

Abstract

Incorporation of polar functionality into a series of highly potent calcitonin gene-related peptide (CGRP) receptor antagonists was explored in an effort to improve pharmacokinetics. This strategy identified piperazinone analogues that possessed improved solubility at acidic pH and increased oral bioavailability in monkeys. Further optimization led to the discovery of the clinical candidate 2-[(8R)-8-(3,5-difluorophenyl)-10-oxo-6,9-diazaspiro[4.5]dec-9-yl]-N-[(2R)-2′-oxo-1,1′,2′,3-tetrahydrospiro[indene-2,3′-pyrrolo[2,3-b]pyridin]-5-yl]acetamide (MK-3207) (4), the most potent orally active CGRP receptor antagonist described to date.
Keywords: MK-3207, calcitonin gene-related peptide, receptor antagonist, pharmacokinetics
Migraine is a common, neurovascular disorder characterized by attacks of severe headache that are highly disabling.1 The gold standard for acute treatment of migraine is the triptan class of 5-HT1B/1D receptor agonists.2 While these agents are effective for the majority of migraineurs, they are contraindicated in patients with cardiovascular disease because of their vasoconstrictive effects.3
Calcitonin gene-related peptide (CGRP) is a 37 amino acid neuropeptide that is widely distributed in the central and peripheral nervous system.4 The CGRP receptor is composed of the calcitonin receptor-like receptor (CLR), a family B G protein-coupled receptor (GPCR), in association with receptor activity modifying protein 1 (RAMP1) and receptor component protein.5 A number of lines of evidence implicated CGRP in migraine pathophysiology, and this led to interest in the potential utility of CGRP receptor antagonists as novel therapeutics for migraine.6 Importantly, CGRP receptor antagonists are not direct vasoconstrictors, so this approach may offer an advantage over the use of triptans.7,8
Clinical evidence for the utility of CGRP receptor antagonists for the acute treatment of migraine was initially obtained with olcegepant (1, Chart 1).9 This highly potent CGRP receptor antagonist (Ki = 10 pM) is not orally bioavailable but has shown compelling efficacy in a phase II clinical trial when administered via intravenous infusion.10 More recently, orally dosed telcagepant11 (3, Chart 1) was shown to be effective for acute treatment of migraine in a phase III study.12 In this study, telcagepant 300 mg demonstrated efficacy comparable to zolmitriptan 5 mg but with fewer associated adverse effects. These and other clinical studies highlight the potential of CGRP receptor antagonists as a new class of antimigraine therapeutics.13
Chart 1. Selected CGRP Receptor Antagonists.

The evolution of telcagepant from high-throughput screening lead 2 (Chart 1) has been described.14 We have also reported the identification of spiroindane-based CGRP receptor antagonists from lead compound 2.15 More recently, we described the discovery of novel antagonists including the high affinity compound 5 (Table 1).16 Herein, we report the preliminary structure−activity relationship (SAR) studies on 5, which led to the discovery of the clinical candidate MK-3207 (4).
Table 1. CGRP Receptor Antagonist Data.
