

Abstract
Protein carbamylation is of great concern both in vivo and in vitro. Here, we report the first structural characterization of a protein carbamylated at the N-terminal proline. The unexpected carbamylation of the α-amino group of the least reactive codified amino acid has been detected in high-resolution electron density maps of a new crystal form of the HIV-1 protease/saquinavir complex. The carbamyl group is found coplanar to the proline ring with a trans conformation. The reaction of N-terminal with cyanate ion derived from the chaotropic agent urea was confirmed by mass spectra analysis on protease single crystals. Implications of carbamylation process in vitro and in vivo are discussed.
Keywords: Carbamylation, N-terminal proline, HIV-1 protease/saquinavir complex, single crystals
The HIV protease (PR) is an aspartic protease that shares sequence homology around the active site with other retroviral proteases, as the conserved Asp-Thr-Gly catalytic triad.1 PR is required for proteolytic cleavage of viral Gag and Gag-Pol polyproteins into individual structural and functional proteins during viral maturation.2 In the absence of this proteolysis, immature noninfectious virions are produced, thus making PR a prime target for structure-assisted drug design in antiviral therapy.3,4
The structure-assisted drug design and discovery process utilizes techniques such as protein crystallography, nuclear magnetic resonance (NMR), and computational biochemistry to guide synthesis of potential drugs. PR has been widely characterized biochemically and structurally, which has led to the discovery of HIV protease inhibitors (PIs). Their utilization in highly active antiretroviral therapy (HAART) has been a major turning point in the management of HIV/acquired immune-deficiency syndrome (AIDS).5
Recently, we have demonstrated that high-resolution X-ray crystallography can be used as a powerful method to identify the most potent PR inhibitor present in an epimeric mixture.6 The pseudosymmetric peptidomimetic inhibitor based on a novel Phe−Pro isostere core matched the 2-fold symmetry of the homodimeric structure of the PR. Fourier maps obtained by high-resolution diffraction data (1.3 Å) clearly showed the catalytic site fully occupied by a single ordered stereoisomer. On the contrary, several X-ray crystal structures of PR complexed with more asymmetric FDA-approved inhibitors, like darunavir, nelfinavir, and saquinavir (SQV), have the catalytic site occupied by the inhibitor oriented in two almost equivalent positions related by a pseudo-2-fold symmetry.7−12
To investigate this order/disorder phenomenon and its possible relationship with crystal packing, we have undertaken a systematic search for new crystal forms of wild-type PR in complex with SQV. Single crystals of the PR/SQV complex were obtained by a vapor diffusion technique and analyzed by X-ray diffraction. These crystals belong to a new monoclinic crystal form, which contains two dimers of the complex in the asymmetric unit. Other crystal structures of PR/SQV, including variants of PR, so far reported are orthorhombic and cubic forms.7,9,11−14 This new PR/SQV crystal structure is isomorphic with the structure reported for PR in complex with indinavir (IDV) (pdb code = 2AVV).15 The electron density maps of PR/SQV, obtained at 1.39 Å resolution, show the catalytic sites of both crystallographically independent dimers, each fully occupied by a SQV molecule statistically distributed in two different orientations, related by a pseudo-2-fold symmetry (Figure 1), with occupancies of 38−62% (AB dimer) and 25−75% (CD dimer), respectively. A similar report with statistical disorder of ligand (52 and 48%) has been obtained for the isomorphic structure of PR/IDV.15
Figure 1.
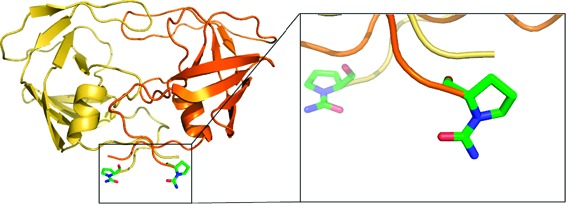
Dimeric structure of HIV-1 protease (ribbon/loop rendering) viewed parallel to the 2-fold symmetry axis. The electron density 2Fo−Fc map (contour level at 1.0σ) of the catalytic channel of HIV-1 protease shows the double orientation (green and cyan sticks) of SQV.
