

Abstract
Targeting allosteric protein sites is a promising approach to interfere selectively with cellular signaling cascades. We have discovered a novel class of allosteric insulin-like growth factor-I receptor (IGF-1R) inhibitors. 3-Cyano-1H-indole-7-carboxylic acid {1-[4-(5-cyano-1H-indol-3-yl)butyl]piperidin-4-yl}amide (10) was found with nanomolar biochemical, micromolar, cellular IGF-1R activity and no relevant interference with cellular insulin receptor signaling up to 30 μM. The allosteric binding site was characterized by X-ray crystallographic studies, and the structural information was used to explain the unique mode of action of this new class of inhibitors.
Keywords: IGF-1R, kinase inhibitor, allosteric inhibition, indole-butyl-amine
Long-term medical indications require long-term drug treatments with a well-tolerated toxicity profile. This has been understood for the treatments of conditions such as high blood pressure and is now also realized even in life-threatening diseases, such as cancer.1 Selectivity and specificity of the corresponding drugs are therefore essential. Kinases are involved in numerous crucial cell-signaling cascades and have been implicated in oncogenesis and tumor progression through their aberrant regulation or overexpression. Many drugs on the market or in clinical development inhibit kinases.2 Kinase inhibition by ATP competition (type I) is associated with selectivity hurdles due to the high sequence conservation in the ATP-binding pocket within the kinome. However, the conformational variability of kinases allows for allosteric inhibition by targeting non-ATP sites (type II/III). Such allosteric binding sites are less frequent in protein kinase structures through the kinome, offering a very promising approach to achieve the selectivity objective.3
There are only a few published reports of kinase inhibitors that are non-ATP and/or substrate competitive. After the initial identification of SB-203580, a specific inhibitor of the serine/threonine kinase p38 MAPK (mitogen-activated protein kinase),4 which interacts with amino acid residues outside of the ATP binding pocket, additional compounds for p38,5 PLK1,6 GSK-3,7 Akt,8 and MEK9 were reported, but only for a few of them could the allosteric interaction be demonstrated experimentally.
The insulin-like growth factor-I receptor (IGF-1R) is a member of the receptor tyrosine kinase family.10 The peptides IGF-1 and IGF-2 are the cognate activating ligands of IGF-1R.11 Several lines of epidemiological and mechanistic evidence link IGF-1R activation and signaling to tumor biology.12 Recent reports of Pfizer's IGF-1R antibody CP-751871 (currently in clinical trials) support the hypothesis that IGF-1R inhibition has a beneficial effect on the disease progression of cancer patients and might provide a proof of concept for this target.13−16
The insulin receptor (IR) is a related receptor tyrosine kinase and shares a high sequence identity of 84% in the tyrosine kinase domain with IGF-1R.17 This homology makes it very difficult to target one of the receptors selectively and means a special challenge for IGF-1R inhibitor design.18 In this context, it is interesting that the overall folding of the catalytic domains of protein kinases is very similar in the fully activated form, while striking differences exist among the various inactive conformations of different kinase subfamilies.19,20
This is also true for IGF-1R and IR, which have been structurally characterized in the unactivated, nonphosphorylated (0P), and fully activated, triply phosphorylated (3P) forms.21,22 Overall, the structures of unactivated IGFR-0P closely resemble those of the IR-0P, which is also true for the fully active forms IGFR-3P and IR-3P. However, the unactivated and activated forms are structurally very distinct from each other, especially regarding the positions of the activation loop and αC helix.
