

Abstract

Amalgamation of the structure−activity relationship of two series of GlyT1 inhibitors developed at Merck led to the discovery of a clinical candidate, compound 16 (DCCCyB), which demonstrated excellent in vivo occupancy of GlyT1 transporters in rhesus monkey as determined by displacement of a PET tracer ligand.
Keywords: Inhibitor, GlyT1, structure−activity relationship, DCCCyB, PET tracer ligand
The hypofunction of N-methyl-d-aspartate (NMDA) receptors has been implicated in the pathophysiology of schizophrenia, with evidence coming from both preclinical models1−3 and the limited clinical data available.4−10 The latter includes the reversible psychosis induced in nonschizophrenics by NMDA antagonists such as phencyclidine (PCP), and the clinical efficacy observed when antipsychotic medication was supplemented with the obligatory NMDA coagonist glycine or with sarcosine, a weak endogenous inhibitor of type 1 glycine uptake transporters (GlyT1). Most importantly, this adjunctive therapy has been shown to give significant improvements in the negative and cognitive symptoms of stable schizophrenics, for which significant unmet medical need remains due to the lack of efficacy of conventional antipsychotics against these symptoms.6 In the brain, glycine levels are thought to be maintained tonically at submaximal concentrations in the synapse by GlyT1.11 This suggests that the pharmacological manipulation of synaptic glycine concentration using a GlyT1 inhibitor may be a viable method of potentiating NMDA receptor function in vivo, hence ameliorating the negative and cognitive symptoms of schizophrenia. To test this hypothesis, there has been an industry wide effort to identify potent and selective GlyT1 inhibitors.12−15
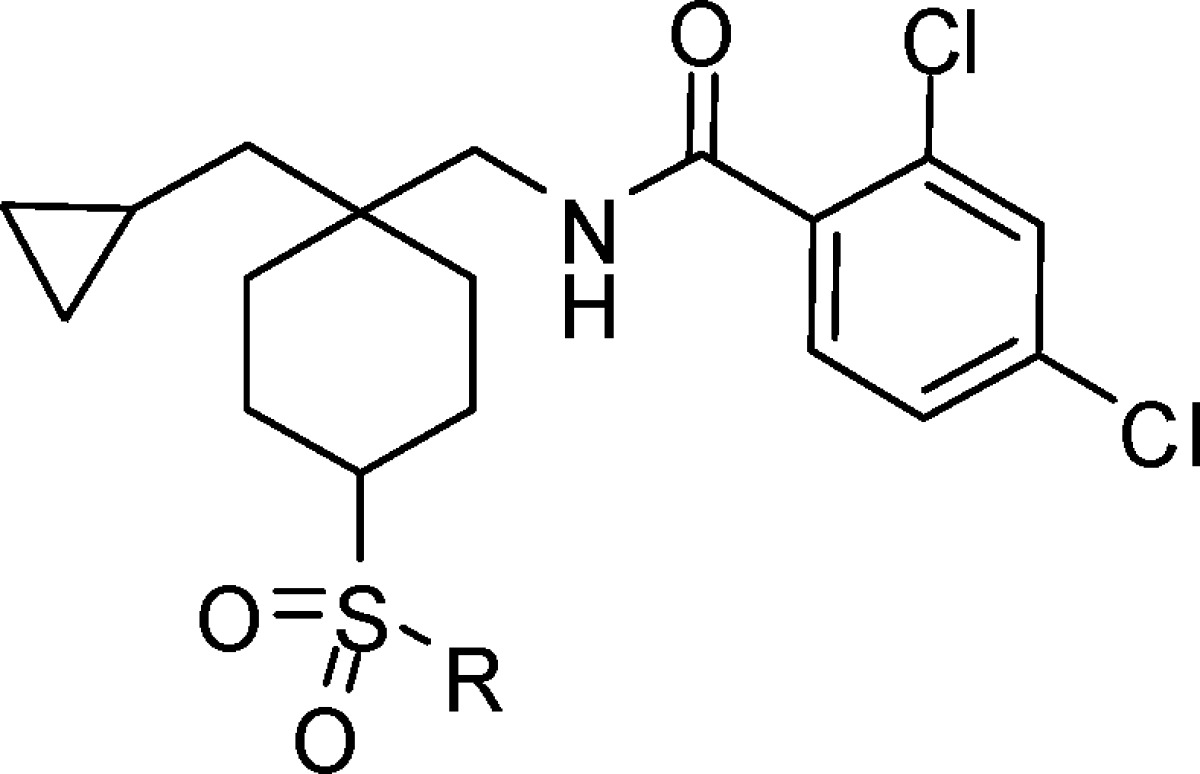
Recent communications from our laboratory have disclosed two related series of hGlyT1 inhibitors, typified by the piperidine sulfonamide 1(16) and cyclohexyl sulfone 2,17 which demonstrate excellent selectivity over the related GlyT2 and TauT transporters (Figure 1). Early issues for the piperidine series were poor oral bioavailability in rat and dog due to high plasma clearance, although this was resolved in the 2,4-dichlorophenyl analogue 1.