A working hypothesis on the presence of an ordered or disordered inhibitor in the catalytic site is that the asymmetry of the dimeric protein induced by the inhibitor binding can either be sufficient or not to influence the crystal growing in an ordered manner (presence of only one single orientation of the inhibitor). The disorder of the ligand into two equivalent orientations is observed if the overall structure of the complexed dimer substantially maintains its 2-fold symmetry. In this case, the molecular association process (crystal growth) produces an equiprobable statistical distribution over two almost equivalent protein orientations. This process is governed not only by the inhibitor-induced asymmetry of the complex (even observed with symmetric inhibitors16) but also by the specific intermolecular contacts present in the crystal packing (crystal form) and the crystal growth rate.
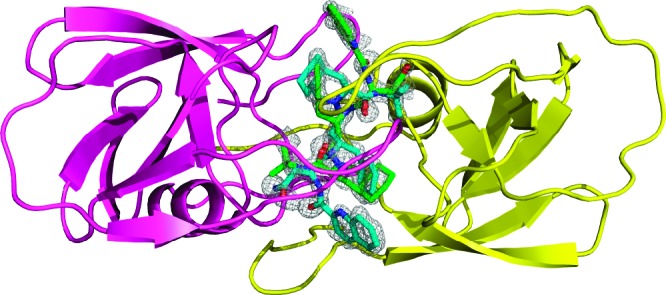
Behind the order/disorder phenomenon of the catalytic site, the most striking feature observed in the present PR/SQV structure is the modification of the N-terminal proline. Fourier maps obtained by high-resolution diffraction data from synchrotron radiation clearly show that both N-terminals of one of the two crystallographically independent dimers are involved in extra covalent bonds with groups having a trigonal geometry. The hydrogen-bonding network formed by these groups suggested the presence of an H-bond acceptor (possibly an oxygen atom) and a double H-bond donor (possibly an NH2 group) bound to the central atom (possibly a carbon atom) involved in the N-terminal modification (Figure 2).
Figure 2.
Electron density 2Fo−Fc map (contour level at 1.0σ) of a carbamylated N-terminal proline. The carbamyl group, coplanar with the proline ring, assumes a trans conformation and forms hydrogen bonds with a neighboring dimer.
This suggested the presence of carbamylation of N-terminal proline, which was confirmed by liquid chromatography/mass spectra analysis on single crystals of the PR/SQV complex. The carbamyl group is coplanar with the proline ring and assumes a trans conformation (NH2−C−N−CA torsion angle of 172°). The mass spectra show that the most abundant species present in the crystal is a monocarbamylated PR, with minor fractions of “native” PR and double carbamylated PR also present (Figure 3).
Figure 3.

Mass spectra of a PR/SQV single crystal.
The purification method of PR employed in this study made use of urea (8 M solution) as a chaotropic agent. The carbamylation of protein by residual cyanate ions derived from urea has long been established,17 and the cyanate present in 8 M urea solution is sufficient to react with amino groups of protein.17,18 The chaotrope urea is commonly used during recombinant protein expression as a denaturant/solubilizing agent, and cyanate formation in the urea buffer cannot be prevented under the condition of normal protein purification.19 For example, transformation of insulin by reaction with cyanate slowly formed in a freshly prepared urea buffer solution during chromatography experiments carried out at room temperature has also been reported.19 Protein modification by carbamylation of ε-NH2 group of lysine has been particularly reported.17,20,21 α-Amino groups of alanine, cysteine, and glycine have also been reported as possible carbamylation sites.22−24 To the best of our knowledge, the present communication represents the first experimental report on carbamylation of N-terminal proline in protein. It is interesting to note that the logarithm of the rate constants for carbamylation reaction of cyanate with a series of peptides has been found linearly related to the pKa values of the amino groups.25 Thus, considering the Ka values of amino acids, the secondary α-amino group of proline is expected to be the least reactive N-terminal in proteins, with reactivity very close to that of the primary amino group of lysine side chains. However, in the X-ray structure, the carbamylation was not observed in any of the seven lysine ε-NH2 groups present in PR.