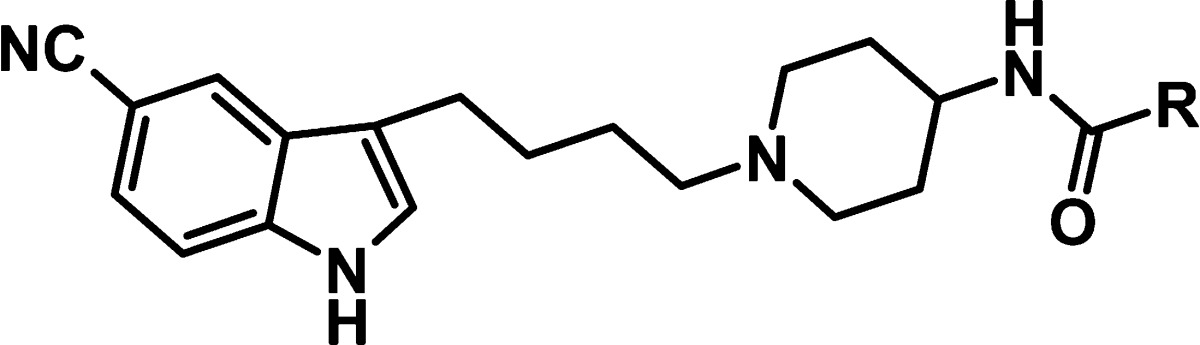
In a high-throughput screening (HTS) campaign of the Merck Serono compound collection against IGF-1R, compound 1 was found to inhibit the enzyme (Figure 1). Although the inhibitory activity with an IC50 of 10 μM was only moderate, we decided to further characterize this hit. Subsequent cellular testing in a phospho-IGF-1R enzyme-linked immunosorbent assay (ELISA) revealed an IC50 value of 18 μM for 1.
Figure 1.

Structure of HTS hit 1.
Structures from this indolealkylamine class are known to bind to G-protein-coupled receptors in the central nervous system (CNS) with nanomolar affinity. They have been previously shown to be very selective binders of the serotonin receptor subtype 1A and the serotonin reuptake transporter,23 but it was not known before that, in addition to their CNS activity, some of these compounds also inhibit protein kinases such as IGF-1R.
The chemical structure of the screening hit 1 does not obviously represent a typical hinge-binding motif.24 Therefore, we started with structure−activity relationship (SAR) expansion around this hit (Table 1).
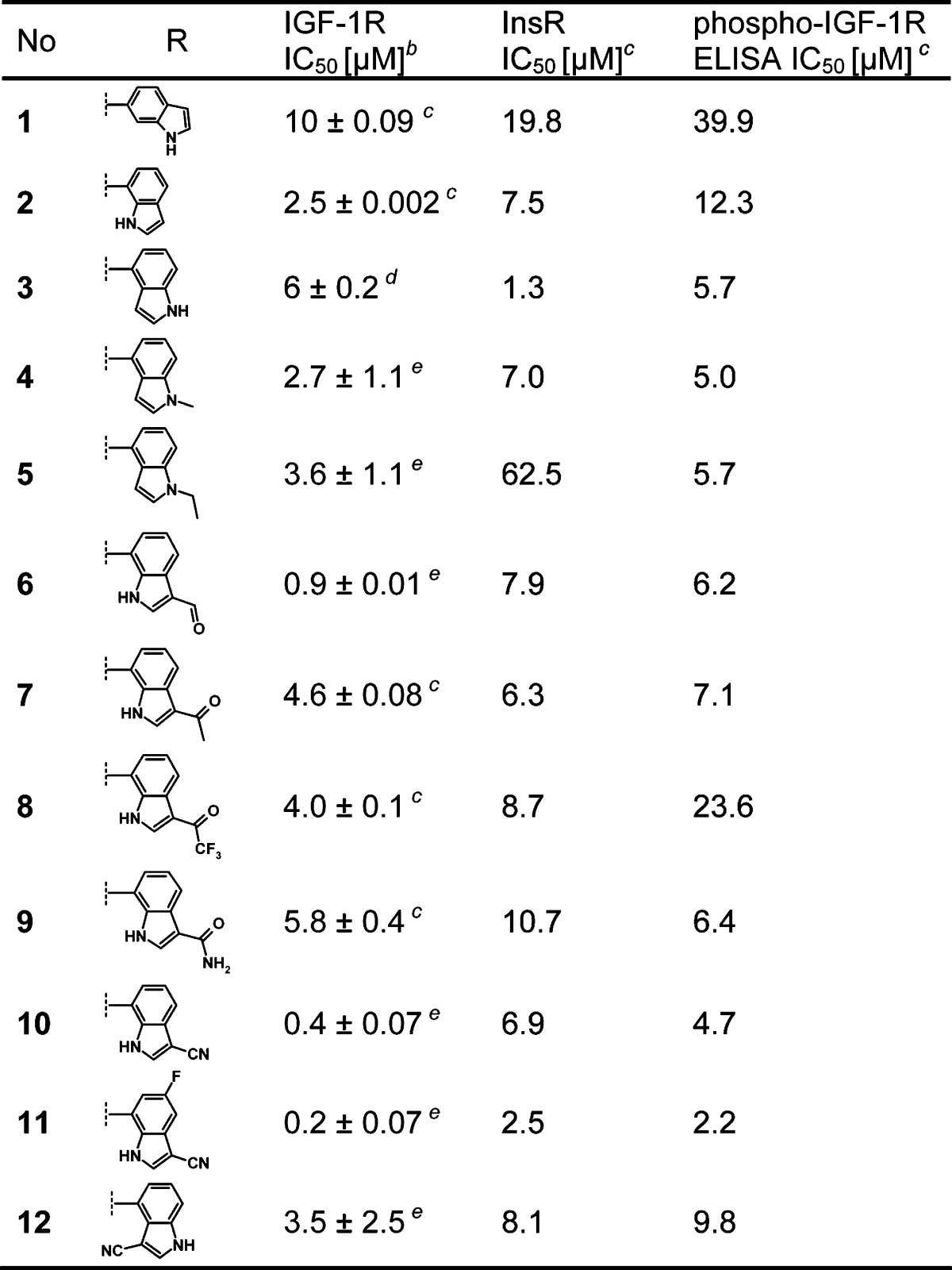
Table 1. Biochemical and Cellular Activity of Indole-butyl-aminesa.
 |
Biochemical assays for the SAR determination were performed using Caliper LifeSciences electrophoretic mobility shift technology. IGF-1R (0.1 ng/μL, BPS BioScience, CA) or InsR (0.02 ng/μL, Carna Biosciences, Kobe, Japan) was incubated with a dose titration of inhibitor (final 1% DMSO), 1 μM substrate peptide (FITC-KKSRGDYMTMQIG-NH2), and ATP at the respective Kms of 15 and 50 μM ATP, for 90 min at 25 °C.
Mean ± standard deviation; n = 2,c 3,d and 4.e
Compounds 1−12 were prepared as previously described.23 The synthesis of building block 15 starts with the acylation of 5-cyano-indole 13 with chloro butyric acid chloride, and afterward, the carbonyl carbon is completely reduced with a complex aluminum hydride reagent to 14. The chlorine is substituted with 4-(BOC amino)piperidine under basic conditions, the protecting group is cleaved with acid, and the free amine 15 is subjected to standard amide coupling conditions to give the target compounds (Scheme 1).