Figure 1.

Structures of hGlyT1 inhibitors.
However, compound 1 was shown to have unacceptably high covalent binding in vivo to both rat liver and plasma proteins after oral dosing at 20 mg/kg (6 h postdose: liver, 153 pmol equiv/mg protein; plasma, 214 pmol equiv/mg protein). Metabolite identification experiments using radiolabeled piperidine sulfonamide analogues, or trapping experiments with radiolabeled cyanide, led us to hypothesize that the observed covalent binding was due to oxidation of the piperidine ring leading to the formation of a reactive species. High levels of covalent binding have been linked to increased incidence of idiosyncratic toxicities of compounds in the clinic;18 therefore, further chemical optimization was undertaken in the sulfone series. The required cyclohexyl sulfone derivatives were accessed as delineated in Scheme 1.19 The cis and trans isomers of the key alcohol intermediate A were separated chromatographically and assigned based upon nuclear Overhauser effect NMR experiments.
Scheme 1.

Where the required thiol was readily available, formation of the mesylate of A followed by displacement gave the thioether. Alternatively, the thioether was accessed via Mitsunobu chemistry with thioacetate followed by a one-pot deprotection−alkylation protocol. Oxidation of the thioether with oxone gave the required final compound.
The inhibition at hGlyT1 transporters and microsomal turnover in rat and human microsomes for a selection of heterocyclic sulfone compounds is given in Table 1. Compound 2 exhibited excellent oral bioavailability in the rat and occupied GlyT1 transporters in vivo, as adjudged by our previously reported in vivo binding assay in the rat20 using a proprietary GlyT1 radiolabel, with an Occ50 of 3.4 mg/kg.17 Analysis of the plasma and brain drug levels required to achieve Occ50 (1.2 and 0.2 μM, respectively) revealed a low brain to plasma ratio of 0.16. A similarly low brain to plasma ratio of 0.1 was determined from a 10 mg/kg oral dose of compound 3. A subsequent study in mdrla +/+ and −/− mice determined the ratio between the brain:blood ratios of the −/− and +/+ animals to be 8.7, suggesting compound 3 to be a P-gp substrate.
Table 1. hGlyT1 Potency and Human and Rat Liver Microsomal Turnover of Selected Heterocyclic Sulfone Analogues.
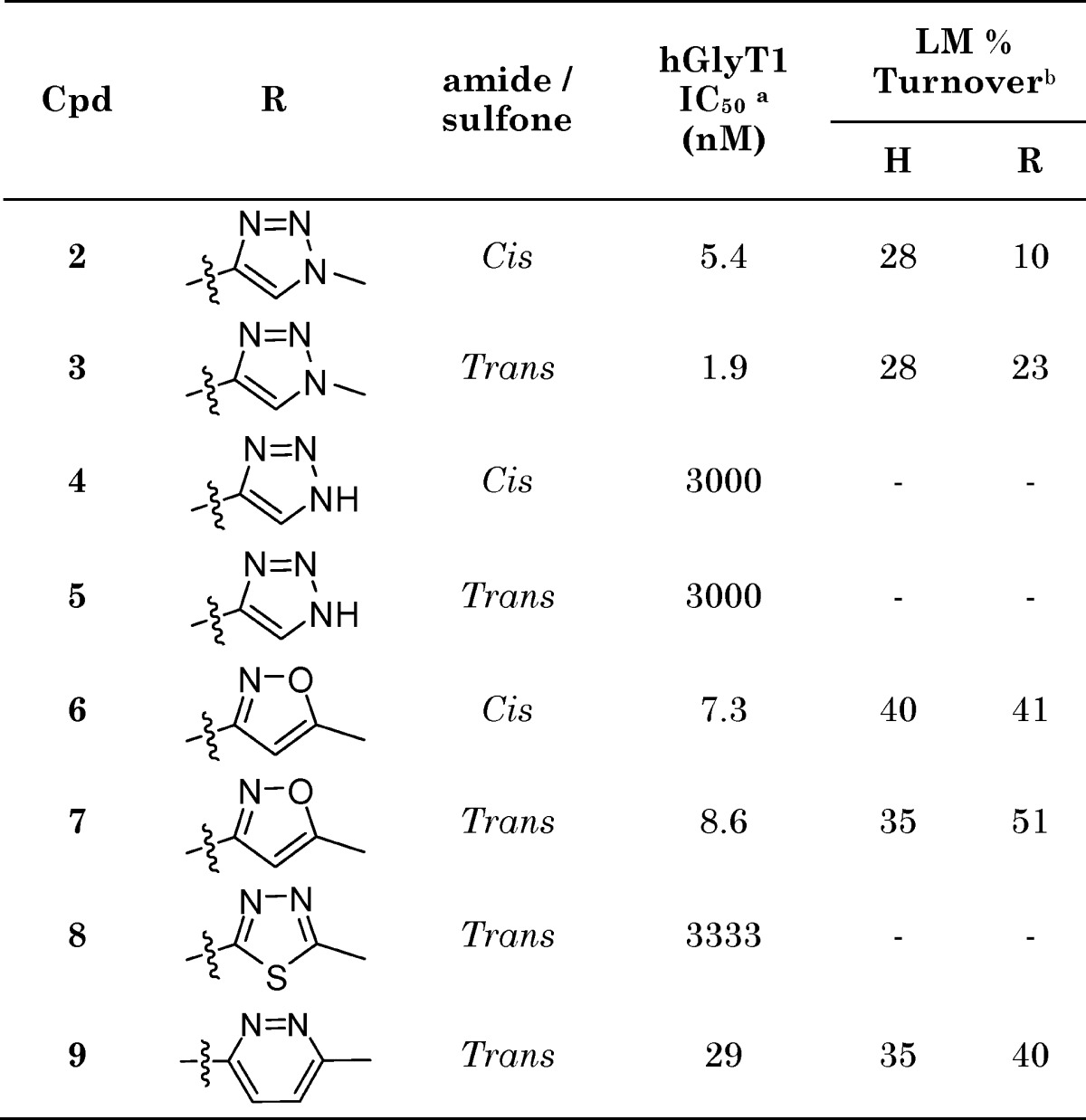 |
IC50 values are averages of at least two measurements. The hGlyT1a isoform was used for the assay.
Turnover of GlyT1 compounds (1 μM) in rat and human liver microsomes. All incubations were carried out at 37 °C for 15 min. Protein concentration = 0.5 mg/mL; cosolvent = 0.99% MeCN + 0.01% DMSO. For compounds in this series, LM % turnover ≤40 was considered acceptable for further compound progression.
Compound 2 did not inhibit common Cyp isoforms (2D6, 2C9, and 3A4: IC50 > 10 μM); however, the NH-triazole analogues 4 and 5, potential metabolites of compounds 2 and 3, respectively, proved to be extremely potent inhibitors of Cyp 2C9 (compound 5 Cyp 2C9: IC50 = 10 nM). The potent Cyp inhibition, in