMed] [Google Scholar]
- Mabiala-Bassiloua C.-G.; Zwolinska M.; Therisod H.; Sygusch J.; Therisod M. Separate synthesis and evaluation of glucitol bis-phosphate and mannitol bis-phosphate, as competitive inhibitors of fructose bis-phosphate aldolases. Bioorg. Med. Chem. Lett. 2008, 18, 1735–1737. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.