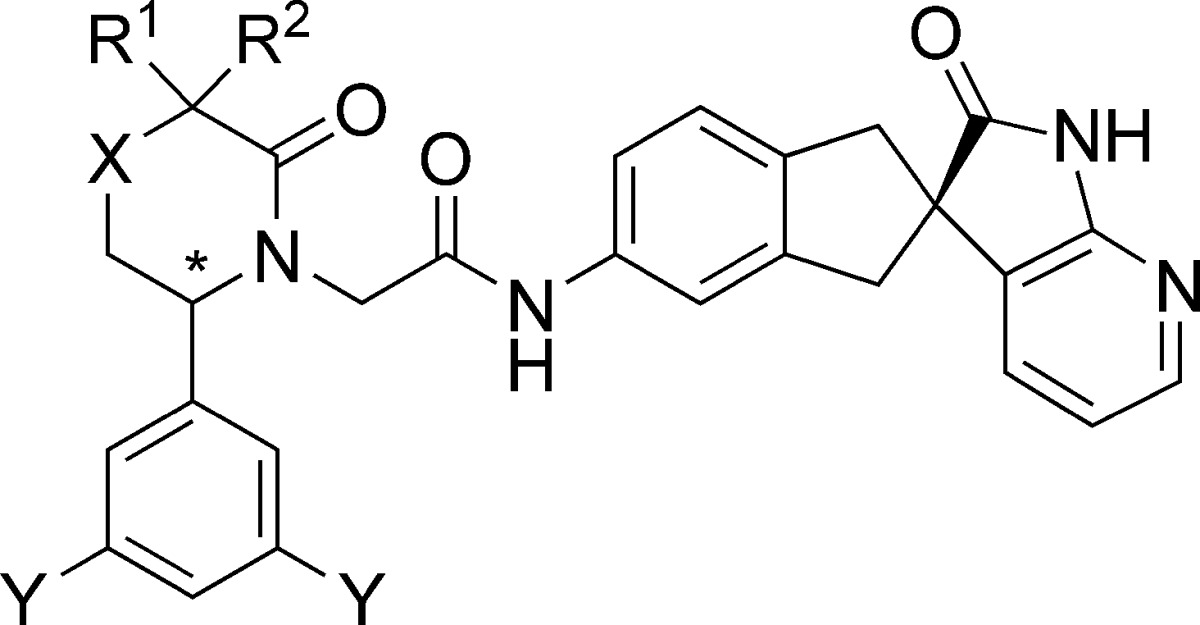
| compd | R1 | R2 | X | Y | * | CGRP Kia,b (pM) | cAMP IC50a,c (nM) | cAMP + HS IC50a,d (nM) |
|---|---|---|---|---|---|---|---|---|
| 3 | 770 ± 70 (13) | 2.2 ± 0.3 (8) | 11 ± 2 (10) | |||||
| 5 | Me | Me | CH2 | F | S | 39 ± 12 (13) | 0.16 ± 0.06 (5) | 0.35 ± 0.10 (5) |
| 6 | Me | Me | CH2 | H | S | 120 (2) | 0.59 (2) | 1.4 (2) |
| 7 | Me | Me | O | H | R | 82 ± 30 (5) | 0.22 (2) | 0.27 (2) |
| 8 | Et | Et | O | H | R | 67 ± 18 (6) | 0.23 ± 0.04 (3) | 0.37 ± 0.05 (3) |
| 9 | Et | Et | O | F | R | 18 ± 6 (3) | 0.32 ± 0.08 (3) | 0.44 ± 0.12 (3) |
| 10 | Me | Me | NH | F | R | 34 ± 11 (27) | 0.17 ± 0.03 (7) | 0.25 ± 0.07 (7) |
| 11 | Et | Et | NH | F | R | 20 ± 4 (4) | 0.14 (2) | 0.18 (2) |
| 4 | −CH2CH2CH2CH2− | NH | F | R | 21 ± 6 (15) | 0.12 ± 0.05 (6) | 0.17 ± 0.07 (6) | |
| 12 | −CH2CH2CH2CH2CH2− | NH | F | R | 17 ± 5 (7) | 0.22 ± 0.05 (3) | 0.33 ± 0.12 (3) | |
| 13 | −CH2CH2CH2CH2CH2− | NH | F | S | 570 ± 110 (5) | 10 (2) | 11 (2) |
Mean value ± standard deviation, where appropriate; the number of replicates is in parentheses.
The Ki value for inhibition of 125I-hCGRP binding was determined using membranes from HEK293 cells stably expressing human CLR/RAMP1.17
Inhibition of CGRP-induced cAMP production in HEK293 cells stably expressing human CLR/RAMP1.17
Lactam 5 had high receptor affinity (Ki = 39 pM) and subnanomolar potency in a cell-based functional assay (cAMP IC50 = 0.16 nM). It maintained similar potency in the same functional assay run in the presence of 50% human serum (cAMP + HS IC50 = 0.35 nM), indicating that the activity of 5 would not be dramatically affected by plasma protein binding in vivo. When the pharmacokinetic profile of compound 5 was investigated, however, a key liability was discovered. While 5 possessed modest oral bioavailability in rats (F = 12%) and dogs (F = 44%), it had no oral bioavailability in monkeys even when dosed in a solubilizing 90% PEG400 vehicle (Table 2). This was a significant problem because most small molecule CGRP receptor antagonists exhibited species-selective pharmacology. Telcagepant, for example, exhibited approximately 1500-fold higher affinity for the human and rhesus CGRP receptors as compared to the rat and dog receptors.17 Thus, oral bioavailability in monkeys was an important requirement for the comprehensive evaluation of the in vivo pharmacology of a compound.