combination with the high plasma Occ50 due to the P-gp issue, precluded the further development of triazole analogues 2 and 3. Heterocyclic sulfone analogues in which the pendant alkyl group was linked through carbon exhibited either increased microsomal turnover (6 and 7) or reduced potency at hGlyT1 (8 and 9).
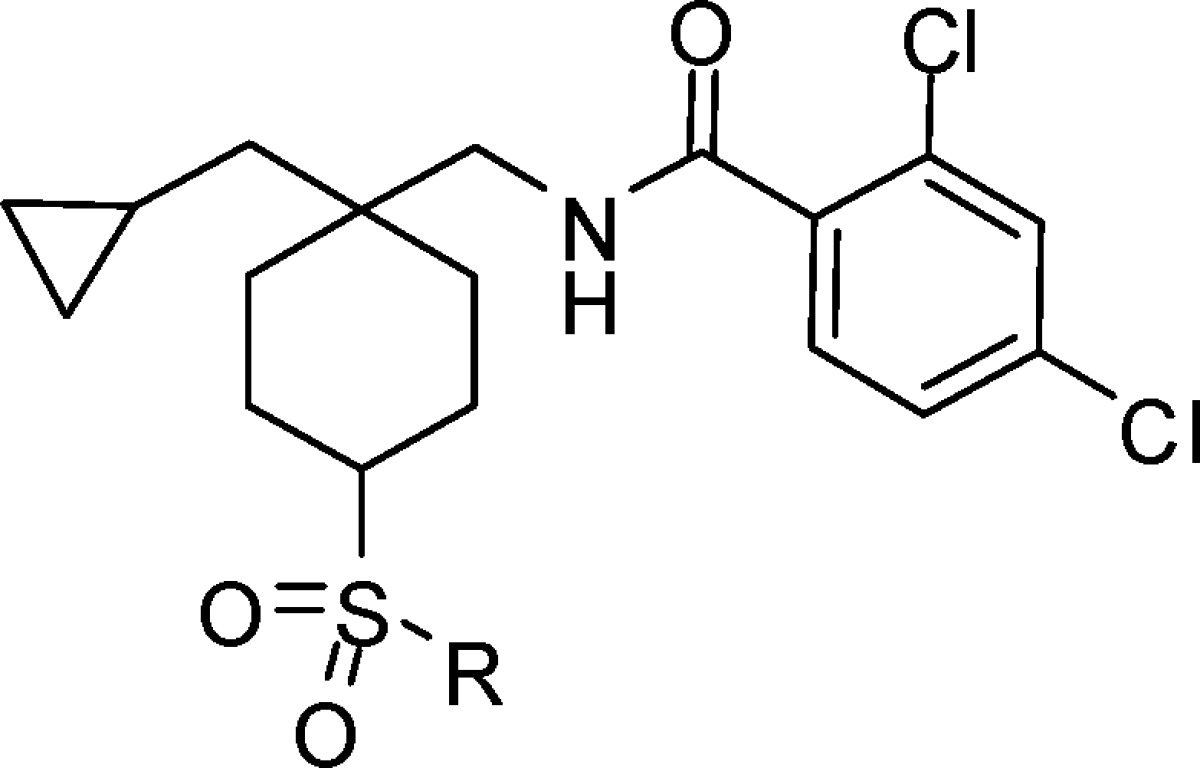
Investigation of simple alkyl sulfone derivatives related to compound 1 (Table 2) established that the structure−activity relationship (SAR) was reminiscent of the previously described 4-pyridyl piperidine series16 with a significant reduction in potency observed in the series propyl 10, ethyl 11, and methyl 12. In the alkyl sulfone series, a more stringent requirement for the cis relationship between the sulfone and the amide was observed than in the heterocyclic series, with compounds 10 and 11 demonstrating >10-fold greater potency relative to 13 and 14. Although the cyclobutylmethyl compound 15 demonstrated a loss in potency relative to propyl analogue 10, the cyclopropylmethyl compound 16 (DCCCyB) retained potency but with improved microsomal stability. Compound 16 demonstrated an acceptable level of in vivo covalent binding (<25 pmol equiv/mg after a 20 mg/kg oral dose) in the rat and was selected for further profiling.
Table 2. hGlyT1 Potency and Human and Rat Liver Microsomal Turnover of Selected Alkyl Sulfone Analogues.
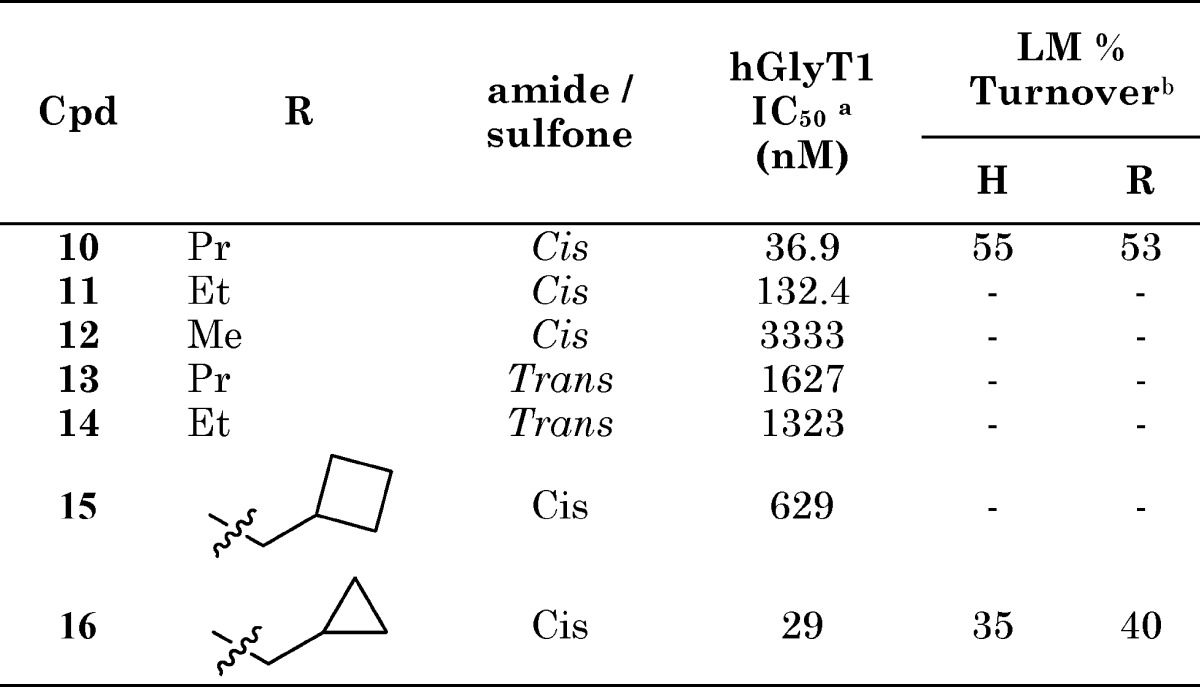 |
IC50 values are averages of at least two measurements. The hGlyT1a isoform was used for the assay.