Protein carbamylation is of great concern both in vivo and in vitro. Lippincott and Apostol23 reported in their studies that, in the presence of urea, hemoglobins have a significant level of carbamylated cysteines as an artifact of protein digestion. In fact, in vivo carbamylation of proteins in various diseases has been reported. Carbamylation of α-crystallins (caused by cigarette smoking) plays an important role in the development of cataract by generating conformational changes that lead to lens opacity.26,27 Carbamylation of extracelullar matrix proteins in kidneys of uremic patients have been substantiated,28 and it has been shown that cyanate can induce hemolysis by carbamylation of erythrocytes.29 Carbamyl modification of amino acid side chains results in changes in protein charge state, which can lead to conformational alterations, variations in analytical profiles, and modification of bioactivity.30−32
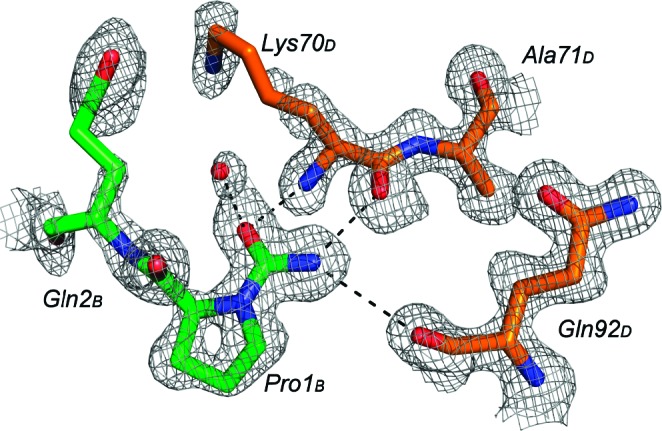
Of the four crystallographically independent N termini, only the AB dimer showed a convincing electron density for a similar modification at both proline residues. These carbamylated residues are involved in special dimer−dimer interactions. The carbamyl group on Pro 1B forms two hydrogen bonds with Lys 70D and one weak hydrogen bond with Gln 92D of the neighboring unmodified CD dimer (Figure 2). The carbamyl group on Pro 1A interacts trough a water molecule with Arg 57B of a symmetry-related AB dimer. The unit cell analysis shows that the monoclinic crystal form obtained in this work is related by group−subgroup relations33 to the orthorhombic crystal form of the PR/SQV complex obtained at 1.16 Å of resolution.9 The recognition of group−subgroup relations between space groups and unit cells of different crystal structures can provide a direct insight into protein packing.34 The asymmetric unit of the orthorhombic form is composed of one crystallographically independent dimer having a 50−50% statistical disorder of SQV ligand. A similar packing (close in energy) is observed for the nonisomorphic monoclinic crystal structure here reported. This structure has a lower crystal symmetry (the P21 space group is a maximal nonisomorphic subgroup of the P212121 space group), and as a consequence, the number of crystallographically independent dimers doubles. The asymmetric unit in this case is formed by two nonequivalent dimers: the carbamylated AB dimer and the unmodified CD dimer. The loss of crystallographic symmetry elements can be related to the peculiar dimer−dimer interaction exhibited by the carbamoylated prolines. The SQV inhibitor is statistically distributed in two orientations with occupancies of 38−62% (AB carbamylated dimer) and 25−75% (CD unmodified dimer), while the orthorhombic form is 50−50%. Thus, the reduction of the crystal symmetry seems to be related to a reduced disorder of the inhibitor (the estimated error on occupancy factors evaluated by SHELXH least-squares refinement of the SQV molecule having the same isotropic thermal factors for equivalent atom pairs is about 3%). Furthermore, a small difference is observed in occupancies among modified and unmodified dimers. This could be associated with the slight increase of the intermolecular contacts observed in the carbamylated N termini, which are probably not influenced by the orientation of the inhibitor. However, on the basis of the present results, no definitive conclusion on ordered/disordered phenomenon can be drawn, and further experimental and theoretical studies are needed to investigate the problem.
Acknowledgments
We acknowledge Prof. Gianluca Tell of the University of Udine (Italy) for provision of PR plasmid and the beamline scientists at the Elettra Synchrotron for their technical assistance.
Supporting Information Available
Protocols for expression and purification of PR, crystallization, structure determination, and liquid chromatography/mass spectrometry analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
We are grateful to the Schlumberger Foundation for the Doctoral fellowship awarded to F.M.O. and the International Centre for Theoretical Physics (Trieste, Italy) for the STEP Fellowship. We thank MIUR (FIRB RBRN062BCT) and Friuli-Venezia-Giulia region (DPReg. 120/2007/Pres.) for economic and scientific support.
The coordinates for the structure described here have been submitted to the Protein Data Bank as entry 3K4V.