Table 2. Pharmacokinetic Data.
| compd | species | Fa (%) | Clb (mL/min/kg) |
|---|---|---|---|
| 5 | rat | 12 | 7.4 |
| dog | 44 | 17 | |
| monkey | 0c | 16 | |
| 7 | rat | 9 | 23 |
| 8 | rat | 33 | 5 |
| 9 | rat | 59 | 10 |
| dog | 72c | 3.5 | |
| monkey | 0c | 20 | |
| 10 | rat | 9 | 48 |
| dog | 16c | 4.6 | |
| monkey | 7 | 15 | |
| 11 | rat | 28 | 8.2 |
| 4 | rat | 74 | 11 |
| dog | 67d | 8.0 | |
| monkey | 9 | 15 | |
| 12 | rat | 61 | 1.8 |
| dog | 38 | 6.2 | |
| 13 | rat | 44 | 41 |
Determined after dosing in 1% methocel at 10 (rat), 1 (dog), or 2 mpk (monkey) except as noted.
Determined after dosing in DMSO at 2 (rat) or 0.5 mpk (dog and monkey).
Dosed in 90% PEG400.
Dosed at 2 mpk.
It was known that for telcagepant, intestinal first-pass metabolism played a significant role in the low oral bioavailability in monkeys.18 We suspected that a similar intestinal first-pass effect was also contributing to the lack of rhesus bioavailability for lactam 5. In vitro metabolism studies indicated that 5 was subject to extensive oxidative metabolism on the δ-valerolactam and its substituents. To reduce the extent of such metabolism and also with the goal of improving aqueous solubility, we sought to incorporate polar functionality into this region of the molecule.
One strategy was insertion of a heteroatom in the lactam ring to afford, for example, a morpholinone analogue. The first such example was compound 7 (Table 1), which maintained similar receptor affinity (Ki = 82 pM) to the corresponding lactam 6 (Ki = 120 pM). The 3,3-diethylmorpholinone analogue 8 performed similarly to its dimethyl progenitor 7 in both the binding assay and the cAMP functional assay (Table 1) but exhibited superior rat pharmacokinetics (Table 2).
Compound 8 possessed lower plasma clearance (5 vs 23 mL/min/kg) and higher F (33 vs 9%). This was initially surprising in the context of our strategy to improve oral bioavailability by increasing polarity: Morpholinone 8 was more lipophilic (cLogP = 3.63) than 7 (cLogP = 2.58) and contained more potential sites for metabolism; yet, it possessed reduced plasma clearance in rats.
Morpholinone 9, the 3,5-difluorophenyl analogue of 8, had similar cell-based activity to the other morpholinones in Table 1 combined with good rat bioavailability (F = 59%, Table 2). On the basis of these encouraging results, a more detailed investigation of the pharmacokinetic profile of 9 was conducted. In dogs, 9 was found to have low clearance (Cl = 3.5 mL/min/kg) and excellent oral bioavailability (F = 72%). However, in monkeys, the compound was not orally bioavailable (Table 2). In part, this discrepancy between the behavior of 9 in monkeys and the other species could be explained by moderate−high clearance in monkeys as compared with low clearance in rats and dogs (Table 2). Moreover, it appeared that plasma protein binding contributed to the observed differences in clearance: The unbound fraction of 9 (Table 3) was significantly higher in monkeys (fu = 2.2%) than in rats (fu = 0.1%) or dogs (fu = 0.3%).