Turnover of GlyT1 compounds (1 μM) in rat and human liver microsomes. All incubations were carried out at 37 °C for 15 min. Protein concentration = 0.5 mg/mL; cosolvent = 0.99% MeCN + 0.01% DMSO. For compounds in this series, LM % turnover ≤40 was considered acceptable for further compound progression.
The pharmacokinetic parameters of compound 16 in preclinical species are given in Table 3. Clearance is low in dogs and moderate in rats and rhesus monkey, and this combined with moderate Vd(ss) in all species gave acceptable half-life values. Oral bioavailability of 65 and 48% in rat and dog, respectively, was obtained using the 0.5% methocel suspension dosing vehicle.
Table 3. Pharmacokinetic Parameters of Compound 16 in Preclinical Species.
| rat | rhesus | dog | ||
|---|---|---|---|---|
| dose (iv and po) | mg/kg | 1.0 | 1.0 | 1.0 |
| Cl | mL/min/kg | 36 | 24 | 4.9 |
| Vd(ss) | L/kg | 4.1 | 2.3 | 3.1 |
| T1/2 | h | 2.4 | 1.5 | 10 |
| F | % | 65 | 2 | 48 |
| Cmax | μM | 0.14 | 0.04 | 1.39 |
| Tmax | h | 0.8 | 2.7 | 1.0 |
Compound 16 was not a substrate for human or mouse P-gp, had a significantly increased brain to plasma ratio of 2.3, and exhibited a lower plasma Occ50 of 0.35 μM in the rat GlyT1 in vivo binding assay as compared to compound 2. No significant off-target activity was observed for compound 16 in a broad ancillary pharmacology panel screen.
A GlyT1 inhibitor would be expected to lead to an increase in the levels of extracellular glycine in the brain. This has been demonstrated in the literature, and at Merck1 using proof of concept compounds, by in vivo dialysis through a probe inserted into the rat frontal cortex. Glycine levels were determined up to 4 h postdose with compound 16 at 20 and 3 mg/kg po. Both doses significantly elevated extracellular glycine levels above basal concentrations (mean % peak glycine efflux as a % basal ± SEM; 20 mg/kg = 184.0 ± 17.0%; 3 mg/kg = 151.0 ± 25.0%). The increase in glycine levels at the 3 mg/kg po dose of compound 16 is consistent with that observed for other GlyT1 inhibitors shown to be efficacious in animal models of schizophrenia.21
Impairments in executive function have long been considered to be a core feature of schizophrenic illness,22 with the attentional set shifting aspect of executive function commonly assessed in patients using the Wisconsin Card Sorting Test.23 The intra/extra dimensional (ED/ID) rodent model of executive function can be used, following impairment in perceptual attentional set shifting by PCP administration, as a model for the set shifting deficits observed in schizophrenic patients.24 In the rat ED/ID assay,25 compound 16 dosed at 3 mg/kg po reversed the PCP-induced cognitive deficit in the ED shift (Figure 2). Although all discriminations were performed, for clarity, only the ED data are shown as subchronic dosing of PCP induced a deficit exclusively in this aspect of the assay.
Figure 2.
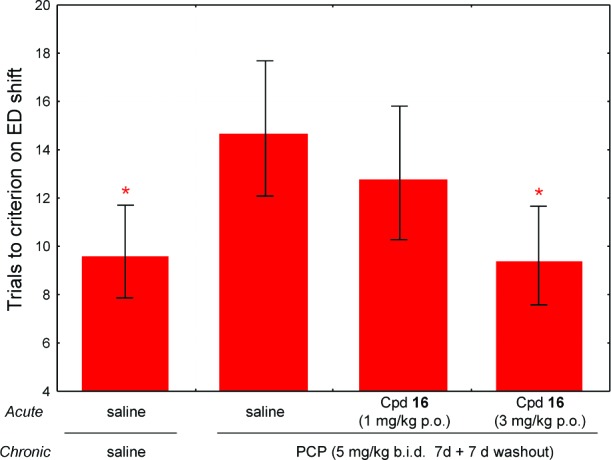
Trials to criterion for ED shift. Asterisk-marked columns denote significance at p < 0.05 vs PCP and vehicle for ED shift.