Scheme 1. Synthesis of Compounds 1−12.

Conditions: (a) (i) DCM, AlCl3, 4-chlorobutyric acid chloride; (ii) THF, sodium bis(2-methoxyethoxy)aluminum hydride. (b) (i) 4-(tert-Butoxycarbonyl amino) piperidine, DIPEA, AcCN; (ii) 1 N HCl, dioxane. (c) THF, NMM, HOBt, EDCI, corresponding indole acid.25
A significant potency gain on IGF-1R by a factor of 4 was observed by shifting the amide bond attachment on the indole ring from position 6 (1, IC50 = 10 μM) into position 7 (2, IC50 = 2.5 μM). In line with this trend, although less pronounced, was the 4-indolyl derivative 3 (IC50 = 6.0 μM), indicating a certain influence of the angular attachment of the amide to the indole ring. Hence, we focused on 4- and 7-yl indoles bearing electron-withdrawing and -donating substituents to further explore the SAR. As summarized in Table 1, slight modifications have a dramatic impact on the compounds' potency. The attachment of electron-donating substituents such as methyl (4, IC50 = 2.7 μM) and ethyl (5, IC50 = 3.6 μM) on the 4-indolyl nitrogen resulted in a distinct reduction of inhibition as compared to the unsubstituted indole 3. The decrease in potency was also observed in a subseries of 7-indolyl derivatives with carbonyl substitution in the 3-position. In comparison with the unsubstituted derivative 2, a 2−3-fold drop of potency was observed for the methyl (7, IC50 = 4.6 μM) and the trifluoromethyl (8, IC50 = 4.0 μM) ketons as well as for the amide 9, while the less voluminous aldehyde (6, IC50 = 0.9 μM) was slightly more potent. The trend that straight electron-withdrawing substitution in position 3 of the indole increases biochemical activity was confirmed with the 3-cyano derivatives 10 (IC50 = 0.4 μM) and 11 (IC50 = 0.2 μM). Both compounds showed a submicromolar IC50 and were up to 50-fold more potent than starting point 1. In addition to the electronic properties, also steric aspects of indolyl substituents had an influence on IGF-1R inhibition. This was demonstrated with the 3-cyano indol-4-yl isomer 12 (IC50 = 3.5 μM), which is by a factor of 18 less active than the most potent 3-cyano-5-fluoro-indol-7-yl molecule (11).