Table 3. Plasma Protein Binding Dataa.
|
fu (%) |
||||
|---|---|---|---|---|
| compd | human | rat | dog | monkey |
| 9 | 1.7 | 0.1 | 0.3 | 2.2 |
| 10 | 41.1 | 18.1 | 38.3 | 42.9 |
| 4 | 9.4 | 0.8 | 6.1 | 9.6 |
| 12 | 2.8 | 0.2 | 1.1 | 6.1 |
| 13 | 5.4 | 3.4 | 2.5 | 7.4 |
Determined using equilibrium dialysis methodology.
Nonetheless, it seemed likely that intestinal first-pass metabolism also contributed to the lack of monkey oral bioavailability. It was known that the first-pass effect observed with telcagepant could be saturated at higher oral doses. However, the extremely low aqueous solubility of 9 (at pH 4, solubility <0.0001 mg/mL) would probably make it difficult to achieve the concentrations needed to saturate the intestinal first-pass effect. To improve aqueous solubility, the morpholinone ether functionality was replaced with an amino moiety to give a piperazinone ring. An early example of such analogues was compound 10, which gratifyingly exhibited improved aqueous solubility as compared with morpholinone 9, especially at acidic pH (at pH 4, solubility = 1.95 mg/mL; at pH 7, solubility = 0.13 mg/mL). Importantly, the incorporation of this solubilizing amine functionality in 10 was compatible with affinity for the CGRP receptor (Ki = 34 pM) and with functional potency in the presence of serum (cAMP + HS IC50 = 0.25 nM).
The improvements in solubility observed with 10 translated into measurable, albeit low, monkey oral bioavailability (F = 7%). Similar results were obtained in rats and dogs for compound 10 (Table 2), and this demonstration of modest oral bioavailability in three preclinical species led to a significant focus on such piperazinone analogues.
The plasma clearance of 10 was low in dogs, moderate in monkeys, and high in rats (Table 2). The plasma unbound fraction of the compound was relatively high in all species, ranging from 18.1% in rats to 42.9% in monkeys (Table 3). Because replacement of the gem-dimethyl substituents in morpholinone 7 with gem-diethyl groups led to reduced clearance and improved F in rats for compound 8, a similar strategy was explored for piperazinone 10.
The 3,3-diethylpiperazinone 11 had slightly higher affinity for the CGRP receptor as compared with 10 (Table 1), and as anticipated, this modification afforded lower clearance (Cl = 8.2 mL/min/kg) and higher oral bioavailability (F = 28%) in rats. In analogy with the gem-diethyl analogues like 11, constrained versions containing spirocycloalkyl moieties were also synthesized. Both the spirocyclopentyl analogue 4 and the spirocyclohexyl version 12 were found to be potent CGRP receptor antagonists in vitro (Table 1). Importantly, as compared with the 3,3-dimethylpiperazinone 10, both 4 and 12 exhibited improved oral bioavailability in rats and dogs (Table 2).
In rats, plasma protein binding was apparently a significant factor in attenuating the clearance of these piperazinone analogues. As the size of the 3-position substituents increased, rat free fraction and plasma clearance decreased as follows: compound 10 (fu = 18.1%; Cl = 48 mL/min/kg); compound 4 (fu = 0.8%; Cl = 11 mL/min/kg); and compound 12 (fu = 0.2%; Cl = 1.8 mL/min/kg). Further evidence for the key role of plasma protein binding was provided by compound 13, the epimer of 12. Inversion of the piperazinone stereocenter in 12 led to a significant (> 30-fold) loss in affinity for the CGRP receptor (Table 1). Interestingly, the rat free fraction and clearance also increased significantly for 13 (fu = 3.4%; Cl = 41 mL/min/kg) as compared with 12. The fact that epimers 12 and 13 exhibit 17-fold different rat free fractions strongly suggests that the high level of plasma protein binding observed for compound 12 in rats is due, at least in part, to molecular recognition. There is apparently a protein in rat plasma that binds 12 with much higher affinity than 13, and this interaction has an important influence on the free fraction and, consequently, plasma clearance.