The availability of the GlyT1 PET tracer [18F]MK-657726 facilitated the evaluation of the plasma occupancy relationship for compound 16 in higher species. Figure 3 shows the time−activity curves and PET/MRI coregistered images from the PET scans.27 The differences in tracer uptake under baseline and blockade conditions can be used to determine GlyT1 occupancy at different doses/plasma concentrations. A plasma occupancy curve for 16 was generated giving rise to an estimated plasma Occ50 concentration of 120 nM in rhesus monkey, which is in good agreement with the plasma Occ50 concentration generated in rat.
Figure 3.
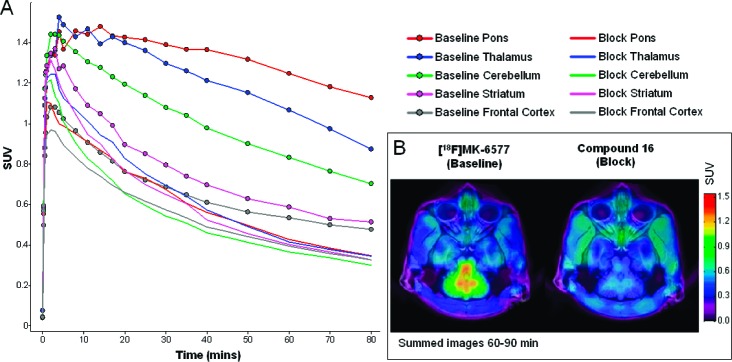
(A) Time−activity curves for baseline ([18F]MK-6577 treated; filled markers) and for blockade ([18F]MK-6577 and cpd 16 treated; no markers) conditions. (B) PET/MRI coregistered images in rhesus monkey brain under baseline (left) and blockade (right) conditions with cpd 16.
In summary, replacement of the piperidine ring of compound 1 generated a series of cyclohexyl sulfone inhibitors of hGlyT1 and removed the in vivo covalent binding observed with the former series. Reoptimization of the sulfone moiety removed the P-gp liability observed for the heterocyclic sulfone derivatives and led to the identification of compound 16. The clinical evaluation of compound 16 will be published in the near future.
Acknowledgments
W.B. thanks Scott Wolkenberg for his help in the preparation of this manuscript.
Supporting Information Available
Experimental procedures for the preparation of compound 16 including analytical and spectral characterization data (1H and 13C NMR, HR-MS, and HPLC). This material is available free of charge via the Internet at http://pubs.acs.org.
Department of Medicinal Chemistry, BioFocus, Chesterford Research Park, CB10 1Xl, United Kingdom.
Supplementary Material
References
- Lindsley C. W.; Zhao Z.; Leister W. H.; O'Brien J.; Lemaire W.; Williams D. L. Jr.; Chen T.-B.; Chang R. S. L.; Burno M.; Jacobson M. A.; Sur C.; Kinney G. G.; Pettibone D. J.; Tiller P. R.; Smith S.; Tsou N. N.; Duggan M. E.; Conn P. J.; Hartman G. D. Design, synthesis, and in vivo efficacy of glycine transporter-1 (GlyT1) inhibitors derived from a series of [4-phenyl-1-(propylsulfonyl)piperidin-4-yl]methyl benzamides. ChemMedChem 2006, 1, 807–811. [DOI] [PubMed] [Google Scholar]
- Bridges T. M.; Williams R.; Lindsley C. W. Design of potent GlyT1 inhibitors: in vitro and in vivo profiles. Curr. Opin. Mol. Ther. 2008, 10, 591–601. [PubMed] [Google Scholar]
- Black M. D.; Varty G. B.; Arad M.; Barak S.; De Levie A.; Boulay D.; Pichat P.; Griebel G.; Weiner I. Procognitive and antipsychotic efficacy of glycine transport 1 inhibitors (GlyT1) in acute and neurodevelopmental models of schizophrenia: latent inhibition studies in the rat. Psychopharmacology 2009, 202, 385–396. [DOI] [PubMed] [Google Scholar]
- Olney J. W.; Newcomer J. W.; Farber N. B. J. NMDA receptor hypofunction model of schizophrenia. Psychiatry Res. 1999, 33, 523–533. [DOI] [PubMed] [Google Scholar]
- Javitt D. C.; Zylberman I.; Zukin S. R.; Heresco-Levy U.; Lindenmayer J. P. Amelioration of negative symptoms in schizophrenia by glycine. Am. J. Psychiatry 1994, 151, 1234–1236. [DOI] [PubMed] [Google Scholar]
- Tsai G.; Lane H. Y.; Yang P.; Chong M. Y.; Lange N. Glycine transporter I inhibitor, N-methylglycine (sarcosine), added to antipsychotics for the treatment of schizophrenia. Biol. Psychiatry 2004, 55, 452–456. [DOI] [PubMed] [Google Scholar]
- Heresco-Levy U. N-Methyl-D-aspartate (NMDA) receptor-based treatment approaches in schizophrenia: The first decade. Int. J. Neuropsychopharmacol. 2000, 3, 243–258. [DOI] [PubMed] [Google Scholar]
- Jentsch J. D.; Roth R. H. The neuropsychopharmacology of phencyclidine: From NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 1999, 20, 201–225. [DOI] [PubMed] [Google Scholar]
- Leiderman E.; Zylberman I.; Zukin S. R.; Cooper T. B.; Javitt D. C. Preliminary investigation of high-dose oral glycine on serum levels and negative symptoms in schizophrenia: An open-label trial. Biol. Psychiatry 1996, 39, 213–215. [DOI] [PubMed] [Google Scholar]
- The recent report of positive phase II results obtained with GlyT1 inhibitor RG1678 in treating the negative symptoms and the personal and social functioning of patients with schizophrenia lends further weight to this hypothesis.