Supplementary Material
References
- Weber I. T.; Miller M.; Jaskolski M.; Leis J.; Skalka A. M.; Wlodawer A. Molecular modeling of the HIV-1 protease and its substrate binding site. Science 1989, 243, 928–931. [DOI] [PubMed] [Google Scholar]
- Kohl N. E.; Emini E. A.; Schleif W. A.; Davis L. J.; Heimbach J. C.; Dixon R. A.; Scolnick E. M.; Sigal I. S. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. U.S.A. 1988, 85, 4686–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temesgen Z. Current status of antiretroviral therapies. Expert Opin. Pharmacother. 2001, 2, 1239–1246. [DOI] [PubMed] [Google Scholar]
- Wlodawer A.; Vondrasek J. Inhibitors of HIV-1 protease: a major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 249–284. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Chapsal B. D.; Weber I. T.; Mitsuya H. Design of HIV protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance. Acc. Chem. Res. 2008, 41, 78–86. [DOI] [PubMed] [Google Scholar]
- Geremia S.; Demitri N.; Wuerges J.; Benedetti F.; Berti F.; Tell G.; Randaccio L. A potent HIV protease inhibitor identified in an epimeric mixture by high-resolution protein crystallography. ChemMedChem 2006, 1, 186–188. [DOI] [PubMed] [Google Scholar]
- Liu F.; Kovalevsky A. Y.; Tie Y.; Ghosh A. K.; Harrison R. W.; Weber I. T. Effect of flap mutations on structure of HIV-1 protease and inhibition by saquinavir and darunavir. J. Mol. Biol. 2008, 381, 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kožíšek M.; Bray J.; Řezáčová P.; Šašková K.; Brynda J.; Pokorná J.; Mammano F.; Rulíšek L.; Konvalinka J. Molecular analysis of the HIV-1 resistance development: enzymatic activities, crystal structures, and thermodynamics of nelfinavir-resistant HIV protease mutants. J. Mol. Biol. 2007, 374, 1005–1016. [DOI] [PubMed] [Google Scholar]
- Tie Y.; Kovalevsky A. Y.; Boross P.; Wang Y.-F.; Ghosh A. K.; Toszer J.; Harrison R. W.; Weber I. T. Atomic resolution crystal structures of HIV-1 protease and mutants V82A and I84V with saquinavir. Proteins: Struct., Funct., Bioinf. 2007, 67, 232–242. [DOI] [PubMed] [Google Scholar]
- Kovalevsky A. Y.; Liu F.; Leshchenko S.; Ghosh A. K.; Louis J. M.; Harrison R. W.; Weber I. T. Ultra-high resolution crystal structure of HIV-1 protease mutant reveals two binding sites for clinical inhibitor TMC114. J. Mol. Biol. 2006, 363, 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong L.; Zhang X. C.; Hartsuck J. A.; Tang J. Crystal structure of an in vivo HIV-1 protease mutant in complex with saquinavir: insights into the mechanisms of drug resistance. Protein Sci. 2000, 9, 1898–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munshi S.; Chen Z.; Yan Y.; Li Y.; Olsen D. B.; Schock H. B.; Galvin B. B.; Dorsey B.; Kuo L. C. An alternate binding site for the P1-P3 group of a class of potent HIV-1 protease inhibitors as a result of concerted structural change in the 80s loop of the protease. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2000, 56, 381–388. [DOI] [PubMed] [Google Scholar]
- Prabu-Jeyabalan M.; Nalivaika E. A.; King N. M.; Schiffer C. A. Viability of a drug-resistant human immunodeficiency virus type 1 protease variant: structural insights for better antiviral therapy. J. Virol. 2003, 77, 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulkes J. E.; Prabu-Jeyabalan M.; Cooper D.; Henderson G. J.; Harris J.; Swanstrom R.; Schiffer C. A. Role of invariant Thr80 in human immunodeficiency virus type 1 protease structure, function, and viral infectivity. J. Virol. 2006, 80, 6906–6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Boross P. I.; Wang Y. F.; Tozser J.; Louis J. M.; Harrison R. W.; Weber I. T. Kinetic, stability, and structural changes in high-resolution crystal structures of HIV-1 protease with drug-resistant mutations L24I, I50V, and G73S. J. Mol. Biol. 2005, 354, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer G. B; Boehm J. C.; Chenera B.; DesJarlais R. L.; Hassell A. M.; Meek T. D.; Tomaszek T. A. Jr.; Lewis M. A symmetric inhibitor binds HIV-1 protease asymmetrically. Biochemistry 1993, 32, 937–947. [DOI] [PubMed] [Google Scholar]