In contrast to the observations in rats, the clearance of these piperazinone compounds did not seem to be highly influenced by plasma protein binding in dogs and monkeys, as illustrated by compounds 10 and 4. In dogs, the free fraction drops significantly from 38.3% for 10 to 6.1% for 4, but the plasma clearance is actually higher for 4 (Table 2). In monkeys, the unbound fraction in plasma differs by 5-fold for the two compounds (Table 3), but they exhibit the same clearance (Table 2). On the basis of its favorable profile in terms of in vitro potency and pharmacokinetics, compound 4 was selected for more detailed evaluation. A summary of its pharmacokinetics in preclinical species is shown in Table 4.
Table 4. Preclinical Pharmacokinetics of Compound 4.
| species | PO dose (mpk) | Fa (%) | PO Cmaxa (μM) | IV t1/2b (h) | Clb (mL/min/kg) | Vdssb (L/kg) |
|---|---|---|---|---|---|---|
| rat | 10 | 74 | 14 | 0.6 | 11 | 0.3 |
| 50 | 87 | 52 | ||||
| dog | 2 | 67 | 2.6 | 1.0 | 8.0 | 0.6 |
| 10 | 87 | 10 | ||||
| monkey | 2 | 9 | 0.13 | 1.5 | 15 | 1.7 |
| 20 | 41 | 3.5 |
Determined after dosing in 1% methocel.
Determined after dosing in DMSO at 2 (rat) or 0.5 mpk (dog and monkey).
In rats and dogs, 4 exhibited low plasma clearance and a low volume of distribution, resulting in plasma half-lives of about 1 h. Exposure following oral dosing in rats and dogs was approximately dose-proportional with excellent bioavailability. In rhesus monkeys, moderate clearance and a higher volume of distribution were observed with compound 4. It appeared that a saturable first-pass metabolism contributed to low monkey oral bioavailability, and the oral bioavailability increased at higher doses (F = 9% at 2 mpk; F = 41% at 20 mpk).
Compound 4 had excellent in vitro potency in the radioligand binding assay (Ki = 21 pM) and the cell-based functional assay (cAMP IC50 = 0.12 nM). There was a small shift in the functional assay in the presence of human serum (cAMP + HS IC50 = 0.17 nM), consistent with the significant unbound fraction of 4 in human plasma (fu = 9.4%). In this serum-shifted functional assay, 4 was about 65-fold more potent than telcagepant (cAMP + HS IC50 = 11 nM, Table 2). When tested in a diverse panel of more than 160 enzymes, receptors, channels, and transporters, 4 was found to be >50000-fold selective. In this test panel, 4 showed the highest affinity for the calcitonin receptor (Ki = 1.09 μM).
Gratifyingly, the high level of in vitro potency of 4 against the CGRP receptor translated into a pharmacodynamic model run in anesthetized rhesus monkeys: the capsaicin-induced dermal vasodilation (CIDV) model.17 In this model, the plasma concentration that inhibited 90% of the CIDV response (EC90) was 7 nM. For telcagepant, the CIDV EC90 = 994 nM, indicating that 4 was approximately 100-fold more potent than telcagepant as a CGRP receptor antagonist in vivo. On the basis of its potency, selectivity, pharmacokinetics, and overall profile, compound 4 was selected as a development candidate.
The methodology used to synthesize compounds 4−13 has been described.19 The synthesis of compound 4 is outlined in Scheme 1. Methyl 1-aminocyclopentanecarboxylate was alkylated with 3,5-difluorophenacyl bromide (14) to give amino ketone 15. Interestingly, 15 was somewhat unstable and could undergo oxidative dimerization. This decomposition was minimized by storing intermediate 15 as the mesylate salt. The key step involved reductive amination of ketone 15 with glycine ethyl ester and cyclization of the resulting amine in situ to afford, after Boc protection, piperazinone 16.20 Chiral high-performance liquid chromatography (HPLC) resolution of 16 and saponification gave the carboxylic acid 17, which was coupled to (R)-5-amino-1,3-dihydrospiro[indene-2,3′-pyrrolo[2,3-b]pyridin]-2′-(1′H)-one19,21 under standard conditions to give, after deprotection, the desired compound 4. The other piperazinone compounds were synthesized analogously.