- Zafra F.; Gomeza J.; Olivares L.; Aragon C.; Gimenez C. Regional distribution and developmental variation of the glycine transporters GlyT1 and GlyT2 in the rat CNS. Eur. J. Neurosci. 1995, 7, 1342–1352. [DOI] [PubMed] [Google Scholar]
- Lowe J. A. III. Novel inhibitors of the type 1 transporter for glycine (GlyT1) as antipsychotic agents. Expert Opin. Ther. Pat. 2005, 15, 1657–1662. [Google Scholar]
- Lowe J. A. III; DeNinno S. L.; Drozda S. E.; Schmidt C. J.; Ward K. M.; David T., F.; Sanner M.; Tunucci D.; Valentine J. An octahydro-cyclopenta[c]pyrrole series of inhibitors of the type 1 glycine transporter. Bioorg. Med. Chem. Lett. 2010, 20, 907–911. [DOI] [PubMed] [Google Scholar]
- Smith G.; Mikkelsen G.; Eskildsen J.; Bundgaard C. The synthesis and SAR of 2-arylsulfanylphenyl-1-oxyalkylamino acids as GlyT1 inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 3981–3984. [DOI] [PubMed] [Google Scholar]
- Jolidon S.; Alberati D.; Dowle A.; Fischer H.; Hainzl D.; Narquizian R.; Norcross R.; Pinard E. Design, synthesis and structure-activity relationship of simple bis-amides as potent inhibitors of GlyT1. Bioorg. Med. Chem. Lett. 2008, 18, 5533–5536. [DOI] [PubMed] [Google Scholar]
- Wolkenberg S. E.; Zhao Z.; Wisnoski D. D.; Leister W. H.; O'Brien J.; Lemaire W.; Williams D. L. Jr.; Jacobson M. A.; Sur C.; Kinney G. G.; Pettibone D. J.; Tiller P. R.; Smith S.; Gibson C.; Ma B. K.; Polsky-Fisher S. L.; Lindsley C. W.; Hartman G. D. Discovery of GlyT1 inhibitors with improved pharmacokinetic properties. Bioorg. Med. Chem. Lett. 2009, 19, 1492–1495. [DOI] [PubMed] [Google Scholar]
- Thomson J. L.; Blackaby W. P.; Jennings A. S. R.; Goodacre S. C.; Pike A.; Thomas S.; Brown T. A.; Smith A.; Pillai G.; Street L. J.; Lewis R. T. Optimisation of a series of potent, selective and orally bioavailable GlyT1 inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 2235–2239. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Kassahun K.; Tschirret-Guth R. A.; Mitra K.; Baillie T. A. Minimizing metabolic activation during pharmaceutical lead optimization: progress, knowledge gaps and future directions. Curr. Opin. Drug Discovery Des. 2008, 11, 43–52. [PubMed] [Google Scholar]
- Blackaby W. P.; Castro Pineiro J. L.; Lewis R. T.; Naylor E. M.; Street L. J.. WO2006/131711, 2006.