Nevertheless, the successful biochemical optimization of screening hit 1 prompted us to analyze the series in more detail. Compounds from Table 1 were tested in the capture ELISA assay to assess their inhibitory activity on IGF-1-induced IGF-1R phosphorylation in MCF-7 breast cancer cells. These IGF-1R inhibitors had moderate cellular activity with IC50 values ranging from 2.2 up to 40 μM.
The IGF-1R inhibitors 1−12 were also tested in recombinant enzyme and cellular phospho-insulin receptor kinase (InsR) assays to assess their activity in this context. In the recombinant enzyme assay, all of the compounds (except one) show a higher activity on IGF-1R by factors ranging from 1.4 (7) to 23 (10). The exception is the unsubstituted 4-indolyl derivative 3 (which shows a higher activity on the InsR by a factor of 6). The cellular assays highlight the IGF-1R selectivity of the compounds. In comparison to the activities against the IGF-1R, all of the inhibitors demonstrated cellular InsR IC50 values greater than 30 μM.26 The most potent derivatives 10 and 11 showed a difference in activity against the IGF-1R and InsR by factors of 6 and 14, respectively.
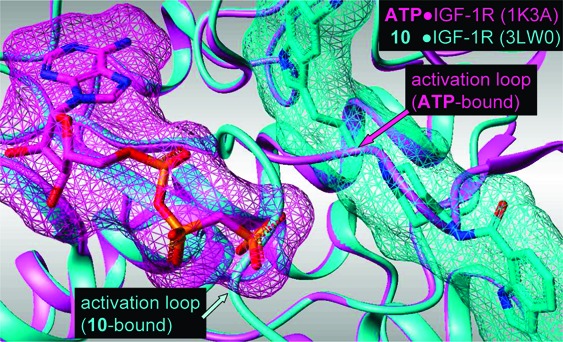
To further characterize the binding mode of these compounds, 10 was cocrystallized with the kinase domain of human IGF-1R. The crystal structure was solved at 1.8 Å and shows the typical bilobal kinase fold with the helical C-terminal lobe and the N-terminal lobe comprised of 5-stranded β-sheets and the αC helix. On the basis of published data, a nonphosphorylated IGF-1R construct containing two mutations (E1097A and E1099A) was used.27 There are four molecules in the asymmetric unit with slightly different overall conformations. Minor differences exist mainly regarding the opening of N- and C-terminal lobes, which correlates with the conformation of the flexible activation loop. Because of this flexibility, some parts of the activation loop are disordered. The binding position of the ligand is clearly defined in the electron density map except for the 3-cyano indolyl carboxamide substituent, which has been included in a most reasonable conformation in the final model, but with occupancy set to 0. In contrast to other published IGF-1R inhibitors,28,2910 did not bind in the ATP binding site but to an adjacent binding pocket next to the DFG motif and the activation loop (type III binding). The 5-cyano indole ring is sandwiched between the side chains of Met 1054 and Met 1079 and recognized via an H-bond between the indole NH and the main chain carbonyl oxygen atom of Val 1063 (Figure 2).
Figure 2.