Scheme 1. Synthesis of Compound 4.

Conditions: (a) Methyl 1-aminocyclopentanecarboxylate hydrochloride, Na3PO4, DMF. (b) Glycine ethyl ester hydrochloride, NaCNBH3, AcOH, MeOH, 50 °C. (c) Boc2O, DIEA, CH3CN, 60 °C. (d) Chiralcel OD, hexane:i-PrOH:Et2NH (60:40:0.1), (R)-enantiomer is second major peak. (e) LiOH, H2O, THF. (f) (R)-5-amino-1,3-dihydrospiro[indene-2,3′-pyrrolo[2,3-b]pyridin]-2′-(1′H)-one,21 HATU, NMM, DMF. (g) HCl, EtOAc, 0 °C.
The quest for orally acting drugs that target the CGRP receptor is nontrivial. In part, this may be a reflection of the difficulty of identifying orally bioavailable antagonists that target family B GPCRs, such as CLR, for which the endogenous ligands are large peptides.14 A consideration of the physicochemical properties of clinical candidates 1, 3, and 4 highlights some of the progress made to date (Table 5).
Table 5. Ligand Efficiency and Physicochemical Parameters.
| compd | Δg (kcal/mol) | MW | HBD | HBA | cLogPa | PSAb (Å2) |
|---|---|---|---|---|---|---|
| 1 | 0.27 | 869 | 8 | 5 | 1.67 | 181 |
| 2 | 0.18 | 522 | 4 | 5 | 3.43 | 147 |
| 3 | 0.31 | 566 | 2 | 4 | 2.74 | 103 |
| 4 | 0.35 | 557 | 3 | 4 | 2.17 | 113 |
Determined using ALogP98.
PSA calculated by method of Clark.24
From the perspective of Lipinski's rule-of-five guidelines for orally active drugs,22 all three clinical compounds have high MW (>500), although 3 and 4, which are both orally bioavailable, satisfy the other RO5 criteria. Both 3 and 4 also meet the oral drug guidelines of Veber et al.,23 specifically rotatable bond count ≤10 and polar surface area (PSA) ≤ 140 Å2. During the evolution of 4 from the original HTS lead 2, there was a focus on the improvement of potency without a significant increase in MW and an effort to reduce PSA, to increase the probability of good oral absorption.24 These goals are reflected in improvements in ligand efficiency from 2 (Δg = 0.18 kcal/mol) to 4 (Δg = 0.35 kcal/mol) and a reduced count of H-bond donors and acceptors, correlating with reduced PSA. The overall result is that 4 possesses similar receptor affinity to 1 but is orally active.
In conclusion, a series of highly potent CGRP receptor antagonists have been optimized. To improve the pharmacokinetic profile, polar functionality was incorporated, leading to piperazinone analogues that possessed improved solubility at acidic pH and increased oral bioavailability in monkeys. Further modification focused on balancing antagonist potency, plasma protein binding, and in vivo clearance. These efforts led to the discovery of the clinical candidate 4, the most potent orally bioavailable CGRP receptor antagonist described to date. The detailed pharmacological and clinical evaluation of 4 will be reported elsewhere.
Acknowledgments
We thank the MS and NMR groups for spectroscopic data; Joe Pawluczyk for synthesis of key intermediates; Ken Anderson and Mary Beth Young for pharmacokinetic analysis; Jennifer Adelsberger, Michael Lyman, Kim Michel, Maria Michener, and Tamara Pittman for animal dosing; Matt Zrada for plasma protein binding and solubility determinations; Constantine Kreatsoulas for molecular modeling; and Sam Graham for helpful discussions.