- Zeng Z.; O'Brien J. A.; Lemaire W.; O'Malley S. S.; Miller P. J.; Zhao Z.; Wallace M. A.; Raab C.; Lindsley C. W.; Sur C.; Williams D. L. Jr. A novel radioligand for glycine transporter 1: Characterization and use in autoradiographic and in vivo brain occupancy studies. Nucl. Med. Biol. 2008, 35, 315–325. [DOI] [PubMed] [Google Scholar]
- Depoortere R.; Dargazanli G.; Estenne-Bouhtou G.; Coste A.; Lanneau C.; Desvignes C.; Poncelet M.; Heaulme M.; Santucci V.; Decobert M.; Cudennec A.; Voltz C.; Boulay D.; Terranova J. P.; Stemmelin J.; Roger P.; Marabout B.; Sevrin M.; Vige X.; Biton B.; Steinberg R.; Francon D.; Alonso R.; Avenet P.; Oury-Donat F.; Perrault G.; Griebel G.; George P.; Soubrie P.; Scatton B. Neurochemical, Electrophysiological and Pharmacological Profiles of the Selective Inhibitor of the Glycine Transporter-1 SSR504734, a Potential New Type of Antipsychotic. Neuropsychopharmacology 2005, 30, 1963–1985. [DOI] [PubMed] [Google Scholar]
- Sullivan E. V.; Shear P. K.; Zipursky R. B.; Sagar H. J.; Pfefferbaum A. A deficit profile of executive, memory, and motor functions in schizophrenia. Biol. Psychiatry 1994, 36, 641–653. [DOI] [PubMed] [Google Scholar]
- Perry W.; Heaton R. K.; Potterat E.; Roebuck T.; Minassian A.; Braff D. L. Working memory in schizophrenia: Transient “online” storage versus executive functioning. Schizophr. Bull. 2001, 27, 157–176. [DOI] [PubMed] [Google Scholar]
- Egerton A.; Reid L.; McKerchar C. E.; Morris B. J.; Pratt J. A. Impairment in perceptual attentional set-shifting following PCP administration: A rodent model of set-shifting deficits in schizophrenia. Psychopharmacology 2005, 179, 77–84. [DOI] [PubMed] [Google Scholar]
- Goetghebeur P; Dias R. Comparison of haloperidol, risperidone, sertindole, and modafinil to reverse an attentional set-shifting impairment following subchronic PCP administration in the rat—A back translational study. Psychopharmacology 2009, 202, 287–293. [DOI] [PubMed] [Google Scholar]
- Hamill T. G.; Eng W.; Jennings A. S. R.; Lewis R. T.; Thomas S.; Wood S.; Street L. J.; Wisnoski D. D.; Wolkenberg S. E.; Lindsley C. W.; Sanabria-Bohorquez S. M.; Patel S.; Ryan C.; Cook J.; Sur C.; Burns H. D.; Hargreaves R.. The Synthesis and Preclinical Evaluation in Rhesus Monkey of [18F]MK-6577 and [11C]CMPyPB, Glycine Transporter 1 (GlyT1) PET Radiotracers Synapse, in press. [DOI] [PubMed] [Google Scholar]
- PET Imaging Studies: All studies were conducted under the guiding principles of the American Physiological Society and the Guide for the Care and Use for Laboratory Animals published by the Institute of Laboratory Animal Resources, National Research Council (1996), and were approved by the West Point Institutional Animal Care and Use Committee at Merck Research Laboratories. Rhesus monkeys (∼10 kg) were initially anesthetized with ketamine (10 mg/kg i.m.) and then induced with propofol (5 mg/kg iv), intubated, and respired with medical grade air. Anaesthesia was maintained with propofol (0.4 mg/kg/min) for the duration of the study. PET scans were performed on an ECAT EXACT HR+. This scanner acquires 63 planes of data over a 15.5 cm axial field of view, thus allowing the whole brain to be imaged. Emission data were acquired in 3D (retracted septa) mode; transmission data (for subsequent attenuation correction) were acquired in 2D mode before injection of the radiopharmaceutical. Dynamic emission scans were performed following injection of ∼5 mCi of the PET tracer. The scans were initiated at the time of tracer injection. The emission scans were corrected for attenuation, scatter, and dead time and reconstructed with a ramp filter, resulting in transverse and axial spatial resolution of approximately 5 mm at FWHM. For blockade studies of [18F]MK-6577, 16 was injected (10:40:50 EtOH:H2O:PEG400) as an iv bolus plus infusion. The tracer was administered 1 h after the start of the infusion. PET Regions of Interest: For each scan, a static (or summed) PET image was obtained by summing the dynamic frames acquired during the acquisition. Regions of interest (ROIs) were drawn on the summed images in the striatum (caudate and putamen), thalamus, cortical regions (mainly occipital cortex), and white matter. Then, ROIs were projected into the dynamic scans to obtain the corresponding time−activity curves (TACs). TACs were expressed in standard uptake value (SUV) units using the monkey body weight and the tracer injected dose as:
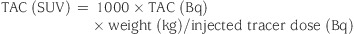
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.