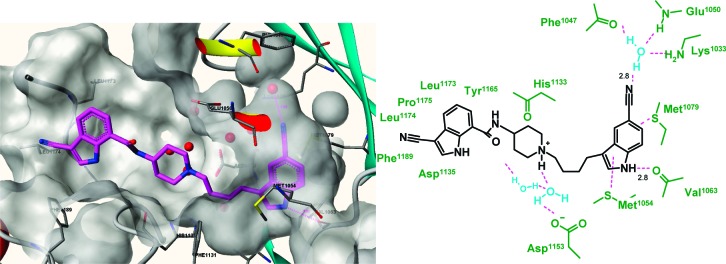
X-ray structure of 10 in IGF-1R and schematic description of the interactions of 10 with the allosteric IGF-1R binding site.
In addition, the 5-nitrile group is found in the H-bond distance to a water molecule located in the interface of the side chains of Lys 1033 and Glu 1050 and the backbone carbonyl of Phe 1047. These results are in clear agreement with the activity of further screening hits regarding the importance of the indole NH donor functionality and the role of the nitrile.30 The n-butyl chain forms van der Waals interactions to the side chains of Met 1054, His 1133, Ile 1151, and Phe 1131 on one side. On the opposite site, the n-butyl chain is in contact with a vast cluster of water molecules. Two water molecules of this cluster mediate H-bonds between the piperidine N-atom and the side chain of Asp 1153. Apart from van der Waals interactions to His 1133, the piperidine ring is mainly surrounded by the aforementioned cluster of water molecules. As previously mentioned, the attached second 3-cyano indole ring is not defined in the electron density map, reflected by a position already exposed to the protein surface. However, several polar (Arg 1134 and Asp 1135) and lipophilic amino acids (Leu 1174, Pro 1175, Phe 1189, and Tyr 1165) are in the vicinity of this distal indole ring, indicating a degree of influence of this region on inhibitor binding as seen in the SAR.
A superimposition of the crystal structures of IGF-1R in complex with 10 and the apo structure of the unphosphorylated form (PDB code 1P40) revealed significant conformational changes in the activation loop, especially regarding Phe 1154 from the DFG motif (“Phe-out”), but only moderate changes in the relative positions of the GC loop and αC helix (Figure 3A). This “Phe-out” conformation is similar to the corresponding Phe 1151 in the apo structure of the unphosphorylated InsR (Figure 3B). However, the GC loop and αC helix are significantly repositioned as observed before for the IGF-1R-0P and InsR-0P apo structures.21 The amino acids directly interacting with 10 in IGF-1R are conserved in InsR, but the binding site of the indole ring of 10 is partially blocked by Met 1051 and Met 1076. These conformational differences result in altered shapes of the allosteric pockets, which might explain the observed selectivity of 1−12 against InsR on a cellular screening level. These detailed insights into the binding mode of 10 to IGF-1R are helpful to explain the results of our synthetic work and give hints about the mode of action of these allosteric IGF-1R inhibitors.
Figure 3.

(A) Comparison of the X-ray structures of 10 in IGF-1R (magenta) with the apo structure of unphosphorylated IGF-1R (dark gray) showed an induced fit of the activation loop including Phe 1154 but only slight changes regarding the GC loop and αC helix. (B) The position of Phe 1154 in the X-ray structures of 10 in IGF-1R (magenta) corresponds to that in InsR (green), but significant conformational differences are observed regarding the GC loop and αC helix. The binding site for the indole ring of 10 is blocked by Met 1051 and Met 1076.
In conclusion, the structural analysis of hit 10 (MSC1609119A-1) has revealed an allosteric binding mode, and contributing amino acid contacts were identified. The most potent biochemical IGF-1R compound 11 is by a factor of 50 more potent than the original screening hit 1. The compounds have cellular activity in the phospho-IGF-1R ELISA assay and, as indicated, do not influence IR signaling significantly up to concentrations of 30 μM. These hits and their binding mode to IGF-1R could serve as a starting point to generate even more pronounced selectivity against InsR.
Acknowledgments
We thank Proteros, Munich, for the preparation of the IGF-1R X-ray cocrystal structure, Uwe Eckert and Nicole Lange for the preparation of the compounds, Irina Bersch for the performance of the cellular assays, and Ben Askew for valuable discussions.
Supporting Information Available
Representative experimental procedures, melting points, and NMR data for all new test compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The coordinates of the cocrystal structure of IGF1-R in complex with 10 have been deposited with the RCSB Protein Data Bank under the accession code 3LW0.