- Stark G. R.; Stein W. H.; Moore S. J. Reactions of cyanate present in aqueous urea with amino acids and proteins. J. Biol. Chem. 1960, 235, 3177–3181. [Google Scholar]
- Stark G. R.Modification of proteins with cyanate. Methods in Enzymol. Enzyme Structure; Volume 11 ed.; Academic Press, 1967; pp 590−594. [Google Scholar]
- Cole R. D. The chromatography of insulin in urea-containing buffer. J. Biol. Chem. 1960, 235, 2294–2299. [PubMed] [Google Scholar]
- Kraus L. M.; Elberger A. J.; Handorf C. R.; Pabst M. J.; Kraus A. P. Jr. Urea-derived cyanate forms epsilon-aminocarbamoyl-lysine (homocitrulline) in leukocyte proteins in patients with end-stage renal disease on peritoneal dialysis. J. Lab. Clin. Med. 1994, 123, 882–891. [PubMed] [Google Scholar]
- Wang Z.; Nicholls S. J.; Rodriguez E. R.; Kummu O.; Horkko S.; Barnard J.; Reynolds W. F.; Topol E. J.; DiDonato J. A.; Hazen S. L. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nature Med. 2007, 13, 1176–1184. [DOI] [PubMed] [Google Scholar]
- Slebe J. C.; Martinez-Carrion M. Carbamylation of aspartate transaminase and the pK value of the active site lysyl residue. J. Biol. Chem. 1976, 251, 5663–5669. [PubMed] [Google Scholar]
- Lippincott J.; Apostol I. Carbamylation of cysteine: a potential artifact in peptide mapping of hemoglobins in the presence of urea. Anal. Biochem. 1999, 267, 57–64. [DOI] [PubMed] [Google Scholar]
- Martin S.; Harding J. J. Site of carbamoylation of bovine gamma-II-crystallin by potassium [14C]cyanate. Biochem. J. 1989, 262, 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark G. R. Reactions of cyanate with functional groups of proteins. 3. Reactions with amino and carboxyl groups. Biochemistry 1965, 4, 1030–1036. [DOI] [PubMed] [Google Scholar]
- Derham B. K.; Harding J. J. Effects of modifications of alpha-crystallin on its chaperone and other properties. Biochem. J. 2002, 364, 711–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M.; Rixon K. C.; Harding J. J. Aspirin prevents carbamylation of soluble lens proteins and prevents cyanate-induced phase separation opacities in vitro: a possible mechanism by which aspirin could prevent cataract. Exp. Eye Res. 1985, 40, 297–311. [DOI] [PubMed] [Google Scholar]
- Kraus L. M.; Kraus A. P. Jr. Carbamoylation of amino acids and proteins in uremia. Kidney Int. Suppl. 2001, 78, S102–S107. [DOI] [PubMed] [Google Scholar]
- Hasuike Y.; Nakanishi T.; Maeda K.; Tanaka T.; Inoue T.; Takamitsu Y. Carbamylated hemoglobin as a therapeutic marker in hemodialysis. Nephron 2002, 91, 228–234. [DOI] [PubMed] [Google Scholar]
- Cole E. G.; Mecham D. K. Cyanate formation and electrophoretic behavior of proteins in gels containing urea. Anal. Biochem. 1966, 14, 215–222. [DOI] [PubMed] [Google Scholar]
- Salinas M.; Fando J. L.; Grisolia S. A sensitive and specific method for quantitative estimation of carbamylation in proteins. Anal. Biochem. 1974, 62, 166–172. [DOI] [PubMed] [Google Scholar]
- Frantzen F. Chromatographic and electrophoretic methods for modified hemoglobins. J. Chromatogr., B: Biomed. Sci. Appl. 1997, 699, 269–286. [DOI] [PubMed] [Google Scholar]
- Di Costanzo L.; Forneris F.; Geremia S.; Randaccio L. Phasing protein structures using the group-subgroup relation. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2003, 59, 1435–1439. [DOI] [PubMed] [Google Scholar]
- Geremia S.; Di Costanzo L.; Randaccio L.; Engel D. E.; Lombardi A.; Nastri F.; DeGrado W. F. Response of a designed metalloprotein to changes in metal ion coordination, exogenous ligands, and active site volume determined by X-ray crystallography. J. Am. Chem. Soc. 2005, 127, 17266–17276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.