Supporting Information Available
Representative experimental procedures, NMR, MS, and HPLC data for all new test compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Goadsby P. J.; Lipton R. B.; Ferrari M. D. Migraine−current understanding and treatment. N. Engl. J. Med. 2002, 346, 257–270. [DOI] [PubMed] [Google Scholar]
- Tfelt-Hansen P.; De Vries P.; Saxena P. R. Triptans in Migraine. Drugs 2000, 60, 1259–1287. [DOI] [PubMed] [Google Scholar]
- Dodick D.; Lipton R. B.; Martin V.; Papademetriou V.; Rosamond W.; VanDenBrink A. M.; Loutfi H.; Welch K. M.; Goadsby P. J.; Hahn S.; Hutchinson S.; Matchar D.; Silberstein S.; Smith T. R.; Purdy R. A.; Saiers J. Consensus statement: Cardiovascular safety profile of triptans (5-HT1B/1D agonists) in the acute treatment of migraine. Headache 2004, 44, 414–425. [DOI] [PubMed] [Google Scholar]
- van Rossum D.; Hanisch U.-K.; Quirion R. Neuroanatomical localization, pharmacological characterization and functions of CGRP, related peptides and their receptors. Neurosci. Biobehav. Rev. 1997, 21, 649–678. [DOI] [PubMed] [Google Scholar]
- Recober A.; Russo A. F. Calcitonin gene-related peptide: An update on the biology. Curr. Opin. Neurol. 2009, 22, 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham P. L. Inhibition of calcitonin gene-related peptide function: A promising strategy for treating migraine. Headache 2008, 48, 1269–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brain S. D.; Grant A. D. Vascular actions of calcitonin gene-related peptide and adrenomedullin. Physiol. Rev. 2004, 84, 903–934. [DOI] [PubMed] [Google Scholar]
- Petersen K. A.; Birk S.; Lassen L. H.; Kruuse C.; Jonassen O.; Lesko L.; Olesen J. The CGRP-antagonist, BIBN4096BS does not affect cerebral or systemic haemodynamics in healthy volunteers. Cephalalgia 2005, 25, 139–147. [DOI] [PubMed] [Google Scholar]
- Rudolf K.; Eberlein W.; Engel W.; Pieper H.; Entzeroth M.; Hallermayer G.; Doods H. Development of human calcitonin gene-related peptide (CGRP) receptor antagonists. 1. Potent and selective small molecule CGRP antagonists. 1-[N2-[3,5-Dibromo-N-[[4-(3,4-dihydro-2(1H)-oxoquinazolin-3-yl)-1-piperidinyl]carbonyl]-d-tyrosyl]-l-lysyl]-4-(4-pyridinyl)piperazine: the first CGRP antagonist for clinical trials in acute migraine. J. Med. Chem. 2005, 48, 5921–5931. [DOI] [PubMed] [Google Scholar]
- Olesen J.; Diener H.-C.; Husstedt I. W.; Goadsby P. J.; Hall D.; Meier U.; Pollentier S.; Lesko L. M. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N. Engl. J. Med. 2004, 350, 1104–1110. [DOI] [PubMed] [Google Scholar]
- Paone D. V.; Shaw A. W.; Nguyen D. N.; Burgey C. S.; Deng J. Z.; Kane S. A.; Koblan K. S.; Salvatore C. A.; Mosser S. D.; Johnston V. K.; Wong B. K.; Miller-Stein C. M.; Hershey J. C.; Graham S. L.; Vacca J. P.; Williams T. M. Potent, orally bioavailable calcitonin gene-related peptide receptor antagonists for the treatment of migraine: Discovery of N-[(3R,6S)-6-(2,3-difluorophenyl)-2-oxo-1-(2,2,2-trifluoroethyl)azepan-3-yl]-4-(2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridine-1-yl)piperidine-1-carboxamide (MK-0974). J. Med. Chem. 2007, 50, 5564–5567. [DOI] [PubMed] [Google Scholar]
- Ho T. W.; Ferrari M. D.; Dodick D. W.; Galet V.; Kost J.; Fan X.; Leibensperger H.; Froman S.; Assaid C.; Lines C.; Koppen H.; Winner P. K. Efficacy and tolerability of MK-0974 (telcagepant), a new oral antagonist of calcitonin gene-related peptide receptor, compared with zolmitriptan for acute migraine: A randomized, placebo-controlled, parallel-treatment trial. Lancet 2008, 372, 2115–2123. [DOI] [PubMed] [Google Scholar]