Supplementary Material
References
- Ciardiello F.; Tortora G. Anti-epidermal growth factor receptor drugs in cancer therapy. Expert Opin. Invest. Drugs 2002, 11, 755–768. [DOI] [PubMed] [Google Scholar]
- Renhowe P. A. Inhibitors of growth factor receptor kinase-dependent signaling pathways in anticancer chemotherapy—Clinical progress. Curr. Opin. Drug Discovery Dev. 2002, 5, 214–224. [PubMed] [Google Scholar]
- Kiselyov A.; Balakin K. V.; Tkachenko S. E.; Savchuk N. P. Recent progress in development of non-ATP competitive small-molecule inhibitors of protein kinases. Mini-Rev. Med. Chem. 2006, 6, 711–717. [DOI] [PubMed] [Google Scholar]
- Dumas J.; Sibley R.; Riedl B.; Monahan M. K.; Lee W.; Lowinger T. B.; Redman A. M.; Johnson J. S.; Kingery-Wood J.; Scott W. J.; Smith R. A.; Bobko M.; Schoenleber R.; Ranges G. E.; Housley T. J.; Bhargava A.; Wilhelm S. M.; Shrikhande A. Discovery of a new class of p38 kinase inhibitors. Bioorg. Med. Chem. Lett. 2000, 10, 2047–2050. [DOI] [PubMed] [Google Scholar]
- Yong H.-Y.; Koh M.-S.; Moon A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin. Invest. Drugs 2009, 18, 1893–1905. [DOI] [PubMed] [Google Scholar]
- Gumireddy K.; Reddy M.V. R.; Cosenza S. C.; Boominathan R.; Baker S. J.; Papathi N.; Jiang J.; Holland J.; Reddy E. P. ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell 2005, 7, 275–286. [DOI] [PubMed] [Google Scholar]
- Martinez A.; Alonso M.; Castro A.; Pérez C.; Moreno F. J. First non-ATP competitive glycogen synthase kinase 3 β (GSK-3β) inhibitors: Thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer's disease. J. Med. Chem. 2002, 45, 1292–1299. [DOI] [PubMed] [Google Scholar]
- Ghayad S. E; Cohen P. A. Inhibitors of the PI3K/ Akt/mTOR pathway: New hope for breast cancer patients. Recent Pat. Anti-Cancer Drug Discovery 2010, 5, 29–57. [DOI] [PubMed] [Google Scholar]
- Austin D.; Shivaani K. Targeting mitogen-activated protein kinase kinase (MEK) in solid tumors. Targeted Oncol. 2009, 4, 267–273. [DOI] [PubMed] [Google Scholar]
- Denley A.; Cosgrove L. J.; Booker G. W.; Wallace J. C.; Forbes B. E. Molecular interactions of the IGF system. Cytokine Growth Factor Rev. 2005, 16, 421–439. [DOI] [PubMed] [Google Scholar]
- Frasca F.; Pandini G.; Scalia P.; Sciacca L.; Mineo R.; Costantino A.; Goldfine I. D.; Belfiore A.; Vigneri R. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol. 1999, 19, 3278–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Sun Y. Insulin-like growth factor receptor-1 as an anti-cancer target: Blocking transformation and inducing apoptosis. Curr. Cancer Drug Targets 2002, 2, 191–207. [DOI] [PubMed] [Google Scholar]
- Pollak M. N.; Eisenberg P.; Karp D.; Cohen R.; Kreisman H.; Adjei A.; Langer C.; Melvin C.; Yin D.; Gualberto A.. Safety and tolerability of the anti-IGF-I receptor antibody CP-751,871 in combination with paclitaxel and carboplatin in patients with advanced solid tumors AACR Annual Meeting 2007, 98th; April 17; Absract LB-343.