- Tepper S. J.; Stillman M. J. Clinical and preclinical rationale for CGRP-receptor antagonists in the treatment of migraine. Headache 2008, 48, 1259–1268. [DOI] [PubMed] [Google Scholar]
- Williams T. M.; Burgey C. S.; Salvatore C. A. Calcitonin gene-related peptide antagonists for the treatment of migraine. Prog. Med. Chem. 2009, 47, 1–35. [DOI] [PubMed] [Google Scholar]
- Bell I. M.; Bednar R. A.; Fay J. F.; Gallicchio S. N.; Hochman J. H.; McMasters D. R.; Miller-Stein C.; Moore E. L.; Mosser S. D.; Pudvah N. T.; Quigley A. G.; Salvatore C. A.; Stump C. A.; Theberge C. R.; Wong B. K.; Zartman C. B.; Zhang X.-F.; Kane S. A.; Graham S. L.; Vacca J. P.; Williams T. M. Identification of novel, orally bioavailable spirohydantoin CGRP receptor antagonists. Bioorg. Med. Chem. Lett. 2006, 16, 6165–6169. [DOI] [PubMed] [Google Scholar]
- Wood M. R.; Schirripa K. M.; Kim J. J.; Quigley A. G.; Stump C. A.; Bell I. M.; Bednar R. A.; Fay J. F.; Bruno J. G.; Moore E. L.; Mosser S. D.; Roller S.; Salvatore C. A.; Kane S. A.; Vacca J. P.; Selnick H. G. Novel CGRP receptor antagonists through a design strategy of target simplification with addition of molecular flexibility. Bioorg. Med. Chem. Lett. 2009, 19, 5787–5790. [DOI] [PubMed] [Google Scholar]
- Salvatore C. A.; Hershey J. C.; Corcoran H. A.; Fay J. F.; Johnston V. K.; Moore E. L.; Mosser S. D.; Burgey C. S.; Paone D. V.; Shaw A. W.; Graham S. L.; Vacca J. P.; Williams T. M.; Koblan K. S.; Kane S. A. Pharmacological characterization of MK-0974 [N-[(3R,6S)-6-(2,3-difluorophenyl)-2-oxo-1-(2,2,2-trifluoroethyl)azepan-3-yl]-4-(2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridine-1-yl)piperidine-1-carboxamide], a potent and orally active calcitonin gene-related peptide receptor antagonist for the treatment of migraine. J. Pharmacol. Exp. Ther. 2008, 324, 416–421. [DOI] [PubMed] [Google Scholar]
- Roller S.; Cui D.; Laspina C.; Miller-Stein C.; Rowe J.; Wong B.; Prueksaritanont T. Preclinical pharmacokinetics of MK-0974, an orally active calcitonin gene-related peptide (CGRP)-receptor antagonist, mechanism of dose dependency and species differences. Xenobiotica 2009, 39, 33–45. [DOI] [PubMed] [Google Scholar]
- Wood M. R.; Bell I. M.; Selnick H. G.; Stump C. A.; Gallicchio S. N.; Zartman C. B.. WO 2008/020902.
- Gallicchio S. N.; Bell I. M. Convenient synthesis of 1,3-substituted-6-phenylpiperazin-2-ones. Tetrahedron Lett. 2009, 50, 3817–3819. [Google Scholar]
- Stump C. A.; Bell I. M.; Bednar R. A.; Bruno J. G.; Fay J. F.; Gallicchio S. N.; Johnston V. K.; Moore E. L.; Mosser S. D.; Quigley A. G.; Salvatore C. A.; Theberge C. R.; Zartman C. B.; Zhang X.-F.; Kane S. A.; Graham S. L.; Vacca J. P.; Williams T. M. The discovery of highly potent CGRP receptor antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 214–217. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discovery Today: Technol. 2004, 1, 337–341. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H.-Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
- Clark D. E. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 1. Prediction of intestinal absorption. J. Pharm. Sci. 1999, 88, 807–814. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.