- Kusada Y.; Fujita-Yamaguchi Y. An overview of currently available anti- insulin-like growth factor I receptor antibodies. BioSci. Trends 2007, 1, 128–133. [PubMed] [Google Scholar]
- Atzori F.; Traina T. A.; Ionta M. T.; Massidda B. Targeting insulin-like growth factor type 1 receptor in cancer therapy. Targeted Oncol. 2009, 4, 255–266. [DOI] [PubMed] [Google Scholar]
- Weroha S. J.; Haluska P. IGF-1 receptor inhibitors in clinical trials-early lessons. J. Mammary Gland Biol. Neoplasia 2008, 13, 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich A.; Gray A.; Tam A. W.; Yang-Feng T.; Tsubokawa M.; Collins C.; Henzel W.; Le Bon T.; Kathuria S.; Chen E.; Jacobs S.; Francke U.; Ramachandran J.; Fujita-Yamaguchi Y. Insulin-like growth factor I receptor primary structure: Comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986, 5, 2503–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IR inhibition would induce diabetes in treated patients sooner or later, what is not compatible with the strategy to deal with cancer as with blood preasure in a live long medication context.
- Schindler T.; Bornmann W.; Pellicena P.; Miller W. T.; Clarkson B.; Kuriyan J. Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science 2000, 289, 1938–1942. [DOI] [PubMed] [Google Scholar]
- Engh R. A.; Bossemeyer D. The protein kinase activity modulation sites: Mechanisms for cellular regulation—Targets for therapeutic intervention. Adv. Enzyme Regul. 2001, 41, 121–149. [DOI] [PubMed] [Google Scholar]
- Munshi S.; Kornienko M.; Hall D. L.; Reid J. C.; Waxman L.; Stirdivant S. M.; Darke P. L.; Kuo L. C. Crystal structure of the apo, unactivated insulin-like growth factor-1 receptor kinase. Implication for inhibitor specificity. J. Biol. Chem. 2002, 277, 38797–38802. [DOI] [PubMed] [Google Scholar]
- Li W.; Favelyukis S.; Yang J.; Zeng Y.; Yu J.; Gangjee A.; Miller W. T. Inhibition of insulin-like growth factor I receptor autophosphorylation by novel 6−5 ring-fused compounds. Biochem. Pharmacol. 2004, 68, 145–154. [DOI] [PubMed] [Google Scholar]
- Heinrich T.; Boettcher H.; Gericke R.; Bartoszyk G. D.; Anzali S.; Seyfried C. A.; Greiner H. E.; van Amsterdam C. Synthesis and structure−activity relationship in a class of indolebutylpiperazines as dual 5-HT1A receptor agonists and serotonin reuptake inhibitors. J. Med. Chem. 2004, 47, 4684–4692. [DOI] [PubMed] [Google Scholar]
- A lot of different derivatives with modified indole cores and butyl chains were prepared. Unfortunately, all of these derivatizations did not result in any improvement, so we decided not to show these compounds explicitly.
- Crassier H.; Boettcher H.; Eckert U.; Bathe A.; Emmert S.. Procedure for the production of (3-cyano-1H-indol-7-yl)-[4-(4-fluorophenylethyl)piperazin-1-yl]methanone and its salts. DE 10102944, 2002.
- See the Supporting Information.
- Munshi S.; Hall D. L.; Kornienko M.; Darke P. L.; Kuo L. C. Structure of apo, unactivated insulin-like growth factor-1 receptor kinase at 1.5 Å resolution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2003, D59, 1725–1730. [DOI] [PubMed] [Google Scholar]
- Wittman M. D.; Balasubramanian B.; Stoffan K.; Velaparthi U.; Liu P.; Krishnanathan S.; Carboni J.; Li A.; Greer A.; Attar R.; Gottardis M.; Chang C.; Jacobson B.; Sun Y.; Hansel S.; Zoeckler M.; Vyas D. M. Novel 1H-(benzimidazol-2-yl)-1H-pyridin-2-one inhibitors of insulin-like growth factor I (IGF-1R) kinase. Bioorg. Med. Chem. Lett. 2007, 17, 974–977. [DOI] [PubMed] [Google Scholar]
- Warshamana-Greene G. S.; Litz J.; Buchdunger E.; Garcia-Echeverria C.; Hofmann F.; Krystal G. W. The insulin-like growth factor-I receptor kinase inhibitor, NVP-ADW742, sensitizes small cell lung cancer cell lines to the effects of chemotherapy. Clin. Cancer Res. 2005, 11, 1563–1571. [DOI] [PubMed] [Google Scholar]
- Unpublished results.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.